
Abstract
Background
To understand the ecological and evolutionary consequences of species interactions in food webs necessitates that interactions are properly identified. Genetic analyses suggest that many supposedly generalist parasitoid species should rather be defined as multiple species with a more narrow diet, reducing the probability that such species may mediate indirect interactions such as apparent competition among hosts. Recent studies showed that the parasitoid Asecodes lucens mediate apparent competition between two hosts, Galerucella tenella and G. calmariensis, affecting both interaction strengths and evolutionary feedbacks. The same parasitoid was also recorded from other species in the genus Galerucella, suggesting that similar indirect effects may also occur for other species pairs.
Methods
To explore the possibility of such interactions, we sequenced mitochondrial and nuclear genetic markers to resolve the phylogeny of both host and parasitoid and to test the number of parasitoid species involved. We thus collected 139 Galerucella larvae from 8 host plant species and sequenced 31 adult beetle and 108 parasitoid individuals.
Results
The analysis of the Galerucella data, that also included sequences from previous studies, verified the five species previously documented as reciprocally monophyletic, but the Bayesian species delimitation for A. lucens suggested 3–4 cryptic taxa with a more specialised host use than previously suggested. The gene data analyzed under the multispecies coalescent model allowed us to reconstruct the species tree phylogeny for both host and parasitoid and we found a fully congruent coevolutionary pattern suggesting that parasitoid speciation followed upon host speciation.
Conclusion
Using multilocus sequence data in a Bayesian species delimitation analysis we propose that hymenopteran parasitoids of the genus Asecodes that infest Galerucella larvae constitute at least three species with narrow diet breath. The evolution of parasitoid Asecodes and host Galerucella show a fully congruent coevolutionary pattern. This finding strengthens the hypothesis that the parasitoid in host search uses cues of the host rather than more general cues of both host and plant.
Similar content being viewed by others

Background
Consumer species feeding on multiple resources may indirectly mediate interactions among their resources [1–3], and thereby affect both trait evolution of interacting species and eventually speciation. A classic example is apparent competition, where one species affects the density of other species through an increased density of the shared consumer [4–9]. We are interested in the potential for such interactions among a set of chrysomelid beetles (Galerucella spp.) and their parasitoids, including the ecological and evolutionary consequences. Previous studies within this system show that parasitism rates may be very high, and that parasitoids may mediate apparent competition between hosts [10–12]. The literature suggests that the same parasitoid species Asecodes lucens (Nees) (Hymenoptera: Eulophidae) attacks all Galerucella species [10, 11, 13, 14], and parasitoid mediated interaction could therefore involve additional species. However, cryptic species are common in parasitoid systems and diet breadth may be more narrow than literature information suggest [15–18]. For instance, Smith et al. [15] showed that 17 tachinid fly species that were thought to be broad generalist parasitoids on lepidopteran larvae consist of 32 species with a more narrow diet breadth. Similarly, Apanteles leucostigmus, thought to be a generalist parasitoid on at least 32 species of Hesperiidae caterpillars, revealed 36 distinct Barcode clusters interpreted as provisional specialist species by Smith et al. [19].
DNA-based species delimitation provides a novel method for discovering cryptic species where supposedly generalist consumers are shown to be morphologically more or less identical specialist consumers. However, the standard DNA barcoding protocols used to infer putative species are based on single loci and do not test species limits with models tracking speciation and population genetic processes e.g., [20]. An improved protocol such as the General Mixed Yule Coalescent model (hereafter GMYC) treats species-to-population transitions in a gene tree [21]. However, GMYC also assumes reciprocal monophyly of species and that the gene tree is known without error, ignoring that non-monophyletic species in gene trees are common [22, 23] and that the time needed for complete lineage sorting of ancestral polymorphism is substantial ([24], see also [25, 26]). When deriving the species tree, one approach is to concatenate multiple loci into a superlocus by assuming that gene trees are consistent among each other and with the species tree. An alternative approach is to explicitly model the coalescent process of each gene tree separately under the constraint of a common species tree [27–31]. This later approach is implemented in BPP, a Bayesian species delimitation method [32], that in a recent evaluation came out as the most accurate species delimitation method under varying conditions of divergence time, number of loci and migration [33]. BPP offers an exciting new tool to detect recent speciation events and cryptic species and is particularly suitable for host-parasitoid systems due to the prerequisite of an informed prior (a “guide tree”; Yang and Rannala 2010, see material and methods).
Cryptic species are thought to be more common in groups where chemical senses are more highly developed than vision, such as most insects, because changes in chemical communication do not necessitate morphological changes for the evolution of reproductive barriers between sibling species [34]. Cryptic speciation in parasitic hymenoptera is particularly interesting as this group is often reported to mediate indirect interactions between host species [5, 35–37], but also because host finding in parasitoids involves both host volatiles and volatiles from the plant associated to the herbivore host [38]. Host finding is often described along a detectability-reliability axis, where plant volatiles associated to the host are more abundant than host volatiles, but also have a lower reliability. Parasitoids using a combination of host and plant volatiles may then face greater difficulties in finding hosts on different host plants compared to parasitoid species that only use host volatiles for host finding. As a consequence, generalist parasitoid species may have to use more general cues for host finding and this may, in turn, result in less efficient host search cf. [39]. Due to such trade-offs, specialisation and speciation cannot be separated from the chemical ecology underlying host finding.
The chrysomelid beetle genus Galerucella contains a mixture of monophagous and polyphagous herbivore species that occur commonly in wetlands across the northern hemisphere. Within this genus, in contrast to the generally accepted dogma for speciation in herbivore insects, host shifts have not necessarily involved closely related host plant species. Speciation in Galerucella has instead occurred between co-occurring wetland plants of such distant relations as Salicaceae, Primulaceae, Lythraceae, Rosaceae, Polygonaceae, Nymphaeaceae and Betulaceae. It is not likely that such distantly related plant species share much volatile profiles beyond green leaf volatiles [40], and even these are likely to occur in very different proportions (supported by unpublished chemical analyses in our research group). One may then ask how parasitoids face this chemical diversity and yet manage to find hosts and do host shifts. Most Galerucella species are only parasitized during the larval stage by Asecodes spp, but some species are also attacked by other hymenopteran taxa. The female parasitoids mainly attack the early larval stages, and lay multiple eggs in one larvae. In our study area, hosts and parasitoids typically have one generation per year but exceptions do occur.
In this paper, we use multilocus sequence data to test whether A. lucens attacking Galerucella larvae represent one generalist or multiple specialist species. For this purpose, we collected Galerucella larvae from most of their respective host plant species, reared them to adults to collect the parasitoids and sequenced them. For comparative purposes, we also sequenced three genes of the collected Galerucella species, to complement previous analyses [41]. Bayesian species delimitation suggested that A. lucens consists of 3–4 cryptic taxa with a more specialised host use than previously suggested. The gene data analyzed under the multispecies coalescent model allowed us to construct the species tree for both host and parasitoid and we found a fully congruent coevolutionary pattern suggesting that parasitoid speciation followed upon host speciation.
Methods
Study species and sampling
Specimens of Galerucella (Coleoptera) and Asecodes lucens (Nees) (Hymenoptera) were collected in Sweden and Finland during 2011 (Figure 1, for details see Additional file 1: Table SI-1). Larvae of Galerucella were collected on their host plant, and subsequent rearings provided either adult beetle or parasitoid specimens in the lab. One species, G. nymphaeae, was collected but was not found to be parasitized and is therefore excluded from the data set. All specimens were stored in 99% ethanol. The sampling in Sweden was mainly from Uppland and Västmanland but parasitoids infesting G. tenella and G. calmariensis were also sampled from northern localities. In particular, a systematic sampling was performed in Skeppsvik archipelago in Umeå, where parasitism rates are typically much higher than in southern localities (> 70% vs. < 10%).

Sampling locations for Galerucella and Asecodes specimens in Sweden and Finland. Circles refer to sites were Galerucella were sampled and triangles refer to the collection sites of Asecodes.
As outgroups to the Galerucella dataset we downloaded sequences of Trirhabda bacharidis, Pyrrhalta spp. and Ophraella communa (Galerucinae, Chrysomelidae) from GenBank. Galerucella sequences available on GenBank were also downloaded and included in our dataset (see Additional file 1: Table SI-1). In the Asecodes dataset we included part of the CO1 sequence of Horismenus missouriensis, H. petiolatus and Pediobius spp. (Entedoninae, Eulophidae) as well as Quadrastichus haitiensis (Tetrastichinae, Eulophidae) as outgroup species (downloaded from GenBank). Two Pediobius specimens were also reared from G. sagittariae, an host association that was not previously recorded (Additional file 1: Table SI-1).
DNA extraction, amplification and sequencing
Extractions
Specimens for DNA extraction were chosen to represent samples from different collection sites and, in the case of G. sagittariae, to represent different host plant preferences. We used QIAamp® DNA Mini Kit (QiaGen) for the DNA extraction, where one to two legs per specimen were used for Galerucella while two to four legs were used for Asecodes. Legs were put in ATL lysis buffer and proteinase K and left in 56°C overnight. The purification procedure was carried out according to the manufacturer’s protocol except that the Asecodes extracts were eluted in 100 μl AE buffer.
DNA amplification
For both Asecodes and Galerucella we amplified the mitochondrial DNA of cytochrome oxidase 1 (CO1) gene and two nuclear genes; the internal transcribed spacer (ITS2) from the nuclear ribosomal region and the D2 region of the 28S ribosomal subunit. CO1 was amplified in two fragments without overlap using the primer pairs LCO-1490 & HCO-2198 [42] and C1-J-2183 (Jerry) & TL2-N-3014 (Pat) [43]. For the amplification of the nuclear genes we used the primer pairs ITS2f & ITS2r [44] and 28S_D2_F & 28S_D2_R [44] respectively. For Asecodes we also amplified the nuclear fragment of phosphogluconate dehydrogenase (PGD) using the primer pair PGD_hym_3F & PGD_hym_intRb (Malm unpubl). PCR reactions were performed with Ready-ToGo™ PCR beads (Amersham Biosciences) in a total reaction volume of 25 μl only adding 1 μl each of forward- and reverse primers (10 μM), 2 μl template and dH2O. The cycling profile started with a 5 min denaturation step at 95°C, followed by 38 cycles of 30 s at 95°C, annealing for 30 s at 49°C (ITS2), 50°C (CO1), 60°C (28S), extension for 1 min at 72°C, and a final extension step of 8 min at 72°C. For amplification of PGD, 40 cycles were run, 45 s was used in the denaturation step and annealing was performed for 40 s at 54°C in each cycle. The results were visualized on an agarose gel and stained with GelRed™ (Life Technologies). The PCR products were cleaned using Exonuclease I and FastAP (Fermentas) and the sequencing reactions were performed with BigDye™ Terminator ver. 3.1 Cycle Sequencing Kit (Applied Biosystems). The same primers as for the PCR reactions were used. The sequencing reactions were then cleaned with DyeEx 96 kit (QIAGEN) and run on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems). Genetic fragments were sequenced in both forward and reverse directions. To align the ITS sequences we used the program Mafft [45] and the iterative refinement method which incorporates global pairwise alignment information. All other sequences were aligned and edited by eye in BioEdit v7.0.9 [46].
Phylogenetic analyses
Galerucella terminals were assigned to species based on which host plant they were sampled as well as morphological identification. The Asecodes terminals were assigned to which host they parasitized; e.g., Asecodes/G. lineola refers to an Asecodes specimen hatched from G. lineola.
We used MrModeltest2 v.2.3 [47] in conjunction with PAUP [48] to select the best model for the Bayesian analyses, although the substitution rate matrix was not selected a priori (see below). For CO1 the 1st, 2nd and 3rd codon positions were analyzed separately. Substitution model details for the Galerucella and Asecodes datasets are specified in Additional file 1: Table SI-2. The Bayesian phylogenetic analyses were performed with MrBayes 3.1 [49, 50] at the online Bioportal server (University of Oslo, Norway). The Galerucella dataset consisted of 53 terminals representing eleven Galerucella species and two outgroup taxa for the CO1 + nuclear gene dataset and 19 terminals representing five Galerucella species and two outgroup genera for the nuclear gene dataset. The Asecodes dataset (CO1 + nuclear genes) included 107 taxa and six outgroup taxa. Each gene was analyzed separately (except the 28s for Asecodes) as well as combined, to evaluate the relative information content in each gene fragment. In the datasets including more than one partition (CO1) or including several genes, the substitution model was set to “mixed”, which implements reversible-jump MCMC across the entire space of 203 reversible 4 × 4 nucleotide substitution models [51]. All model parameters except branch lengths and topology were unlinked and relative rates between partions were allowed with a rate multiplier. We made two separate runs completing 10 million generations, each with four incrementally heated chains (T=0.2) and where sampling was done every 1000th generation. The first 2 500 trees from each run were discarded as burn-in and the remaining samples pooled before calculating the majority-rule consensus. We checked that the separate analyses had converged using the average deviation of split frequencies diagnostic (< 0.02 in all runs), and the potential scale reduction factor (close to 1.00 for all parameters).
We used the multispecies coalescent model [30] as implemented in *BEAST (‘Starbeast’) to infer the species tree from multiple gene trees for both Galerucella and Asecodes. In the multispecies coalescent model all model parameters are unlinked across loci, including the topology parameter, which allows the gene trees to differ in topology while being constrained by one and the same species tree. The Galerucella dataset consisted of 18 terminals from five species and for one mitochondrial (CO1) and two nuclear loci (28S and ITS). Substitution models were set according to selected model for each loci by MrModeltest [47]. We applied a strict clock model on branch lengths and calibrated the CO1 partition to 0.0177 substitutions per site per million years after a recent CO1 clock rate estimate of another Polyphaga beetle family [52].
The Asecodes dataset consisted of 38 terminals from four species (as identified by the Bayesian species delimitation method, see below) and for one mitochondrial (CO1) and three nuclear loci (PGD, ITS and 28S). We applied a strict clock model on branch lengths and calibrated the species tree by applying a normal distribution prior (mean = 4.4, stdev = 0.7) on the root node according to the 95% HPD interval of the estimated host root node age. The purpose of this calibration was to see if, under the assumption of contemporary speciation at the root node, branching events after the root node predate or postdate respective branching event in the host tree. While the latter would be in agreement with (but does not ascertain) cospeciation, the former would falsify any such hypothesis. Note that the substitution rate calibration in the Galerucella analysis is applied to the CO1 gene tree whereas the root node calibration in the Asecodes analysis is applied to the species tree. The latter was enabled by hand editing of the xml file following McCormack et al. [53].
Both analyses were run four times independently for 100 million MCMC generations and sampled every 1000 generation. Tracer [54] was used to examine the convergence across runs and the ESS values of sampled parameters. After 10% of each run was discarded as a burn-in, the remaining samples from all runs were pooled and the maximum clade credibility species tree was calculated using the mean node heights.
Species delimitation analyses
To test if the Asecodes parasitoid on Galerucella consists of one generalist species parasitizing multiple host species, or several specialists each attacking a single host, we used a Bayesian species delimitation method described by Yang and Rannala [32]. The method as applied in the software BPP uses reversible-jump MCMC to sample different species delimitation models and estimate the posterior probability of each model. BPP accommodates both uncertainty in gene tree estimations by the Bayesian framework, and incomplete lineage sorting of species via the coalescent process model (both in contrast to the GMYC method below). This statistical power increases both with the addition of loci and individuals per species [55]. BPP needs a guide-tree as input for the analysis and both the number of terminals in the guide-tree and its topology can influence the result [56]. BPP only estimates the posterior probability of all possible ways to collapse the nodes in the guide-tree into fewer species. It does not test any alternative groupings of individuals into species that cannot be derived from collapsing a node in the guide-tree, nor does it test to split the dataset into more species than the given terminals in the guide-tree. The dependency on a guide-tree effectively limits the search space and was a practical necessity introduced by Yang and Rannala [32]. As such, BPP is not suitable for blind biodiversity assessment, but explicit a priori species hypotheses need to exist to avoid artificial divisions like the smallest geographical sampling localities [56]. In this respect, parasitoids have the advantage of having natural and biologically relevant units, the host species, which in our analysis serve as delimiters of the maximal number of terminal units (five). We also tested guide-trees with four and three terminals, which only confirmed the relevant subset of results from the five terminal guide-tree analysis and will not be discussed further. The guide-tree topology was taken from the *BEAST analysis, all nodes of which were supported by a posterior probability of 1.0 and hence we did not need to test the effect on the species delimitation of any alternative plausible topology as guide-tree (compare with 56).
To calculate the likelihood of different species delimitations, a model with two types of parameters is needed (the number of parameters depend on the number of species in the model): Θ = 4Neμ and τ. Θ is a product of the effective population size and the mutation rate and τ is the root age measured in expected number of substitutions per site to the tips. Disregarding variations in the mutation rate, a relatively large value of Θ means a large effective population size and a relatively large value of τ specifies an ancient divergence. Each terminal species in the species delimitation model and each ancestral internode (ancestral species) have a Θ parameter whereas each split between species has a τ parameter. In a Bayesian framework both parameters need a prior which is specified by a gamma distribution in BPP. The gamma distribution is defined by two numbers, α and β, and the mean of the distribution is α/β. The mean should not be orders of magnitude away from the posterior of the parameter as estimated from the data, otherwise the prior can cause flawed results [55]. As a diffuse prior we used α = 1 in all analysis and tested various combinations of β for Θ and τ around the estimated posterior for the parameters. The estimated posterior estimates were Θ ≈ 0.05 and τ ≈ 0.02 (see Additional file 1: Table SI-3). All analyses were therefore run in four combinations of prior gamma distributions: 1: Θ: G(1,10), τ: G(1,10), 2: Θ: G(1,10), τ: G(1,100), 3: Θ: G(1,100), τ: G(1,10) and 4: Θ: G(1,100), τ: G(1,100), representing all combinations where the mean is between 0.1 and 0.01 and which includes the posterior estimates for both parameters. We also tested the effect of the number of loci on the power of the method. To do that we sequentially added loci in the order 1 locus (CO1), 2 loci (CO1+PGD), 3 loci (CO1+PGD+ITS), 4 loci (CO1+PGD+ITS+28S). It could be argued that the adjacent ITS and 28S gene segments should be analyzed as linked, but because of the complex pattern of recombination and concerted evolution in the tandemly arrayed rDNA loci [57], we prefer to analyze them as separate loci. If in doubt about the independence between the two loci, the result from the 3 loci analyses can be consulted. Each analysis of number of loci and prior combination was run 4 times independently to check the convergence of results and variation of estimated posterior probability for species delimitation models. Each analysis consisted of 50000 MCMC generations sampled every 5th generation and discarding 10% as burn-in. The two different algorithms for the reversible-jump MCMC described by Yang and Rannala [32] and named “0” and “1” gave similar results and we therefore only report the result from using the algorithm 0.
The GMYC analysis was performed in R statistical package with the help of ape, gee, MASS, paran and splits packages. The GMYC method takes as input an ultrametric gene tree which we derived from calculating the maximum clade credibility tree with mean node heights from the sampled CO1 gene trees in a BEAST analysis using a strict molecular clock and a codon-partitioned model according to results from MrModeltest. The method calculates the likelihood of a mixed Yule-coalescence model applied with a single threshold across the tree at every node. At each threshold, deeper nodes are modeled as speciation events according to a Yule model whereas each group of younger nodes is modeled separately according to the coalescent process model. The maximum likelihood solution generally identifies the ‘kink’ or point of increased diversification rate in a lineage-through-time plot of trees with multiple individuals per species for multiple species. This node serves as a species delimitation point under the assumption of species monophyly and not permitting any speciation event to be younger than the deepest coalescence. The maximum likelihood of the GMYC model was tested with a likelihood ratio test against a null model treating the entire tree as a single coalescent (i.e. against a one-species model).
Results
In total the Galerucella alignment consisted of 2766 basepairs whereas the alignment of Asecodes contained 3089 basepairs (see Additional file 1: Table SI-4 for details).
Phylogenetic analysis
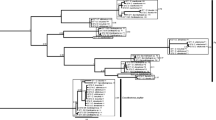
The multispecies coalescent model for Galerucella gave a well supported species tree with the topology (G. sagittariae (G. lineola (G. tenella (G. pusilla + G. calmariensis)))). Subgenus Neogalerucella (including G. lineola, G. tenella, G. calmariensis and G. pusilla) and subgenus Galerucella (including G. sagittariae) were monophyletic as defined by Borghuis et al. (2009), and G. lineola was basal in the Neogalerucella clade. All nodes had a posterior probability support of >0.99 and the age of the root node was estimated to 4.3 my (95% HPD: 3.0 – 5.9) with the mitochondrial clock calibration (Figure 2). The standard concatenated model with unconstrained branch lengths (non-clock model aka time-free model) of all genes combined resolved the species tree as the coalescent model to the same topology with high branch support (>0.99), but is here rooted using outgroups and includes a few additional species in the G. sagittariae group (Figure 3a). Analyzed alone, the mitochondrial CO1 gene tree is rooted differently compared to the nuclear (Figure 3b) and combined tree (CO1 and nuclear genes), which justifies the multispecies coalescent approach where gene trees are allowed to differ. It is interesting to note that the closely related G. calmariensis and G. pusilla were recovered as reciprocally monophyletic in the analyses of the nuclear genes but not by our CO1 data or by the mitochondrial data in Borghuis et al.[41]. The divergence date in the species tree is estimated to 77 000 years ago (95% HPD: 19000–148000).

Topologies of Galerucella and Asecodes species, analysed with multispecies coalescent models in *BEAST. The analyses are based on sequences of CO1, 28S, ITS (Galerucella and Asecodes) and PGD (Asecodes). Branch labels indicate posterior probability values and the bars refer to the confidence interval of the node ages. The Galerucella dataset was rooted using a strict clock while the rooting of Asecodes was calibrated after the estimated age of Galerucella. The Asecodes specimens are coded as “Asecodes /the name of the Galerucella species that is parasitized”.

Phylograms from Bayesian analysis of Galerucella species. (a) Phylogram based on concatenated CO1, 28S and ITS sequences (2677 basepairs). (b) Phylogram based on the nuclear sequences of 28S and ITS (1203 basepairs). Branch labels indicate posterior probability values.
The multispecies coalescent model for Asecodes with species defined according to the BPP analysis resolved the species tree to (Asecodes/G. sagittariae (Asecodes/G. lineola (Asecodes/G. tenella + Asecodes/G. pusilla/calmariensis)) with all posterior probability branch support 1.0 (Figure 2). Note that, as in the Galerucella analysis, the root was inferred from the clock model and not outgroups. As this is in perfect agreement with the phylogeny of the hosts, we calibrated the root node after the estimated age on the equivalent host chronogram node. For this we used the 95% highest posterior density of the host root node height to define a normal distribution (4.3, 0.7) as a prior on the parasite root node that takes the calibration uncertainty into account. This resulted in the node for the split of Asecodes on G. lineola and the node for the split of Asecodes on G. tenella to postdate the equivalent nodes in the host tree. The opposite would have been a potential falsifier of a cospeciation scenario.
With a standard concatenated model time-free (non-clock) model, and using outgroups to root the tree in a Bayesian analysis, three distinct clades were recovered corresponding to parasitoids on G. sagittariae, G. lineola and G. tenella-calmariensis-pusilla. The Asecodes specimens that infest Galerucella sagittariae were recovered as sister to the remaining Asecodes with high posterior probability, in agreement with the multispecies coalescent model. Asecodes on G. sagittariae and on G. lineola were each recovered as reciprocally monophyletic with high posterior probability support (1.0 and 0.91 respectively). Asecodes on G. tenella, G. calmariensis and G. pusilla however were not recovered as reciprocally monophyletic but were mixed in one large cluster, with strong branch support (1.0) (Figure 4). The CO1 gene tree was identical in topology to the combined tree whereas the nuclear dataset alone gave a completely unresolved topology in the ingroup because of few variable sites (Additional file 1: Figure SI-1). This gives an indication of the relative importance of different genes to the combined result. 28S had only a single variable site that separated Asecodes on G. sagittariae and G. lineola from the remaining parasitoids. Two variable positions defined the G. sagittariae parasitoids in the ITS gene fragment.

Phylogram from Bayesian analysis of Asecode specimens. (a) Phylogram based on concatenated CO1, PGD, 28S and ITS sequences (3124 basepairs). Due to short branches and lack of structure within the three clades, they are visualized as triangles codes as “Asecodes /the Galerucella species which are parasitized”. The three clusters include 31 (“Asecodes/G. sagittariae”), 57 “Asecodes/G. tenella-pusilla-calmariensis” and 19 “Asecodes/G. lineola” individuals. (b) Result of the GMYC analysis on an ultrametric CO1 gene tree from BEAST. Splits of thick black branches indicate speciation events whereas splits of thin black branches indicate within-species coalescence events. Grey branches are ambiguous in the GMYC analysis and all solutions from 3 to 7 species are included in a +/− 0.5 Log likelihood confidence interval. The single coalescence one-species model is rejected in a likelihood ratio test p=0.011.
Species delimitation
The BPP analyses yielded conclusive results and multiple runs with the same settings were in close agreement. Independent of the prior combinations used, the species delimitation model with highest posterior probability was the four-species model (Figure 5). The four-species model delimits a separate specialist Asecodes species each for G. sagittariae, G. lineola and G. tenella but a common species for G. pusilla and G. calmariensis. This model ranged in posterior probability from 0.73 to 0.99 (0.83 to 0.99 when analyzing all four genes) (Figure 5). The second best model was the five-species model where also Asecodes on G. pusilla and G. calmariensis was divided into separate species with a posterior probability of 0.002 to 0.27. The three-species model separating only Asecodes on G. sagittariae, G. lineola and remaining Galerucella had a marginal but non-zero support and the posterior probability for the two- and one-species model was 0 across all prior space investigated.

Mean posterior probability value of species delimitation models. The priors Θ and τ refer to the ancestral population size and age of root respectively. The bars indicate standard deviation. a. A four-species model and different number of loci for priors on Θ and τ. Loci: 1 - CO1; 2 - CO1, PGD, 3 - CO1, PGD, ITS; 4 - CO1, PGD, ITS, 28S. b. Analyses using all four genes resulted in highest support for the four-species model followed by the five-species model. c. Analyses using all four genes but only including the Asecodes specimens infesting G. tenella and G. calmariensis from the sympatric population in the northern part of Sweden (but including all other specimen infesting G. sagittaria, G. lineola and G. pusilla).
The effect of the number of loci was most noticeable between one (CO1) and two (CO1 + PGD) loci which yielded increased support for the four-species model. The third (ITS) and fourth (28S) loci contained only three and one variable sites respectively and had therefore marginal discriminatory power between species delimitation models. In fact, with the prior on Θ set to a relatively large population size G(1,10) the addition of ITS and 28S resulted in a decreased probability for the same model (Figure 5a).
The prior on the root age τ had very little effect on the posterior of delimitation models. With four loci and the prior combination Θ: G(1,10), τ: G(1,10) support for the best model was 0.83-0.84 and remained unchanged with Θ: G(1,10), τ: G(1,100). The effect of the prior on Θ was more noticeable and with the same priors on τ but with G(1,100) on Θ instead of G(1,10), the posterior probability increased to 0.97-0.98 for the best model. The support for three separate Asecodes species, on G. sagittariae, on G. lineola and on the remaining Galerucella species, is conclusive and never received a posterior probability of less than 1.0. The support for a fourth species Asecodes on G. tenella was strong but not entirely conclusive, as the three-species model treating Asecodes on G. tenella, G. calmariensis and G. pusilla as a single species also received some support (up to a posterior probability of 0.1 in one analysis). In addition, as the interpretation of allopatric populations with limited gene flow is a controversial issue we tested the BPP method on a sympatric subset of the data with only all Asecodes on G. tenella and G. calmariensis sampled from the same localities in northern Sweden. Used on sympatric populations, species defined unambiguously in the BPP analysis should be uncontroversial [55]. For the same four prior combinations, the posterior probability was more evenly spread across the three-, four- and five-species models with zero probability for the one- and two-species models (Figure 5c). The five-species model received mean posterior support of 0.41-0.72. That the dataset consist of at least three species, as opposed to one, is therefore conclusive, but to be conservative against oversplitting, the three-species model cannot be rejected even though it is the least supported of the three.
The GMYC model delimited five species with the threshold at the maximum likelihood solution (Figure 4b). The log likelihood of the GMYC model at the optimal threshold (247.2) was significantly better than the null model of a single coalescent (241.7) in a likelihood ratio test (p<0.011) and a one-species model could be rejected. However, the delimitation of three, four, five, six and seven species was less than 0.5 log likelihood units away from the maximum likelihood solution and should therefore be included within a reasonable confidence limit. In contrast to the BPP, the GMYC method does not accommodate incomplete lineage sorting, but assumes species monophyly. As the CO1 gene tree does not resolve Asecodes individuals parasitizing G. tenella, G. calmariensis or G. pusilla as reciprocally monophyletic, the delimitation of four or five species is not exactly the same as that assumed and tested in the BPP analysis. The GMYC method applied to the reduced sympatric dataset delimited three species at the maximum likelihood solution (data not shown).
Discussion
The analysis of mitochondrial and nuclear genetic markers indicate that Asecodes lucens is not one generalist species but at least three species with a more narrow host use. Two parasitoid species seem to be specialist on their respective host, Galerucella sagittariae and G. lineola. The three remaining Galerucella species are seemingly attacked by the same parasitoid species, even though these parasitoids also show some population differentiation. There were tendencies that the parasitoids attacking G. tenella, which feeds on F. ulmaria, had diverged from the parasitoids attacking G. calmariensis and G. pusilla, which both feed on L. salicaria. The mitochondrial dating furthermore show that speciation in parasitoids are sequential events following speciation in their host, and a recent but perhaps not completed split between parasitoids attacking G. tenella and G. calmariensis/pusilla, would follow this pattern.
The derived phylogeny of Galerucella matches the previous analysis by Borghuis et al. [41] where the split between what is considered two subgenera, Galerucella and Neogalerucella, is most ancient. In our data set, the subgenus Galerucella only includes G. sagittariae whereas the other species belong to the subgenus Neogalerucella. Borghuis et al.[41] based their analysis only on mitochondrial gene fragments (CO1 and NADH-2), and found strong support for the monophyly of G. tenella and G. lineola but could not resolve G. pusilla and G. calmariensis as reciprocally monophyletic, indicating either a recent divergence or mitochondrial introgression. Our study also included two nuclear DNA fragments (the D2 region in 28S and ITS2), which provides a test of the alternative explanations. Although our nuclear DNA datasets were small, divergence between G. pusilla and G. calmariensis was strongly supported. These two taxa both use Lythrum salicaria (Lythraceae) as host plant, and both ecological and morphological information suggest that the two species are reproductively isolated. The male copulatory organs are distinctly different, the body size differs and both larval and adult colour differs [58, 59]. In these characters, there is quite small overlap. In addition, Nokkala and Nokkala [60] found karyotypic differences which further support species status. In contrast to the conclusion drawn by Borghuis et al.[41], based on the results from mitochondrial gene fragments, the lack of monophyly in the mitochondrial genes for G. pusilla and G. calmarensis might rather indicate some recent “phenomenon” such as introgression, Wolbachia infestation etc. However, a larger sample from each population is necessary to establish if lineage sorting is indeed complete for the nuclear markers, which theoretically should sort slower than mitochondrial counterparts. Due to the maternal inheritage of the mitochondrion, effective population size is lower (1/4th that of nuclear genes) and lineage sorting is therefore faster than for nuclear genes.
The Bayesian species delimitation analysis for the A. lucens group was performed on the same set of nuclear and mitochondrial genes as the analysis for Galerucella, with the addition of the nuclear gene PGD, and provided strong evidence for population differentiation. It is possible to conclude that A. lucens should be split at least into three species but additional data may strengthen the indication of further splits; the species delimitation analysis suggested 3–5 species. The molecular data have been confirmed by morphological studies that found differences in wing patterns among at least 3 taxa [61]. Comparisons with type specimens of available (synonymized) names suggested the identity of two taxa, A. lucens parasitizing G. sagittariae and A. parviclava (Thomson) parasitizing G. tenella, G. calmariensis and G. pusilla. A third species, parasitizing G. lineola, represents a new species named A. lineophagum Hansson & Hambäck [61]. There were no morphological characters supporting a further subdivision of A. parviclava.
The BPP method distinguishes populations as different species if the per generation migration rate Nm << 1 [55], and the interpretation is therefore unambiguous in sympatry but distance-decay patterns in allopatry may alone contribute to partial genetic isolation. Allopatry could explain why the analysis with all data included identified population differentiation between parasitoids collected on G. tenella versus G. pusilla/calmariensis. In the southern area, populations of G. tenella and G. pusilla/calmariensis are often found in slightly different habitats and were typically collected in different localities. In the northern area, where population differentiation was seemingly weaker, G. tenella and G. calmariensis co-occur on Baltic shore lines and were collected in the same localities. It is possible that the southern population of A. parviclava is in an early stage of speciation, but the current data are insufficient to confirm this suspicion.
The possibility of geographic variation in population differentiation of parasitoids is very interesting considering the previously documented parasitoid mediated indirect interactions between G. tenella and G. calmariensis in northern localities. Hambäck et al. [11] found that parasitism rates on G. tenella were higher at sites where this species co-occur with G. calmariensis compared with sites without G. calmariensis. The differences in parasitism rates translated into differences in the strength of interactions between the herbivore and its host plant [12]. In sites with L. salicaria and G. calmariensis, the attack rates on F. ulmaria by G. tenella were lower and the seed set were higher compared to sites without L. salicaria. Other studies show that this apparent competition between the Galerucella species may have evolutionary consequences for F. ulmaria. Galerucella tenella has a very strong impact on plant fitness and studies on differently aged populations suggest that G. tenella causes a shift in the population towards plants with a lower height and with higher concentrations of potential antiherbivore compounds [62]. In the field, the lower quality of F. ulmaria as food for the larvae of G. tenella caused the beetle to expand its diet towards other Rosaceae plants [63]. Preliminary data suggest that these evolutionary changes in F. ulmaria only occur on islands without L. salicaria (Ericson & Stenberg, unpublished data). The hypothesis for these effects was that the parasitoids use both G. tenella and G. calmariensis, even though the behavioural mechanisms are not fully understood [64]. It was therefore important to know whether parasitoids collected from the two host were indeed the same population and this seems to be the case. The potential for a larger population differentiation in southern localities suggest that a similar apparent competition is not likely in these localities, and the parasitism rates in these localities are also typically much lower (<10% vs >70%).
Speciation in the parasitoids for this system seem to follow the identity of the beetle host rather than the identity of the beetle’s host plant. This finding suggests that host finding or recognition cues originate from the larval host rather than from the host plant, such as beetle produced pheromones or beetle specific plant cues. Two contrasts in particular supports this view. First, G. sagittariae feed on multiple host plant species that are not closely related, but we nevertheless found no population differentiation among parasitoids hatching from larvae collected on different plants. Second, G. sagittariae and G. tenella are known to feed on the same host plant, but we find that their respective parasitoid belong to different taxa. It seems less likely that plant produced volatiles provide sufficient information for parasitoids to both differentiate between G. sagittariae and G. tenella, and at the same time to locate G. sagittariae on its different host plants. Beetle produced compounds therefore seem more likely. Previous studies show that at least two Galerucella species (G. pusilla and G. calmariensis) produce aggregation pheromones [65], but these compounds are produced by adult males and seem less likely to provide any information on the whereabouts of Galerucella larvae. To further understand the speciation process in Asecodes parasitoids, we are currently working to identify the compounds involved during the search process.
Speciation in parasitoids may follow different pathways according to published data, and the causes underlying this variability is not well understood. There are cases with host-association differentiation and seemingly tight cospeciation [66, 67], similar to the one suggested in this study for the Galerucella-Asecodes system. In other cases, speciation in parasitoids is less well connected to the phylogeny of their host [68, 69]. The current data both in this and most of the published cospeciation examples cannot differentiate whether this is ecological speciation where hosts or parasitoid species are not geographically isolated or cladogenesis where host and parasitoids are isolated in pairs [70]. Differences in the speciation pattern may arise because the parasitism process involves several steps that create barriers to parasitoids when their host switches diet. Diet switching and speciation in herbivorous insects is often connected to enemy free space where the host is more difficult to either find or exploit on an alternate plant [71–73]. Different plants may produce quite different volatile profiles upon damage and this may cause problems for parasitoid females to either find their host or even to identify the larvae as such. There are cases where a parasitoid species has geographic variability in the type of cues used during host search [74], but this area of research is poorly exploited. Besides affecting host search cues, plant quality is also known to affect herbivore immunocompetence and switching to an alternate plant may affect resistance to parasitoid attack. The Galerucella-Asecodes system seems ideal to study these processes in progress, both because host race formation is common and well documented among Galerucella beetles [75–77] and because interaction strengths between host and parasitoid show quite large geographic differences. Further studies on species interactions in this system however necessitates a better understanding also on the small scale population differentiation, to identify strengths of direct and indirect species interactions and their ecological and evolutionary consequences.
Conclusion
Our analysis, using multilocus sequence data in a Bayesian species delimitation analysis, confirms the phylogenetic structure within the chrysomelid beetle genus Galerucella but also that the parasitoids attacking Galerucella larvae should be subdivided into at least three species. This subdivision has later been confirmed with morphological data. The previous literature suggested that one species of the genus Asecodes attacked Galerucella larvae but our analysis show that the parasitoids have a more narrow diet breadth. The evolution of parasitoid within Asecodes and the host Galerucella show a fully congruent coevolutionary pattern, even though the specific mechanism cannot be identified with the current data. The phylogeny of Asecodes show that host use is not connected to the plant species that host the beetle larvae as different host larvae on different plant species are attacked by different Asecodes species. This finding strengthens the hypothesis that the parasitoid uses cues of the host during search rather than more general cues of both host and plant. Previous studies within the Asecodes-Galerucella system has suggested that parasitoid-mediated interactions may be important for both beetle densities and plant performance. The observation that parasitoids in fact have a more narrow diet breadth suggests that such indirect interactions may be restricted to subsets of the food web involving these two genera.
Availability of supporting data
The gene data set supporting the results of this article is available in Genbank (http://www.ncbi.nlm.nih.gov/genbank/), and accession numbers are found in Additional file 1: Table SI-1. The aligned matrices and trees of the Galerucella analyses (nuclear dataset and COI + nuclear dataset) and the Asecodes analysis (COI + nuclear dataset) are available at TREEBASE under URL http://purl.org/phylo/treebase/phylows/study/TB2:S14144.
References
Holt RD, Kotler BP: Short-term apparent competition. Am Nat. 1987, 130: 412-430. 10.1086/284718.
Brassil CE, Abrams PA: The prevalence of asymmetrical indirect effects in two-host-one-parasitoid systems. Theor Popul Biol. 2004, 66: 71-82. 10.1016/j.tpb.2004.04.001.
van Veen FJF, Morris RJ, Godfray HCJ: Apparent competition, quantitative food webs, and the structure of phytophagous insect communities. Annu Rev Entomol. 2006, 51: 187-208. 10.1146/annurev.ento.51.110104.151120.
Holt RD: Predation, apparent competition, and the structure of prey communities. Theor Popul Biol. 1977, 12: 197-229. 10.1016/0040-5809(77)90042-9.
Holt RD, Lawton JH: Apparent competition and enemy-free space in insect host-parasitoid communities. Am Nat. 1993, 142: 623-645. 10.1086/285561.
Schmitt RJ: Indirect interactions between prey: apparent competition, predator aggregation, and habitat segregation. Ecology. 1987, 68: 1887-1897. 10.2307/1939880.
Bonsall MB, Hassell MP: Parasitoid-mediated effects: Apparent competition and the persistence of host-parasitoid assemblages. Res Popul Ecol (Kyoto). 1999, 41: 59-68. 10.1007/PL00011983.
Hudson PJ, Greenman JV: Apparent competition mediated by parasites: Biological and theoretical progress. Trends Ecol Evol. 1998, 13: 387-390. 10.1016/S0169-5347(98)01475-X.
Tompkins DM, Draycott RAH, Hudson PJ: Field evidence for apparent competition mediated via the shared parasites of two gamebird species. Ecol Lett. 2000, 3: 10-14. 10.1046/j.1461-0248.2000.00117.x.
Waloff N: Observations on the heather beetle Lochmaea suturalis (Thomson) (Coleoptera, Chrysomelidae) and its parasitoids. J Nat Hist. 1987, 21: 545-556. 10.1080/00222938700770321.
Hambäck PA, Stenberg JA, Ericson L: Asymmetric indirect interactions mediated by a shared parasitoid: connecting species traits and local distribution patterns for two chrysomelid beetles. Oecologia. 2006, 148 (3): 475-481. 10.1007/s00442-006-0387-2.
Stenberg JA, Heijari J, Holopainen JK, Ericson L: Presence of Lythrum salicaria enhances the bodyguard effects of the parasitoid Asecodes mento for Filipendula ulmaria. Oikos. 2007, 116: 482-490. 10.1111/j.0030-1299.2007.15357.x.
Hippa H, Koponen S: Parasitism of larvae of Galerucini (Col., Chrysomelidae) by larvae of Asecodes mento (Hym., Eulophidae). Reports from the Kevo Subarctic Research Station. 1984, 19: 63-65.
Hansson C: The status of the genera Asecodes Förster, Ionympha Graham and Teleopterus Silvestri (Hymenoptera: Eulophida), with a review of Nearctic species. Entomologica Scandinavica. 1996, 27: 159-167. 10.1163/187631296X00025.
Smith MA, Woodley NE, Janzen DH, Hallwachs W, Hebert PDN: DNA barcodes reveal cryptic host-specificity within the presumed polyphagous members of a genus of parasitoid flies (Diptera: Tachinidae). Proc Natl Acad Sci USA. 2006, 103 (10): 3657-3662. 10.1073/pnas.0511318103.
Kaartinen R, Stone GN, Hearn J, Lohse K, Roslin T: Revealing secret liaisons: DNA barcoding changes our understanding of food webs. Ecological Entomology. 2010, 35 (5): 623-638. 10.1111/j.1365-2311.2010.01224.x.
Zhang YZ, Si SL, Zheng JT, Li HL, Fang Y, Zhu CD, Vogler AP: DNA barcoding of endoparasitoid wasps in the genus Anicetus reveals high levels of host specificity (Hymenoptera: Encyrtidae). Biol Control. 2011, 58 (3): 182-191. 10.1016/j.biocontrol.2011.05.006.
Stigenberg J, Ronquist F: Revision of the Western Palearctic Meteorini (Hymenoptera, Braconidae), with a molecular characterization of hidden Fennoscandian species diversity. Zootaxa. 2011, 3084: 1-95.
Smith MA, Rodriguez JJ, Whitfield JB, Deans AR, Janzen DH, Hallwachs W, Hebert PDN: Extreme diversity of tropical parasitoid wasps exposed by iterative integration of natural history, DNA barcoding, morphology, and collections. Proc Natl Acad Sci USA. 2008, 105 (34): 12359-12364. 10.1073/pnas.0805319105.
Hebert PDN, Penton EH, Burns JM, Janzen DH, Hallwachs W: Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc Natl Acad Sci USA. 2004, 101 (41): 14812-14817. 10.1073/pnas.0406166101.
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP: Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol. 2006, 55 (4): 595-609. 10.1080/10635150600852011.
Funk DJ, Omland KE: Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu Rev Ecol Syst. 2003, 34: 397-423. 10.1146/annurev.ecolsys.34.011802.132421.
Bergsten J, Bilton DT, Fujisawa T, Elliott M, Monaghan M, Balke M, Hendrich L, Geijer J, Herrmann J, Foster G, Ribera I, Nilsson AN, Barraclough TG, Vogler AP: The effect of geographical scale of sampling on DNA barcoding. Syst Biol. 2012, 61: 851-869. 10.1093/sysbio/sys037.
Hickerson MJ, Meyer CP, Moritz C: DNA barcoding will often fail to discover new animal species over broad parameter space. Syst Biol. 2006, 55 (5): 729-739. 10.1080/10635150600969898.
Rosenberg NA: The shapes of neutral gene genealogies in two species: Probabilities of monophyly, paraphyly, and polyphyly in a coalescent model. Evolution Int J Org Evolution. 2003, 57 (7): 1465-1477.
Hudson RR, Coyne JA: Mathematical consequences of the genealogical species concept. Evolution Int J Org Evolution. 2002, 56 (8): 1557-1565.
Degnan JH, Rosenberg NA: Discordance of species trees with their most likely gene trees. PLoS Genet. 2006, 2 (5): 762-768.
Degnan JH, Rosenberg NA: Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol. 2009, 24 (6): 332-340. 10.1016/j.tree.2009.01.009.
Maddison WP: Gene trees in species trees. Syst Biol. 1997, 46 (3): 523-536. 10.1093/sysbio/46.3.523.
Heled J, Drummond AJ: Bayesian Inference of Species Trees from Multilocus Data. Mol Biol Evol. 2010, 27 (3): 570-580. 10.1093/molbev/msp274.
Liu L, Pearl DK: Species trees from gene trees: Reconstructing Bayesian posterior distributions of a species phylogeny using estimated gene tree distributions. Syst Biol. 2007, 56 (3): 504-514. 10.1080/10635150701429982.
Yang ZH, Rannala B: Bayesian species delimitation using multilocus sequence data. Proc Natl Acad Sci USA. 2010, 107 (20): 9264-9269. 10.1073/pnas.0913022107.
Camargo A, Morando M, Avila LJ, Sites LW: Species delimitation with ABC and other coalescent-based methods: a test of accuracy with simulations and an empirical example with lizards of the Liolaemus darwinii complex (Squamata: Liolaemidae). Evolution Int J Org Evolution. 2012, 66: 2834-2849. 10.1111/j.1558-5646.2012.01640.x.
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, Ingram KK, Das I: Cryptic species as a window on diversity and conservation. Trends Ecol Evol. 2007, 22 (3): 148-155. 10.1016/j.tree.2006.11.004.
Doutt RL, Nakata J: The Rubus leafhopper and its egg parasitoid: An endemic biotic system useful in grape-pest management. Environ Entomol. 1973, 2: 381-386.
Redman AM, Scriber JM: Competition between the gypsy moth, Lymantria dispar, and the northern tiger swallowtail, Papilio canadensis: Interactions mediated by host plant chemistry, pathogens, and parasitoids. Oecologia. 2000, 125: 218-228. 10.1007/s004420000444.
Cronin JT: Shared parasitoids in a metacommunity: Indirect interactions inhibit herbivore membership in local communities. Ecology. 2007, 88 (12): 2977-2990. 10.1890/07-0253.1.
Vet LEM, Dicke M: Ecology of infochemical use by natural enemies in a tritrophic context. Annu Rev Entomol. 1992, 37: 141-172. 10.1146/annurev.en.37.010192.001041.
Janz N, Nylin S: The role of female search behaviour in determining host plant range in plant feeding insects: a test of the information processing hypothesis. Proc Biol Sci. 1997, 264: 701-707. 10.1098/rspb.1997.0100.
Hare JD: Ecological role of volatiles produced by plants in response to damage by herbivorous insects. Annu Rev Entomol. 2011, 56: 161-180. 10.1146/annurev-ento-120709-144753.
Borghuis A, van Groenendael J, Madsen O, Ouborg J: Phylogenetic analyses of the leaf beetle genus Galerucella: Evidence for host switching at speciation?. Mol Phylogenet Evol. 2009, 53 (2): 361-367. 10.1016/j.ympev.2009.07.005.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R: DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994, 3: 294-297.
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P: Evolution, weighting, and phylogenetic utility of mitochondrial gene-sequences and a compilation of conserved polymerase chain-reaction primers. Ann Entomol Soc Am. 1994, 87 (6): 651-701.
Campbell BC, Steffen-Campbell JD, Werren JH: Phylogeny of the Nasonia species complex (Hymenoptera: Pteromalidae): inferred from an internal transcribed spacer (ITS2) and 28S rDNA sequences. Insect Mol Biol. 1994, 2: 225-237. 10.1111/j.1365-2583.1994.tb00142.x.
Katoh K, Misawa K, Kuma K, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acid Research. 2002, 30: 3059-3066. 10.1093/nar/gkf436.
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999, 41: 95-98.
Nylander JAA: MrModeltest v2. Program distributed by the author. 2004, Sweden: Evolutionary Biology Centre, Uppsala University
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. 2002, Sinauer Ass: Sunderland, Massachusetts
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, 17 (8): 754-755. 10.1093/bioinformatics/17.8.754.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19 (12): 1572-1574. 10.1093/bioinformatics/btg180.
Huelsenbeck JP, Larget B, Alfaro ME: Bayesian phylogenetic model selection using reversible jump Markov chain Monte Carlo. Mol Biol Evol. 2004, 21 (6): 1123-1133. 10.1093/molbev/msh123.
Papadopoulou A, Anastasiou I, Vogler AP: Revisiting the Insect Mitochondrial Molecular Clock: The Mid-Aegean Trench Calibration. Mol Biol Evol. 2010, 27 (7): 1659-1672. 10.1093/molbev/msq051.
McCormack JE, Heled J, Delaney KS, Peterson AT, Knowles LL: Calibrating divergence times on species trees versus gene trees: implications for speciation history of Aphelocoma jays. Evolution Int J Org Evolution. 2011, 65: 184-202. 10.1111/j.1558-5646.2010.01097.x.
Rambaut A, Drummond AJ: Tracer v1.4. 200, Available from http://beast.bio.ed.ac.uk/Tracer
Zhang C, Zhang DX, Zhu TQ, Yang ZH: Evaluation of a Bayesian Coalescent Method of Species Delimitation. Syst Biol. 2011, 60 (6): 747-761. 10.1093/sysbio/syr071.
Leache AD, Fujita MK: Bayesian species delimitation in West African forest geckos (Hemidactylus fasciatus). Proc Biol Sci. 2010, 277 (1697): 3071-3077. 10.1098/rspb.2010.0662.
Eickbush TH, Eickbush DG: Finely orchestrated movements: Evolution of the ribosomal RNA genes. Genetics. 2007, 175 (2): 477-485. 10.1534/genetics.107.071399.
Manguin S, White R, Blossey B, Hight SD: Genetics, taxonomy, and ecology of certain species of Galerucella (Coleoptera: Chrysomelidae). Ann Entomol Soc Am. 1993, 86: 397-410.
Hambäck PA: Density-dependent processes in leaf beetles feeding on purple loosestrife: aggregative behaviour affecting individual growth rates. Bull Entomol Res. 2010, 100: 605-611. 10.1017/S000748530999068X.
Nokkala S, Nokkala C: Chromosome numbers and chromosomal polymorphism in Finnish species of Galerucella Crotch (Chrysomelidae, Coleoptera). Hereditas. 1987, 106: 51-58.
Hansson C, Hambäck PA: Cryptic species in the genus Asecodes (Foerster) (Hymemoptera, Eulophidae) parasitizing larvae of Galerucella spp. Journal or Hymenopteran Research. 2013, 30: 51-64. 10.3897/jhr.30.4279.
Stenberg JA, Witzell J, Ericson L: Tall herb herbivory resistance reflects historic exposure to leaf beetles in a boreal archipelago age-gradient. Oecologia. 2006, 148 (3): 414-425. 10.1007/s00442-006-0390-7.
Stenberg JA, Hambäck PA, Ericson L: Herbivore-induced "rent rise" in the host plant may drive a diet breadth enlargement in the tenant. Ecology. 2008, 89 (1): 126-133. 10.1890/07-0252.1.
Stenberg JA, Hambäck PA: Host species critical for offspring fitness and sex ratio for an oligophagous parasitoid: implications for host coexistence. Bull Entomol Res. 2010, 100 (6): 735-740. 10.1017/S0007485310000143.
Bartelt RJ, Cosse AA, Zilkowski BW, Weisleder D, Grode SH, Wiedenmann RN, Post SL: Dimethylfuran-lactone pheromone from males of Galerucella calmariensis and Galerucella pusilla. J Chem Ecol. 2006, 32 (3): 693-712. 10.1007/s10886-005-9026-3.
Kolaczan CR, Heard SB, Segraves KA, Althoff DM, Nason JD: Spatial and genetic structure of host-associated differentiation in the parasitoid Copidosoma gelechiae. J Evol Biol. 2009, 22 (6): 1275-1283. 10.1111/j.1420-9101.2009.01742.x.
Forbes AA, Powell THQ, Stelinski LL, Smith JJ, Feder JL: Sequential sympatric speciation across trophic levels. Science. 2009, 323 (5915): 776-779. 10.1126/science.1166981.
Althoff DM: A test of host-associated differentiation across the 'parasite continuum' in the tri-trophic interaction among yuccas, bogus yucca moths, and parasitoids. Mol Ecol. 2008, 17 (17): 3917-3927. 10.1111/j.1365-294X.2008.03874.x.
Dickey AM, Medina RF: Lack of sequential radiation in a parasitoid of a host-associated aphid. Entomol Exp Appl. 2011, 139 (2): 154-160. 10.1111/j.1570-7458.2011.01118.x.
Feder JL, Forbes AA: Sequential speciation and the diversity of parasitic insects. Ecological Entomology. 2010, 35: 67-76.
Jeffries MJ, Lawton JH: Enemy free space and the structure of ecological communities. Biol J Linn Soc. 1984, 23: 269-286. 10.1111/j.1095-8312.1984.tb00145.x.
Stamp N: Enemy-free space via host plant chemistry and dispersion: assessing the influence of tri-trophic interactions. Oecologia. 2001, 128 (2): 153-163. 10.1007/s004420100679.
Nyman T, Bokma F, Kopelke JP: Reciprocal diversification in a complex plant-herbivore-parasitoid food web. BMC Biol. 2007, 5: 49-10.1186/1741-7007-5-49.
Morehead SA: Geographic variation in host location cues for a dipteran parasitoid of Paraponera clavata. Biotropica. 2001, 33 (3): 495-501.
Stenberg JA, Axelsson EP: Host race formation in the meadowsweet and strawberry feeding leaf beetle Galerucella tenella. Basic Appl Ecol. 2008, 9 (5): 560-567. 10.1016/j.baae.2007.10.007.
Ikonen A, Sipura M, Miettinen S, Tahvanainen J: Evidence for host race formation in the leaf beetle Galerucella lineola. Entomol Exp Appl. 2003, 108: 179-185. 10.1046/j.1570-7458.2003.00079.x.
Pappers SM, van der Velde G, Ouborg NJ: Host preferences and larval performance suggest host race formation in Galerucella nymphaeae. Oecologia. 2002, 130: 433-440. 10.1007/s00442-001-0822-3.
Acknowledgements
We thank T. Malm and R. Bukontaite for providing help with the study, and C. Hansson for identification of parasitoids. The study was supported by the Swedish Research Council (contract no. 621-2009-4943).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contribution
PH designed the study, was involved in the field work and was responsible for the writing, EW carried out the molecular work and was involved in the statistical analysis and in writing the paper, LE, LS and JAS were involved in the field work and in the study design, ACL was involved in the design of the study and in the molecular work, JB was responsible for the statistical analysis and in the writing of the paper. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2012_2335_MOESM1_ESM.doc
Additional file 1: Table SI-1: Information about the specimens included in the analyses. Table SI-2. Substitution model parameters. Table SI-3. The effect of the prior on the posterior estimate of the theta and tau parameters. Table SI-4. Fragment lengths (bp), number of variable and parsimony informative sites and number of ingroup taxa for the Galerucella and Asecodes datasets. Figure SI-1. Relationship between Asecodes specimen based on sequences of the nuclear genes PGD, 28S and ITS with base pair changes of 28S and ITS. (DOC 545 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hambäck, P.A., Weingartner, E., Ericson, L. et al. Bayesian species delimitation reveals generalist and specialist parasitic wasps on Galerucellabeetles (Chrysomelidae): sorting by herbivore or plant host. BMC Evol Biol 13, 92 (2013). https://doi.org/10.1186/1471-2148-13-92
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-13-92