
Abstract
Background
Genes associated with the neurodevelopmental disorder microcephaly display a strong signature of adaptive evolution in primates. Comparative data suggest a link between selection on some of these loci and the evolution of primate brain size. Whether or not either positive selection or this phenotypic association are unique to primates is unclear, but recent studies in cetaceans suggest at least two microcephaly genes evolved adaptively in other large brained mammalian clades.
Results
Here we analyse the evolution of seven microcephaly loci, including three recently identified loci, across 33 eutherian mammals. We find extensive evidence for positive selection having acted on the majority of these loci not just in primates but also across non-primate mammals. Furthermore, the patterns of selection in major mammalian clades are not significantly different. Using phylogenetically corrected comparative analyses, we find that the evolution of two microcephaly loci, ASPM and CDK5RAP2, are correlated with neonatal brain size in Glires and Euungulata, the two most densely sampled non-primate clades.
Conclusions
Together with previous results, this suggests that ASPM and CDK5RAP2 may have had a consistent role in the evolution of brain size in mammals. Nevertheless, several limitations of currently available data and gene-phenotype tests are discussed, including sparse sampling across large evolutionary distances, averaging gene-wide rates of evolution, potential phenotypic variation and evolutionary reversals. We discuss the implications of our results for studies of the genetic basis of brain evolution, and explicit tests of gene-phenotype hypotheses.
Similar content being viewed by others

Background
For over a decade researchers interested in the genetic basis of brain evolution have sought clues in the molecular evolution of genes associated with the neurodevelopmental disorder, primary microcephaly [1–8]. Microcephaly is a congenital disorder characterized by an early cessation of brain growth, specifically affecting cortical development. Mendelian inheritance of microcephaly has now been linked to deleterious mutations at seven unlinked loci [8–15]. These loci encode proteins with central roles in neurogenesis, largely in the formation and function of the centrioles, which in turn control the way in which neural progenitor cells divide [16–18]. Disruption of these crucial functions causes microcephaly, and it is hypothesized that modification of their function through evolutionary time could tip the cell fate switch towards greater neurogenic output, underpinning the evolution of larger brains.
Much of the early focus on the molecular evolution of microcephaly genes centered on the role of two loci (ASPM and MCPH1) in human evolution [19, 20]. Early studies suggested an increase in the rate at which these genes evolved along the lineage leading to humans [1]. Intriguingly, as more microcephaly loci were identified each has been shown to evolve adaptively [5, 6] suggesting they may be a persistent target of positive selection. In addition, as more species were incorporated into analyses the signature of positive selection extended beyond humans first to great apes [4], then to all anthropoid primates [7, 8]. Similarly, the potential phenotypic relevance of this selection was extended from a role in the rapid expansion of human brain size [1–3] to a more widespread, conserved role in primate brain evolution [7, 8]. This shift has been accompanied by more rigorous hypothesis testing on larger datasets [7, 8, 21]. For example, it has previously been found that the molecular evolution of ASPM and CDK5RAP2 co-evolves with brain mass, particularly neonatal brain mass [8]. The association between ASPM evolution and brain mass is particularly interesting as it is found in primate clades which experienced both increases and decreases in brain mass [8, 21]. Elsewhere, tentative evidence has been found linking the evolution of MCPH1 to sexual dimorphism in brain mass in primates [22], a surprising finding supported, in part, by human population studies of sex-specific associations between SNPs in microcephaly genes and brain size [23, 24] and functional analyses of base pair substitutions that may interact with sex-specific developmental pathways [25].
Other studies have extended the taxonomic scope beyond primates to test the hypothesis that microcephaly genes may contribute to the evolution of brain size in other mammals. Notably, so far these studies have been limited to species with relatively large brains. In cetaceans, both MCPH1 and ASPM have been shown to have evolved under positive selection [26, 27]. However, evidence linking selection on either locus to brain size in cetaceans is lacking [26, 28]. In addition, exclusively studying large brained clades inevitably leads to an ascertainment bias. Indeed, there is evidence that both of these genes evolved under positive selection across placental mammals [26, 28]. If this is the case, there are clear implications for our understanding of brain evolution. First, if there is an evolutionary link between microcephaly loci and changes in neurogenesis, such pervasive selection may suggest a conserved genetic basis to some aspects of mammalian brain size evolution. Second, the diversity of mammalian brain sizes could provide a good comparative framework in which to test for gene-phenotype co-evolution. Finally, if microcephaly genes do have a conserved evolutionary role in brain size, or any other phenotype, the results of functional assays within and between more practically tractable species than primates, such as rodents, may generalize to other mammals.
In this study, we examine patterns of molecular evolution in seven microcephaly genes across placental mammals, including the first comprehensive interspecific analysis of three of these loci, STIL, CEP152 and WDR62, and the first mammal-wide analysis of two further loci, CDK5RAP2 and CENPJ. We perform tests for adaptive evolution within primates, across non-primate mammals and across the combined data to test how pervasive the signature of positive selection is. We further test whether the pattern of evolution of these genes differ in primates compared to non-primates, and finally provide a preliminary assessment of the link between microcephaly genes and brain size in non-primate clades. We find a signature of positive selection that is strong and widespread for all but one of the loci and identify intriguing evidence of an association between two loci and neonatal brain mass in two non-primate clades. Greater sampling both at the molecular and phenotypic level are necessary to perform robust tests of gene-phenotype associations to confirm this hypothesis, but this preliminary extension of the role of microcephaly genes in brain evolution beyond primates may have a number of implications.
Results
Tests for positive selection
Full coding sequences for seven loci (ASPM, CDK5RAP2, CENPJ, CEP152, MCPH1, STIL and WDR62) were obtained for 12 primates and 21 non-primate eutherian mammals (Figure 1a). Two site model tests were applied to these datasets, the M1a/M2a and M8a/M8 pairs implemented in PAML [29]. These allow the ω (estimate of dN/dS) to vary among sites but not across lineages [30, 31].

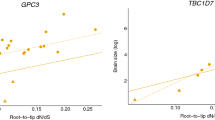
Species included in tests of adaptive evolution and gene-phenotype associations. A) Unrooted phylogeny from Meredith et al. (2011) used in the PAML analyses. P = Primates, E = Euarchontoglires, L = Laurasiatheria, X + A = Xenarthra + Afrotheria. The bracketed clades refer to Glires (B) and Euungulata (C). Panels B) and C) show the relationship between root-to-tip dN/dS and neonatal brain sizeg in Glires and Euungulata respectively, for the two genes with the most consistent pattern across the two groups, ASPM (red circles) and CDK5RAP2 (blue triangles). Regression lines are derived from the PGLS analysis and account for phylogenetic non-independence between the datapoints.
Across primates
Across the 12 primate species for which full coding sequences are available for all 7 loci, four loci - ASPM, CDK5RAP2, CENPJ and CEP152 - show a consistent signature of positive selection (Table 1a). Of the three that do not, one of these, MCPH1, has previously been shown to have evolved under positive selection across a larger dataset of partial coding sequence in anthropoid primates [8] suggesting that the lack of significance may be due to the small sample size. We therefore expanded the anthropoid dataset to include 18 species by amplifying selected exons of the remaining two loci (STIL, WDR62), targeting regions with high dN/dS based on a sliding window analysis of the full coding sequence (Additional file 1: Figure S1). Consistent with an effect of sample size these additional data yield evidence for positive selection acting on WDR62 (M1a/M2a Likelihood ratio (LR) = 11.921, p = 0.003; M8a/M8 LR = 11.903, p = 0.001). The results for STIL are inconsistent between tests (M1a/M2a LR = 3.940, p = 0.139; M8a/M8 LR = 4.688, p = 0.030).
Across mammals
Consistent evidence for positive selection is found across non-primate mammals under both site model tests for 5 loci - ASPM, CDK5RAP2, MCPH1, CENPJ and CEP152 (Table 1b). When the site model tests are repeated after incorporating the 12 primate sequences extensive positive selection is again found, with the M8a/M8 test for STIL also becoming narrowly significant (Table 1c). Among the loci which experienced positive selection across mammals the proportion of sites targeted by selection varies from ~2% to ~10% with dN/dS estimates for these sites typically between 1.5 and 2.5.
Diversifying selection on microcephaly loci across mammals
We next examined patterns of divergent selection in different mammalian clades using branch and clade models. Branch models allow dN/dS to vary across branches in the phylogeny but not across sites [32, 33], whereas clade models allow a proportion of sites to undergo divergent selection pressures in two or more clades defined a priori[34]. Comparisons of the gene-wide average dN/dS between primates and non-primate mammals are significant for all microcephaly loci except WDR62 (Additional file 2: Table S2a). Whilst this may suggest a greater influence of positive selection in primates, the clade models do not support this general conclusion. Here, only two loci, CDK5RAP2 and WDR62, are supported as having a proportion of sites under significantly different selective regimes between primates and other mammals (Additional file 2: Table S2b). It therefore seems likely that the significant branch model results reflect technical issues, such as branch length, or neutral effects such as differences in population size or life history [35–37]. The possibility of divergent selective pressure was next explored on a wider taxonomic scale comparing three major eutherian clades: the Euarchontoglires (n = 20), the Laurasiatheria (n = 9), and the Afrotheria + Xenarthra (n = 4) (see Figure 1a for these clades). Under the branch models there is little evidence for divergent selective regimes between these three clades and clade model tests are only significant for two loci, ASPM and MCPH1 (Additional file 2: Table S2c). The overarching pattern of selection on microcephaly loci is therefore a consistent signature of positive selection across all eutherian mammals, perhaps with the exception of STIL, but with relatively little evidence of clade-specific differences in overall rates of evolution.
Linking molecular and phenotypic evolution
Understanding the phenotypic relevance of this widespread positive selection is clearly of major interest. Given the long held hypothesis that selection on microcephaly genes in primates is linked to brain expansion [1] and the comparative evidence linking rates of evolution of ASPM and CDK5RAP2 to variation in brain mass across anthropoid primates [8, 21], a particular interest is the possibility that microcephaly genes may have a conserved role in mammalian brain evolution [26, 27, 38]. One approach to test a link between molecular evolution of candidate genes and brain size has been to compare pairs of species that differ in brain size (e.g. [39]). In our phylogeny three episodes of brain expansion are relatively well documented in the fossil record and form taxon-pairs with smaller brain species; the expansion of human [40, 41], dolphin [42, 43] and elephant brain sizes [44]. Branch models were used to test for significant differences in dN/dS between the bottlenose dolphin and the cow (divergence c. 50 my), between the elephant and the hyrax (divergence c. 80 mya), and between humans and the bushbaby (chosen to provide a similar branch length, divergence c. 80 mya). Only two loci showed significant differences between elephant and hyrax (Additional file 2: Table S3a), of which only one, MCPH1, had a higher dN/dS in the elephant lineage. Comparing the dolphin and cow branches no loci showed significant differences, though CDK5RAP2 approached significance and had a higher dN/dS on the dolphin branch. Finally, only CEP152 showed a significantly higher dN/dS along the human lineage compared to the bushbaby lineage. Hence, there is no consistent pattern of acceleration of microcephaly evolution along lineages leading to selected large brain species, compared to their smaller brained sister-lineage as represented in this dataset. Again, as these branches are relatively long dN/dS estimates are likely influenced by variation in life history and population size.
An alternative test of a gene-phenotype link is to take a more quantitative approach and test for co-evolution between molecular rates and the trait of interest. We tested for associations between root-to-tip dN/dS and two traits: neonatal and adult brain mass. Comparisons between any associations with these traits are informative as we expect genes involved in the evolution of neurogenesis to show a stronger relationship with neonatal brain mass as mammalian neurogenesis is predominantly prenatal [45–47]. We applied this test to three sub-clades within the mammalian phylogeny; anthropoid primates (n = 8), the focus of previous studies, and two additional grand-orders [48], the Glires (Rodentia + Lagomorpha, n = 5) and Euungulata (Perissodactyla + Cetartiodactyla, n = 5) chosen to reflect a trade-off between a relative consistency in life history parameters, the number of species and sample density. Within primates we found no evidence for an association with any of the seven loci (Additional file 2: Table S3b). Using larger datasets of partial coding sequence the evolution of ASPM and CDK5RAP2 have been linked to brain size [8, 21]. We repeated the tests using only the exons sequenced for previous studies [8] and again found no association suggesting sample size, which was much lower in the current study, and the phenotypic diversity within the dataset contribute to the difference in results between the present analysis and previous studies.
Within the Glires and Euungulata datasets there is an intriguing pattern suggesting a link between brain size and selection on some microcephaly loci. Although based on small samples sizes, these results provide the first evidence of a microcephaly gene-phenotype association outside primates. In Glires only ASPM is significantly associated with neonatal brain mass (t3 = 2.624, p = 0.039), whilst CDK5RAP2 (t3 = 2.235, p = 0.056) and STIL (t3 = 2.192, p = 0.058) show non-significant trends. In all three cases the strength of the association is reduced, or lost, with adult brain mass (Additional file 2: Table S3). Within Euungulata ASPM (t3 = 3.639, p = 0.018), CDK5RAP2 (t3 = 2.859, p = 0.032) and WDR62 (t3 = 2.824, p = 0.033) show significant associations with neonatal brain size and again the significance falls when adult brain size is considered (Additional file 2: Table S3b). In contrast STIL shows a significant association with adult brain size (t3 = 3.785 p = 0.016), which is reduced to a non-significant trend when neonatal brain size is considered (t3 = 1.911, p = 0.076).
Discussion
Our results indicate that the majority of loci linked to microcephaly, a severe neurodevelopmental disorder, have been targeted by positive selection throughout the evolution of eutherian mammals, in both primates and non-primates. Given the large evolutionary time under consideration it is remarkable that such a consistent pattern should be found on a functionally related set of genes that share a key role in neural development. Only STIL shows a weak or inconsistent signature of adaptive evolution. Given the paucity of brain expressed coding-genes with high rates of evolution identified in the majority of genome scans (e.g. [49–52] but see [53]) this raises the intriguing possibility that microcephaly genes are hotspots for positive selection among brain expressed coding genes. Whether this is true or not will require a further examination of genome wide patterns of selection across a greater number of species, as no study has included comparable numbers of species. A recent exome-wide analysis across 7 species of primates, to our knowledge the largest to date, did not report any enrichment for brain-expressed genes or neurodevelopmental processes among positively selected genes [54], nor did a study of six mammalian genomes [52]. However, these sample sizes are towards the lower limit at which site-based models have power to detect positive selection [55]. To fully assess whether microcephaly genes are targeted by positive selection more frequently than other neurodevelopmental genes it will be necessary to perform genome-wide analyses of the selective regimes acting on mammalian protein coding genes with much larger sample sizes than previous studies.
The frequent targeting of microcephaly genes by selection also raises important questions about the functional effects of substitutions in these loci. The majority of the microcephaly genes contribute to the development and function of the spindle poles, or astral microtubule network [56–58], and disruption of this function is linked to changes in spindle or microtubule behavior [59, 60]. The spindle poles play a key role in the cell fate switch of neural progenitor cells. In the developing brain a pool of neural progenitor cells undergo successive symmetric and proliferative divisions, their number increasingly exponentially [61]. After a certain number of divisions these cells begin to divide asymmetrically, with each division contributing a neuron to a radial column of cortical neurons before terminally dividing into two neurons [61, 62]. This switch between proliferative, symmetric divisions to asymmetric neurogenic divisions is controlled by the angle of cell division, which is in turn controlled by the spindle poles [59, 61]. Hence, functional changes in microcephaly genes could conceivably alter the duration of symmetric divisions to ultimately change the number of neurons produced during brain development.
A key role for modification of this cell fate switch is consistent with evo-devo models of brain expansion [45, 62–64]. The Radial Unit Hypothesis suggests a general mechanism for rapidly increasing brain size in mammals is to prolong the period of symmetric divisions, resulting in more radial units of neurons and an expanded volume [45, 63]. Microcephaly genes have precisely the functions the Radial Unit Hypothesis would predict as being targeted during episodes of brain expansion. Notably, another candidate gene, NIN, which functions in the maintenance of asymmetric divisions of neural progenitor cells [65], and has not been linked to microcephaly, also shows a signature of adaptive molecular evolution across anthropoid primates [66]. Maintaining this asymmetric cell division would result in larger numbers of neurons/radial unit and comparative data suggest an association between selection at this locus and interspecific variation in the number of neurons/unit area in the cortex, a suggested proxy for the number of neurons per radial unit [66]. Both cell fate switches highlighted by the Radial Unit Hypothesis therefore involve proteins that were targeted by positive selection across long periods of evolutionary time.
Although there is some variation in cell developmental pathways leading to neuron production among mammals, such as the emergence of additional progenitor cells associated with increased gyrification [67], much of the developmental program is conserved [61, 62, 68, 69]. Comparative data across mammals also suggest the timing of brain development is conserved [70] indicating strong constraints act on brain development, limiting the potential ways in which selection can modify brain size and structure. These constraints may be due to pleiotropic effects of shared developmental pathways or to adaptive, functional co-evolution [70, 71] but regardless, provide little reason to suspect genes targeted by selection in relation to primate brain evolution should differ from those targeted in non-primate mammals. Such convergence, or parallelism, in the genetic basis of mammalian phenotypes may be more widespread than perhaps expected, with examples including sensory perception [72], energy metabolism [39], digestive enzymes [73], immunity genes [74] and coloration [75].
The results of our phylogenetic comparative analyses provide direct evidence that evolution of brain size is indeed linked to four microcephaly genes (ASPM, CDKRAP2, STIL, WDR62) in two mammalian clades, Glires and Euungulata. Furthermore, as predicted by the neurodevelopmental models above, the relationship is generally stronger for neonatal brain size, a time point by which most neurons have already arisen [46, 47], than adult brain size. These results raise the possibility that ASPM and CDK5RAP2 play a consistent role in mammalian brain evolution, as these two loci have previously been implicated in brain evolution in primates [8, 21] (the failure to obtain a positive relationship for these two genes in primates here is most likely related to the sample size).
However, we stress that the sample sizes are small, the significance of our gene-phenotype tests are all >0.01, and it will be necessary to confirm and further explore these results with larger datasets. Regardless, the key implication of our analysis is that diversifying evolutionary studies of brain size beyond enigmatic, large brained clades may offer new avenues for testing evolutionary or functional hypotheses. It is likely however, that testing such gene-phenotype hypotheses over large evolutionary distances will be a challenging endeavor for a number of reasons [28]. First, effects of non-adaptive processes such as variation in life history or population size may affect dN/dS[35–37, 76] introducing noise to any genuine gene-phenotype association making larger, more densely sampled datasets desirable. Similarly, variation in phenotypic structure, such as neuron density, which may affect the linearity of the relationship between brain mass and neuron number [77, 78], may impact upon gene-phenotype comparisons across large evolutionary distances. Careful consideration is therefore needed as to what phenotype is most relevant, and molecular studies should target species where phenotypic data is available, when these data are a limiting factor.
Second, evolutionary reversals may be common in some mammalian clades and this may obscure gene-phenotype associations. For example, if a gene is targeted by selection during both increases and decreases in brain size there could be a mismatch between high rates of evolution at the molecular level and a small perceived difference in brain size. Understanding the evolutionary history of a phenotype then becomes a key component of the search for that phenotype’s molecular basis. An example of this comes from callitrichids, a subfamily of New World Monkeys that experienced a decrease in brain mass [41] and gyrencephaly [79, 80] in association with major episodes of phyletic dwarfism [81, 82]. Callitrichids appear as outliers to the positive association between brain mass and dN/dS for ASPM across anthropoids [8], but when considered alone show a negative association between brain mass and dN/dS for ASPM suggesting functional changes in ASPM may have contributed to decreases in brain mass in this clade [21]. This is also a potential explanation for the lack of an evolutionary association between ASPM and brain mass in cetaceans, where reversals are more common [26–28]. A third problem could be encountered if a gene is associated with a phenotype but has an intermittent role in its evolution. A potential example here is CDK5RAP2 that coevolves with brain mass in primate taxa where it has increased [8], but not when it has decreased [21]. In the present case the positive trends found in Glires and Euungulata would be expected if phenotypic differences within these clades are mostly due to increases in brain size on lineages leading to larger brained species, rather than decreases leading to smaller brained species.
Finally, a major challenge may occur when the gene-phenotype association is limited to a small subset of domains or sites within a gene. Site-based methods for detecting positive selection were developed because it is thought that selection is unlikely to act across a whole gene equally [30, 31]. This positive selection is presumably associated with some phenotypic, functional or fitness-related change, and it is therefore likely that gene-phenotype associations may be limited to a subset of domains or codons. Developing methods, which are capable of testing gene-phenotype associations on a site-by-site basis, or which incorporate a form of sliding window analysis, may be a worthwhile endeavor (e.g. [83]). Again, to gain sufficient statistical power such analyses will require large, densely sampled datasets.
The pervasive signature of positive selection across mammals, combined with the limited evidence of divergence in selection pressures, suggests results from experimentally tractable clades may be applicable to wider taxonomic groups. The link between selection on microcephaly genes and the evolution of brain size has yet to be confirmed (or rejected) by functional data. Mice transgenic for ASPM, and in vitro assays for MCPH1 confirm changes in the coding sequence have functional affects [25, 84]. However, although the human sequence of ASPM rescued the phenotype of transgenic mice with a disrupted copy of ASPM it did not lead to an increase brain size [84]. This has been interpreted as being indicative of functional conservation [84], but without the reciprocal experiments this conclusion may be premature. It could be, for example, that the effects of microcephaly genes are background dependent, or are combinatorial such that changing individual genes has only a minor or no affect in isolation. Given the importance placed in functional confirmation of gene-phenotype associations a shift towards examining phenotypic diversity within experimentally tractable clades may be worthwhile. For example, greater sampling density in rodents could permit comparative analyses to test for positive selection, and subsequently test for gene-phenotype associations. A range of data exists for a number of rodents that vary widely in brain size, neuron number and gyrencephaly [77, 85], and the results presented above for Glires provide some encouragement for pursuing studies in this clade. Both micro and macroevolutionary gene-phenotype associations could be performed and, if an association were found, supporting evidence could be sought through evolutionary developmental studies. Rather than doing one-way human/mouse transgenic experiments [84], it may be just as informative to perform, for example, reciprocal rat/mouse transgenic experiments. Although we note that recent developments in in vitro human organoids may render two-way human/mouse transgenics technically feasible [86], ethical considerations may still limit this approach. Regardless of the results of such a study, greater sampling within clades with smaller brains will provide a useful comparison with larger brained mammalian orders, such as primates and cetaceans, and are clearly necessary to avoid ascertainment biases.
Conclusions
We have shown that microcephaly genes have experienced pervasive positive selection not just across primates but across placental mammals. We find little evidence to suggest that these loci experienced divergent selective pressures in different clades, which may suggest conservation in function and imply a common phenotypic relevance for this wide spread positive selection. Developmental models of cortical expansion and evidence for conservation in brain developmental pathways provide a clear basis with which to hypothesize that the phenotype of relevance is brain mass, or more specifically the number of neurons produced during cortical neurogenesis. We provide evidence to support this hypothesis in Glires and Euungulata that, combined with previous work in primates, suggests that this phenotypic association may be common across mammals. Whilst several challenges face attempts to test gene-phenotype hypotheses, the persistent signal of positive selection should permit useful studies in more experimentally tractable species.
Methods
Data, alignment and phylogeny
Full coding sequence for mammalian species were collected from Ensembl and GenBank. Only species that were available for all seven loci (ASPM, CDK5RAP2, CENPJ, CEP152, MCPH1, STIL and WDR62) were included to permit fair comparisons between loci. Ensembl/Genbank accession IDs for these species are shown in Table S1. In total we obtained full coding sequence for 12 primates and 21 non-primate eutherian mammals. No marsupials were included due to a lack of species with data for all seven loci.
Additional sequence data were generated to test if increased sampling results in stronger evidence for positive selection for two loci in primates. In these cases, regions for amplification were chosen based on peaks of high dN/dS identified using a sliding window analysis performed across the alignment of the full coding sequence from anthropoids using SWAAP [87], the Nei and Gojobori [88] method with a window size of 150 codons and a step size of 15 codons. From this alignment primers were designed in conserved regions using Primer3Plus [89]. Genomic DNA samples were extracted from tissue samples using DNeasy kits (QIAGEN UK, Crawley, UK) for a previous study by the same authors [88]. Ethical approval was not required as all DNA was obtained from archived tissue samples taken from animals that were captive born in the UK and that died of natural causes or that were euthanized for reasons unrelated to the current research. Tissue samples were originally obtained from Andrew Kitchener at the National Museums of Scotland, or Leona Chemnick at the Center for Reproduction of Endangered Species, San Diego Zoo, with permission to use the samples in molecular biology studies.
Polymerase chain reactions (PCR) were performed using standard protocols and BIOTAQ DNA polymerase PCR kits (BIOLINE, London, UK). PCR products were purified using Qiagen QIAquick PCR purification kits. Cycle sequencing on both strands was carried out using BIG DYE v. 3.1 (PE Biosystems) under standard conditions. Precipitated DNA was sent to the Oxford Sequencing Centre (Dept. of Zoology, University of Oxford) for sequencing runs. In total we amplified 5 exons (7, 13, 15, 17 and 18) from STIL, totaling c.2400 bp and one 520 bp exon (30) from WDR62 from an additional 8 species which were added to the 10 available. The site model tests for positive selection were repeated using an alignment of these 18 anthropoids. Sequences were aligned using MUSCLE in MEGA 5.0 [90]. All alignments were filtered to remove poorly aligned sequence, removing short stretches of sequence surrounded by gaps and regions with an excessive number of substitutions in a short sequence. All alignments are available by request from SHM. For the molecular evolution analyses the mammalian phylogeny was taken from two published mammalian phylogenies, which produced comparable results [91, 92].
Molecular evolution analyses
A common measure used to infer selection pressures acting on coding regions of genes is the ratio of rates of non-synonymous to synonymous fixed base changes. Estimation of dN/dS ratios (ω) was carried out using a codon-based maximum likelihood method (codeml in PAML version 4.7 [29]). Several analyses were performed to test the hypothesis that the candidate loci have experienced positive selection or that rates of evolution vary between clades. Nested models were tested by comparing the likelihood ratio statistic (−2(Log[Lh(null model)]–Log[Lh(alternative model)]) to critical values of the Chi-square distribution using degrees of freedom as the difference in the number of parameters estimated by each model. For each test we correct for multiple testing using the sequential Bonferroni method, with n = 7, the number of loci tested.
Tests for positive selection
To detect positive selection we implemented the site models. These allow the ω to vary among sites but not across lineages [30, 31]. The site model tests for positive selection can be carried out using two pairs of models. The first pair compare Model M1a and Model M2a [55, 93]. Model M1a (NearlyNeutral) allows sites to fall into two categories with ω <1 (purifying selection) and ω = 1 (neutral evolution), whilst model M2a (PositiveSelection) allows sites to fall into three categories with ω <1, ω = 1 and ω >1 (positive selection) [55]. The second pair compares Model 8a and Model 8 [93, 94]. These models use the beta distribution to describe the numbers of sites across different categories of ω. M8 has 11 site classes (10 from the beta distribution plus 1 additional class), where one of these classes may have an ω >1. In Model 8a this latter class is restricted to have an ω equal to 1. The critical Likelihood Ratio boundaries of significance for this test are 2.71 at 5% and 5.41 at 1%, but here we calculate significance using a chi squared test with a more conservative one degree of freedom [93]. The M1a-M2a test has slightly lower false positive rates and is more conservative than the M8-M8a test [93]. Tests for positive selection can be affected by missing data, indels and alignment quality [95] and the Ensembl data is incomplete in some cases, with missing data randomly distributed across loci. To assess whether this causes a bias in detecting positive selection we compared the distribution of coverage in sites with significant evidence of positive selection under the Bayes Empirical Bayes method [55] to all other sites. In no case was the distribution significantly different (paired t-tests, all loci p > 0.05) suggesting missing data at some sites does not bias the results.
Tests for rate shifts & diversifying selection
Branch models allow ω to vary across branches in the phylogeny but not across sites. Branch models can also be used to compare whether or not ω varies between clades or pre-defined lineages. We used this model to compare average ω values across primates to non-primate mammals, and to compare the rates between the three major placental clades, the Euarchontoglires, the Laurasiatheria and Xenarthra + Afrotheria which Meredith et al. [92] find to be monophyletic.
A final analysis to detect diversifying selection was performed using Clade model C [34]. Clade models allow a proportion of sites to undergo divergent selection pressures in two or more clades defined a priori. These sites may have any value of ω so do not explicitly test for positive selection, or differing amounts of positive selection but may give an indication of differential selection pressures in different clades. Clade model C was compared to the new null model, M2a_rel [96]. Both clade model C and M2a_rel have a proportion of sites evolving under purifying selection and a proportion evolving neutrally in both clades. Where clade model C has a third category where ω varies between the two clades and may be any value >0, M2a_rel has a third category of ω that may be any value >0, but that is shared between clades. Clade model tests were used for the same comparisons as the branch tests, as an independent assessment of shifts in selection.
Tests for gene-phenotype co-evolution
Branch models can also be used to compare two phenotypically divergent lineages. We use the branch test here to compare whether several large brained/small brained sister lineages have significantly different dN/dS values. A growing number of candidate gene analyses have also sought to explicitly test hypothesised gene-phenotype links by adopting comparative methods to test for an association between dN/dS and the phenotype of interest whilst controlling for phylogeny (e.g. [8, 97, 98]). Here we calculate the root-to-tip dN/dS ratio for each species considered and regress these values against brain size using a Phylogenetic Generalised Least Squares model (PGLS), implemented in Bayes Traits ([99] available from http://www.evolution.rdg.ac.uk), to correct for the non-independence of interspecific data caused by their shared evolutionary history. This approach has been used to test for an association between candidate genes, including microcephaly genes, and brain size in primates [8, 21, 22, 66], and between MCPH1 and ASPM and brain size in cetaceans [26, 28]. A similar approach has also been adopted in other gene-phenotype studies (e.g. [100, 101]). Brain size data were taken from Barton and Capellini [102] and Boddy et al. [85].
Availability of supporting data
The data set supporting the results of this article is included within the article and its additional files.
References
Zhang J: Evolution of the human ASPM gene, a major determinant of brain size. Genetics. 2003, 165 (4): 2063-2070.
Evans PD, Anderson JR, Vallender EJ, Choi SS, Lahn BT: Reconstructing the evolutionary history of microcephalin, a gene controlling human brain size. Hum Mol Genet. 2004, 13 (11): 1139-1145. 10.1093/hmg/ddh126.
Evans PD, Anderson JR, Vallender EJ, Gilbert SL, Malcom CM, Dorus S, Lahn BT: Adaptive evolution of ASPM, a major determinant of cerebral cortical size in humans. Hum Mol Genet. 2004, 13 (5): 489-494. 10.1093/hmg/ddh055.
Kouprina N, Pavlicek A, Mochida GH, Solomon G, Gersch W, Yoon YH, Collura R, Ruvolo M, Barrett JC, Woods CG, Walsh CA, Jurka J, Larionov V: Accelerated evolution of the ASPM gene controlling brain size begins prior to human brain expansion. PLoS Biol. 2004, 2 (5): e126-10.1371/journal.pbio.0020126.
Wang YQ, Su B: Molecular evolution of microcephalin, a gene determining human brain size. Hum Mol Genet. 2004, 13 (11): 1131-1137. 10.1093/hmg/ddh127.
Evans PD, Vallender EJ, Lahn BT: Molecular evolution of the brain size regulator genes CDK5RAP2 and CENPJ. Gene. 2006, 375: 75-79.
Ali F, Meier R: Positive selection in ASPM is correlated with cerebral cortex evolution across primates but not with whole-brain size. Mol Biol Evol. 2008, 25 (11): 2247-2250. 10.1093/molbev/msn184.
Montgomery SH, Capellini I, Venditti C, Barton RA, Mundy NI: Adaptive evolution of four microcephaly genes and the evolution of brain size in anthropoid primates. Mol Biol Evol. 2011, 28 (1): 625-638. 10.1093/molbev/msq237.
Jackson AP, McHale DP, Campbell DA, Jafri H, Rashid Y, Mannan J, Karbani G, Corry P, Levene MI, Mueller RF, Markham AF, Lench NJ, Woods GC: Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am J Hum Genet. 1998, 63 (2): 541-546. 10.1086/301966.
Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, Walsh CA, Woods CG: ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002, 32 (2): 316-320. 10.1038/ng995.
Bond J, Roberts E, Springell K, Lizarraga S, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, Costa SMR, Baralle D, Raponi M, Karbani G, Rashid Y, Jafri H, Bennett C, Corry P, Walsh CA, Woods CG: A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet. 2005, 37 (4): 353-355. 10.1038/ng1539.
Kumar A, Girimaji SC, Duvvari MR, Blanton SH: Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet. 2009, 84 (2): 286-290. 10.1016/j.ajhg.2009.01.017.
Guernsey DL, Jiang H, Hussin J, Arnold M, Bouyakdan K, Perry S, Babineau-Sturk T, Beis J, Dumas N, Evans SC, Ferguson M, Matsuoka M, Macgillivray C, Nightingale M, Patry L, Rideout AL, Thomas A, Orr A, Hoffmann I, Michaud JL, Awadalla P, Meek DC, Ludman M, Samuels ME: Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet. 2010, 87 (1): 40-51. 10.1016/j.ajhg.2010.06.003.
Nicholas AK, Khurshid M, Désir J, Carvalho OP, Cox JJ, Thornton G, Kauser R, Ansar M, Ahmad W, Verloes A, Passemard S, Misson JP, Lindsay S, Gergely F, Dobyns WB, Roberts E, Abramowicz M, Woods CG: WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet. 2010, 42 (11): 1010-1014. 10.1038/ng.682.
Timothy WY, Mochida GH, Tischfield DJ, Sgaier SK, Flores-Sarnat L, Sergi CM, Topçu M, McDonald MT, Barry BJ, Felie J, Sunu C, Dobyns WB, Folkerth RD, Barkovich AJ, Walsh CA: Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat Genet. 2010, 42 (11): 1015-1020. 10.1038/ng.683.
Bond J, Woods CG: Cytoskeletal genes regulating brain size. Curr Opin Cell Biol. 2006, 18 (1): 95-101. 10.1016/j.ceb.2005.11.004.
Cox J, Jackson AP, Bond J, Woods CG: What primary microcephaly can tell us about brain growth. Trends Mol Med. 2006, 12 (8): 358-366. 10.1016/j.molmed.2006.06.006.
Thornton GK, Woods CG: Primary microcephaly: do all roads lead to Rome?. Trends Genet. 2009, 25 (11): 501-510. 10.1016/j.tig.2009.09.011.
Evans PD, Gilbert SL, Mekel-Bobrov N, Vallender EJ, Anderson JR, Vaez-Azizi LM, Tichkoff AS, Hudson RR, Lahn BT: Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science. 2005, 309 (5741): 1717-1720. 10.1126/science.1113722.
Mekel-Bobrov N, Gilbert SL, Evans PD, Vallender EJ, Anderson JR, Hudson RR, Tishkoff SA, Lahn BT: Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science. 2005, 309 (5741): 1720-1722. 10.1126/science.1116815.
Montgomery SH, Mundy NI: Evolution of ASPM is associated with both increases and decreases in brain size in primates. Evolution. 2012, 66 (3): 927-932. 10.1111/j.1558-5646.2011.01487.x.
Montgomery SH, Mundy NI: Microcephaly genes and the evolution of sexual dimorphism in primate brain size. J Evol Biol. 2013, 26 (4): 906-911. 10.1111/jeb.12091.
Wang JK, Li Y, Su B: A common SNP of MCPH1 is associated with cranial volume variation in Chinese population. Hum Mol Genet. 2008, 17 (9): 1329-1335. 10.1093/hmg/ddn021.
Rimol LM, Agartz I, Djurovic S, Brown AA, Roddey JC, Kähler AK, Mattingsdal M, Athanasiu L, Joyner AH, Schork NJ, Halgren E, Sundet K, Melle I, Dale AM, Andreassen OA: Sex-dependent association of common variants of microcephaly genes with brain structure. Proc Natl Acad Sci USA. 2010, 107 (1): 384-388. 10.1073/pnas.0908454107.
Shi L, Li M, Lin Q, Qi X, Su B: Functional divergence of the brain-size regulating gene MCPH1 during primate evolution and the origin of humans. BMC Biol. 2013, 11 (1): 62-10.1186/1741-7007-11-62.
McGowen MR, Montgomery SH, Clark C, Gatesy J: Phylogeny and adaptive evolution of the brain-development gene microcephalin (MCPH1) in cetaceans. BMC Evol Biol. 2012, 11: 98-
Xu S, Chen Y, Cheng Y, Yang D, Zhou X, Xu J, Zhou K, Yang G: Positive selection at the ASPM gene coincides with brain size enlargements in cetaceans. Proc Roy Soc B. 2012, 279 (1746): 4433-4440. 10.1098/rspb.2012.1729.
Montgomery SH, Mundy NI, Bartion RA: ASPM and mammalian brain evolution: A case study in the difficulty in making macroevolutionary inferences about gene-phenotype associations. Proc Roy Soc B. 2013, 281 (1778): 20131743-
Yang Z: PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007, 24 (8): 1586-1591. 10.1093/molbev/msm088.
Nielsen R, Yang Z: Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998, 148 (3): 929-936.
Yang Z, Nielsen R, Goldman N, Pedersen AMK: Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000, 155 (1): 431-449.
Yang Z: Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998, 15 (5): 568-573. 10.1093/oxfordjournals.molbev.a025957.
Yang Z, Nielsen R: Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol. 1998, 46 (4): 409-418. 10.1007/PL00006320.
Bielawski JP, Yang Z: A maximum likelihood method for detecting functional divergence at individual codon sites, with application to gene family evolution. J Mol Evol. 2004, 59 (1): 121-132.
Nikolaev SI, Montoya-Burgos JI, Popadin K, Parand L, Margulies EH, Antonarakis SE: Life-history traits drive the evolutionary rates of mammalian coding and noncoding genomic elements. Proc Natl Acad Sci USA. 2007, 104 (51): 20443-20448. 10.1073/pnas.0705658104.
Welch JJ, Bininda-Emonds OR, Bromham L: Correlates of substitution rate variation in mammalian protein-coding sequences. BMC Evol Biol. 2008, 8 (1): 53-10.1186/1471-2148-8-53.
Lanfear R, Welch JJ, Bromham L: Watching the clock: studying variation in rates of molecular evolution between species. Trends Ecol Evol. 2010, 25 (9): 495-503. 10.1016/j.tree.2010.06.007.
McGowen MR, Grossman LI, Wildman DE: Dolphin genome provides evidence for adaptive evolution of nervous system genes and a molecular rate slowdown. Proc Roy Soc B. 2012, 279 (1743): 3643-3651. 10.1098/rspb.2012.0869.
Goodman M, Sterner KN, Islam M, Uddin M, Sherwood CC, Hof PR, Hou ZC, Lipovich L, Jia H, Grossman L, Wildman DE: Phylogenomic analyses reveal convergent patterns of adaptive evolution in elephant and human ancestries. Proc Natl Acad Sci USA. 2009, 106 (49): 20824-20829. 10.1073/pnas.0911239106.
Jerison H: Evolution of the Brain and Intelligence. 1973, New York: Academic
Montgomery SH, Capellini I, Barton RA, Mundy NI: Reconstructing the ups and downs of primate brain evolution: implications for adaptive hypotheses and Homo floresiensis. BMC Biol. 2010, 8 (1): 9-10.1186/1741-7007-8-9.
Marino L, McShea DW, Uhen MD: Origin and evolution of large brains in toothed whales. Anat Rec A Discov Mol Cell Evol Biol. 2004, 281 (2): 1247-1255.
Montgomery SH, Geisler JH, McGowen MR, Fox C, Marino L, Gatesy J: The evolutionary history of cetacean brain and body size. Evolution. 2013, 67 (11): 3339-3353. 10.1111/evo.12197.
Shoshani J: Understanding proboscidean evolution: a formidable task. Trends Ecol Evol. 1998, 13 (12): 480-487. 10.1016/S0169-5347(98)01491-8.
Rakic P: Specification of cerebral cortical areas. Science. 1988, 241 (4862): 170-176. 10.1126/science.3291116.
Rakic P: Adult neurogenesis in mammals: an identity crisis. J Neurosci. 2002, 22 (3): 614-618.
Bhardwaj RD, Curtis MA, Spalding KL, Buchholz BA, Fink D, Björk-Eriksson T, Nordborg C, Gage FH, Druid H, Eriksson PS, Frisén J: Neocortical neurogenesis in humans is restricted to development. Proc Natl Acad Sci USA. 2006, 103 (33): 12564-12568. 10.1073/pnas.0605177103.
MacKenna MC, Bell SK: Classification of mammals: above the species level. Edited by: Simpson GG. 1997, New York: Columbia University Press
Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, Fiedel-Alon A, Tanenbaum DM, Civello D, White TJ, Sninsky JJ, Adams MD, Cargill M: A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005, 3 (6): e170-10.1371/journal.pbio.0030170.
Shi P, Bakewell MA, Zhang J: Did brain-specific genes evolve faster in humans than in chimpanzees?. Trends Genet. 2006, 22 (11): 608-613. 10.1016/j.tig.2006.09.001.
Wang HY, Chien HC, Osada N, Hashimoto K, Sugano S, Gojobori T, Chou CK, Tsai SF, Wu CI, Shen CKJ: Rate of evolution in brain-expressed genes in humans and other primates. PLoS Biol. 2006, 5 (2): e13-
Kosiol C, Vinař T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, Siepel A: Patterns of positive selection in six Mammalian genomes. PLoS Genet. 2008, 4 (8): e1000144-10.1371/journal.pgen.1000144.
Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, Hobolth A, Lappalainen T, Mailund T, Marques-Bonet T, McCarthy S, Montgomery SH, Schwalie PC, Tang YA, Ward MC, Xue Y, Yngvadottir B, Alkan C, Andersen LN, Ayub Q, Ball EV, Beal K, Bradley BJ, Chen Y, Clee CM, Fitzgerald S, Graves TA, Gu Y, Heath P, Heger A, et al: Insights into hominid evolution from the gorilla genome sequence. Nature. 2012, 483 (7388): 169-175. 10.1038/nature10842.
George RD, McVicker G, Diederich R, Ng SB, MacKenzie AP, Swanson WJ, Shendure J, Thomas JH: Trans genomic capture and sequencing of primate exomes reveals new targets of positive selection. Genome Res. 2011, 21 (10): 1686-1694. 10.1101/gr.121327.111.
Yang Z, Wong WS, Nielsen R: Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005, 22 (4): 1107-1118. 10.1093/molbev/msi097.
Fong KW, Choi YK, Rattner JB, Qi RZ: CDK5RAP2 is a pericentriolar protein that functions in centrosomal attachment of the γ-tubulin ring complex. Mol Biol Cell. 2008, 19 (1): 115-125. 10.1091/mbc.E07-04-0371.
Cormier A, Clément MJ, Knossow M, Lachkar S, Savarin P, Toma F, Sobel A, Gigant B, Curmi PA: The PN2-3 domain of centrosomal P4. 1-associated protein implements a novel mechanism for tubulin sequestration. J Biol Chem. 2009, 284 (11): 6909-6917. 10.1074/jbc.M808249200.
Buchman JJ, Tseng HC, Zhou Y, Frank CL, Xie Z, Tsai LH: CDK5RAP2 interacts with pericentrin to maintain the neural progenitor pool in the developing neocortex. Neuron. 2010, 66 (3): 386-402. 10.1016/j.neuron.2010.03.036.
Fish JL, Kosodo Y, Enard W, Pääbo S, Huttner WB: ASPM specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA. 2006, 103 (27): 10438-10443. 10.1073/pnas.0604066103.
Rujano MA, Sanchez-Pulido L, Pennetier C, le Dez G, Basto R: The microcephaly protein ASP regulates neuroepithelium morphogenesis by controlling the spatial distribution of myosin II. Nat Cell Biol. 2013, 15: 1294-1306. 10.1038/ncb2858.
Götz M, Huttner WB: The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005, 6 (10): 777-788. 10.1038/nrm1739.
Kriegstein A, Noctor S, Martínez-Cerdeño V: Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci. 2006, 7 (11): 883-890. 10.1038/nrn2008.
Rakic P: A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995, 18 (9): 383-388. 10.1016/0166-2236(95)93934-P.
Caviness VS, Takahashi T, Nowakowski RS: Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995, 18 (9): 379-383. 10.1016/0166-2236(95)93933-O.
Wang X, Tsai JW, Imai JH, Lian WN, Vallee RB, Shi SH: Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature. 2009, 461 (7266): 947-955. 10.1038/nature08435.
Montgomery SH, Mundy NI: Positive selection on NIN, a gene involved in neurogenesis, and primate brain evolution. Genes, Brain and Behav. 2012, 11 (8): 903-910.
Fietz SA, Kelava I, Vogt J, Wilsch-Bräuninger M, Stenzel D, Fish JL, Corbeil D, Riehn A, Dister A, Nitsch R, Huttner WB: OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci. 2010, 13 (6): 690-699. 10.1038/nn.2553.
Rakic P: Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci. 2009, 10 (10): 724-735. 10.1038/nrn2719.
Chaplin TA, Yu HH, Soares JG, Gattass R, Rosa MG: A conserved pattern of differential expansion of cortical areas in simian primates. J Neurosci. 2013, 33 (38): 15120-15125. 10.1523/JNEUROSCI.2909-13.2013.
Finlay BL, Darlington RB: Linked regularities in the development and evolution of mammalian brains. Science. 1995, 268 (5217): 1578-1584. 10.1126/science.7777856.
Barton RA, Harvey PH: Mosaic evolution of brain structure in mammals. Nature. 2000, 405 (6790): 1055-1058. 10.1038/35016580.
Parker J, Tsagkogeorga G, Cotton JA, Liu Y, Provero P, Stupka E, Rossiter SJ: Genome-wide signatures of convergent evolution in echolocating mammals. Nature. 2013, 502 (7470): 228-231. 10.1038/nature12511.
Zhang J, Kumar S: Detection of convergent and parallel evolution at the amino acid sequence level. Mol Biol Evol. 1997, 14 (5): 527-536. 10.1093/oxfordjournals.molbev.a025789.
Kriener K, O'hUigin C, Tichy H, Klein J: Convergent evolution of major histocompatibility complex molecules in humans and New World monkeys. Immunogenetics. 2000, 51 (3): 169-178. 10.1007/s002510050028.
Majerus ME, Mundy NI: Mammalian melanism: natural selection in black and white. Trends Genet. 2003, 19 (11): 585-588. 10.1016/j.tig.2003.09.003.
Lartillot N, Poujol R: A phylogenetic model for investigating correlated evolution of substitution rates and continuous phenotypic characters. Mol Biol Evol. 2011, 28 (1): 729-744. 10.1093/molbev/msq244.
Herculano-Houzel S, Mota B, Lent R: Cellular scaling rules for rodent brains. Proc Natl Acad Sci USA. 2006, 103 (32): 12138-12143. 10.1073/pnas.0604911103.
Herculano-Houzel S, Collins CE, Wong P, Kaas JH: Cellular scaling rules for primate brains. Proc Natl Acad Sci USA. 2007, 104 (9): 3562-3567. 10.1073/pnas.0611396104.
Kelava I, Reillo I, Murayama AY, Kalinka AT, Stenzel D, Tomancak P, Matsuzaki F, Lebrand C, Sasaki E, Schwamborn JC, Okano H, Huttner WB, Borrell V: Abundant occurrence of basal radial glia in the subventricular zone of embryonic neocortex of a lissencephalic primate, the common marmoset Callithrix jacchus. Cereb Cortex. 2012, 22 (2): 469-481. 10.1093/cercor/bhr301.
Kelava I, Lewitus E, Huttner WB: The secondary loss of gyrencephaly as an example of evolutionary phenotypical reversal. Front Neuroanat. 2013, 7: 16-
Ford SM: Callitrichids as phyletic dwarfs, and the place of the Callitrichidae in Platyrrhini. Primates. 1980, 21 (1): 31-43. 10.1007/BF02383822.
Montgomery SH, Mundy NI: Parallel episodes of phyletic dwarfism in callitrichid and cheirogaleid primates. J Evol Biol. 2013, 26 (4): 810-819. 10.1111/jeb.12097.
O’Connor TD, Mundy NI: Evolutionary modeling of genotype-phenotype associations, and application to primate coding and non-coding mtDNA rate variation. Evol Bioinform Online. 2013, 9: 301-
Pulvers JN, Bryk J, Fish JL, Wilsch-Bräuninger M, Arai Y, Schreier D, Naumann R, Helppi J, Habermann B, Vogt J, Nitsch R, Tóth A, Enard W, Pääbo S, Hutnner WB: Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc Natl Acad Sci USA. 2010, 107 (38): 16595-16600. 10.1073/pnas.1010494107.
Boddy AM, McGowen MR, Sherwood CC, Grossman LI, Goodman M, Wildman DE: Comparative analysis of encephalization in mammals reveals relaxed constraints on anthropoid primate and cetacean brain scaling. J Evol Biol. 2012, 25 (5): 981-994. 10.1111/j.1420-9101.2012.02491.x.
Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA: Cerebral organoids model human brain development and microcephaly. Nature. 2013, 501 (7467): 373-379. 10.1038/nature12517.
Pride DT: SWAAP: a tool for analyzing substitutions and similarity in multiple alignments. 2000, Distributed by the author
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3 (5): 418-426.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA: Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35 (suppl 2): W71-W74.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28 (10): 2731-2739. 10.1093/molbev/msr121.
Bininda-Emonds OR, Cardillo M, Jones KE, MacPhee RD, Beck RM, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A: The delayed rise of present-day mammals. Nature. 2007, 446 (7135): 507-512. 10.1038/nature05634.
Meredith RW, Janečka JE, Gatesy J, Ryder OA, Fisher CA, Teeling EC, Goodbla A, Aizrik E, Simão TLL, Stadler T, Rabosky DL, Honeycutt RL, Flynn JJ, Ingram CM, Steiner C, Williams TL, Robinson TJ, Burk-Herrick A, Westerman M, Ayoub NA, Springer MS, Murphy WJ: Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science. 2011, 334 (6055): 521-524. 10.1126/science.1211028.
Wong WS, Yang Z, Goldman N, Nielsen R: Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004, 168 (2): 1041-1051. 10.1534/genetics.104.031153.
Swanson WJ, Nielsen R, Yang Q: Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol. 2003, 20 (1): 18-20. 10.1093/oxfordjournals.molbev.a004233.
Fletcher W, Yang Z: The effect of insertions, deletions, and alignment errors on the branch-site test of positive selection. Mol Biol Evol. 2010, 27 (10): 2257-2267. 10.1093/molbev/msq115.
Weadick CJ, Chang BS: An improved likelihood ratio test for detecting site-specific functional divergence among clades of protein-coding genes. Mol Biol Evol. 2012, 29 (5): 1297-1300. 10.1093/molbev/msr311.
Dorus S, Vallender EJ, Evans PD, Anderson JR, Gilbert SL, Mahowald M, Wycoff GJ, Malcom CM, Lahn BT: Accelerated evolution of nervous system genes in the origin of Homo sapiens. Cell. 2004, 119 (7): 1027-1040. 10.1016/j.cell.2004.11.040.
Nadeau NJ, Burke T, Mundy NI: Evolution of an avian pigmentation gene correlates with a measure of sexual selection. Proc Roy Soc B. 2007, 274 (1620): 1807-1813. 10.1098/rspb.2007.0174.
Pagel M: Inferring the historical patterns of biological evolution. Nature. 1999, 401 (6756): 877-884. 10.1038/44766.
Venditti C, Pagel M: Speciation as an active force in promoting genetic evolution. Trends Ecol Evol. 2010, 25 (1): 14-20. 10.1016/j.tree.2009.06.010.
Lüke L, Vicens A, Serra F, Luque-Larena JJ, Dopazo H, Roldan ER, Gomendio M: Sexual selection halts the relaxation of protamine 2 among rodents. PLoS One. 2011, 6 (12): e29247-10.1371/journal.pone.0029247.
Barton RA, Capellini I: Maternal investment, life histories, and the costs of brain growth in mammals. Proc Natl Acad Sci USA. 2011, 108 (15): 6169-6174. 10.1073/pnas.1019140108.
Acknowledgments
We thank Fabian Zimmer for code to quantify per site coverage across species, and Andrew Kitchener and Leona Chemnick for previously providing tissue samples. This work was supported by a BBRSC doctoral studentship and a research fellowship from the Royal Commission for the Exhibition of 1851 to SHM, and Murray Edwards College, Cambridge.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing interests.
Authors’ contributions
SHM and NIM designed the analyses, SHM collected the data, carried out the analyses and wrote the initial draft of the manuscript. SHM and NIM edited the manuscript.
Electronic supplementary material
12862_2014_2602_MOESM1_ESM.pdf
Additional file 1: Figure S1: Sliding window analysis of STIL (A) and WDR62 (B): Ka/Ks = red, Ka = pink, Ks = green. Blue bars indicate regions sequenced from additional anthropoid primates. (PDF 111 KB)
12862_2014_2602_MOESM2_ESM.xlsx
Additional file 2: Table S1: Accession IDs and phenotypic data. Table S2. branch and clade model tests for diversifying selection. Table S3. Linking molecular and phenotypic evolution. (XLSX 26 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Montgomery, S.H., Mundy, N.I. Microcephaly genes evolved adaptively throughout the evolution of eutherian mammals. BMC Evol Biol 14, 120 (2014). https://doi.org/10.1186/1471-2148-14-120
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-14-120