
Abstract
Background
Plasmodium vivax is the most widely distributed human malaria parasite outside of Africa, and its range extends well into the temperate zones. Previous studies provided evidence for vivax population differentiation, but temperate vivax parasites were not well represented in these analyses. Here we address this deficit by using complete mitochondrial (mt) genome sequences to elucidate the broad genetic diversity and population structure of P. vivax from temperate regions in East and Southeast Asia.
Results
From the complete mtDNA sequences of 99 clinical samples collected in China, Myanmar and Korea, a total of 30 different haplotypes were identified from 26 polymorphic sites. Significant differentiation between different East and Southeast Asian parasite populations was observed except for the comparison between populations from Korea and southern China. Haplotype patterns and structure diversity analysis showed coexistence of two different groups in East Asia, which were genetically related to the Southeast Asian population and Myanmar population, respectively. The demographic history of P. vivax, examined using neutrality tests and mismatch distribution analyses, revealed population expansion events across the entire P. vivax range and the Myanmar population. Bayesian skyline analysis further supported the occurrence of ancient P. vivax population expansion.
Conclusions
This study provided further resolution of the population structure and evolution of P. vivax, especially in temperate/warm-temperate endemic areas of Asia. The results revealed divergence of the P. vivax populations in temperate regions of China and Korea from other populations. Multiple analyses confirmed ancient population expansion of this parasite. The extensive genetic diversity of the P. vivax populations is consistent with phenotypic plasticity of the parasites, which has implications for malaria control.
Similar content being viewed by others


Background
Plasmodium vivax malaria threatens 40% of the world's population and causes over 132 million clinical cases every year [1]. Compared with other human malaria parasites, P. vivax has the widest global distribution and occurs in Asia, the Middle East, South and Central America, and parts of Africa [2]. Contrary to its common name "benign tertian malaria", P. vivax is also associated with severe pathology [3]. Furthermore, infection of Duffy-negative patients in Madagascar suggests that this parasite is evolving to exploit alternative invasion pathways, posing new threats to previously resistant human populations [4]. Yet, vivax malaria has been neglected, and the first-line treatments for vivax malaria have remained unchanged for 50 years. As malaria elimination and eradication are once again on the agenda of many malaria-endemic nations, P. vivax research and control is receiving renewed attention.
The history of malaria control provides considerable evidence for the resilience of P. vivax malaria and the underlying genetic diversity of the parasite. Indeed, across its geographic range, P. vivax displays extraordinary phenotypic variation in disease pathology, vectorial preference, and the pattern and frequency of relapse in the host (the time period at which the parasite remains dormant in the liver) [5]. To date, the processes which have led to the diversification of P. vivax are still not well understood.
Most investigations of the evolutionary history and population structure of P. vivax have relied on molecular markers. In an early study, low genetic variability at 13 microsatellite loci suggested little differentiation and a recent (< 10,000 years) world expansion of this parasite [6]. However, several later studies refuted this notion and suggest that Leclerc et al. [6] used a biased set of microsatellite markers [7, 8]. Two recent comparisons of sympatric parasite populations further suggested that P. vivax microsatellites are more polymorphic than those of P. falciparum [9, 10]. Other recent molecular phylogenetic analyses support an ancient demographic history of the P. vivax population, probably as a result of host switch from Asian monkeys [11–14]. While such an opinion is often inferred from the high level of genetic diversity in the parasite in Asia [15] and may not reflect the true origin of this parasite, it is certain that significant subdivision of the parasite populations exists [7, 14, 16]. Whereas most studies agree that the oldest populations appear to have originated in Southeast Asia [12–14], it is also true that the sampling has been opportunistic with poorly defined populations, making it difficult to properly assess the parasite demographic history [14].
The geographic range of P. vivax extends well into the temperate zone. The ability to survive in cool climates is attributed to sporogony in mosquitoes at a much lower temperature and the production of long-term hibernating hypnozoites in human liver. The latter adaptation is especially important for temperate strains, where the primary infection occurs eight months or more following inoculation by an infected mosquito [17]. In recent years, there has been a reemergence of vivax malaria in central China and on the Korean peninsula in areas where malaria control was highly successful during the global malaria eradication campaign of the 1960s [18, 19]. The process of population expansion and spread of temperate strains is not understood. Although earlier evolution and population studies provided evidence about population differentiation of these Asian temperate vivax parasites [12, 13, 20], such analyses would benefit greatly from more extensive and spatially-defined sampling of these populations.
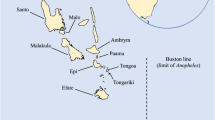
Being genome-level informative, mitochondrial (mt) DNA sequences have been used for population and phylogenetic studies in a wide range of organisms [12, 13, 21–23]. In order to further define the demographic history and population structure of P. vivax in East and Southeast Asia, we sampled four P. vivax populations in a transect across temperate (Korea and central China) and warm temperate (southwestern China and northeastern Myanmar) climates (Figure 1). We obtained sequences of the complete mt genome from 99 parasite isolates. Genetic data were analyzed to look for evidence of genetic structure throughout the range of this parasite in temperate zone. The demographic histories of these populations were inferred in order to illuminate how this parasite species has evolved.

Distribution of P. vivax sampling sites. Circles indicate sampling sites in the present study, while the star marks the location of Guangxi province (a possible earlier sampling site).
Methods
Sample collection, DNA extraction, PCR amplification and DNA sequencing
A total of 99 clinical P. vivax parasite samples were previously collected in four locations of East and Southeast Asia, including 36 samples from Bengbu city, Anhui Province of China in 2004, 26 samples from Luodian county, Guizhou Province of China in 2005, five samples from villages near the northern border of South Korea in 2005, and 32 samples from Laiza township of northeastern Myanmar in 2006-2008 (Figure 1) [20, 24, 25]. Collection sites in China and Korea were malaria hypoendemic with seasonal occurrence of only P. vivax parasites, whereas the Myanmar site was mesoendemic with perennial circulation of both P. vivax and P. falciparum parasites [26]. Ethical clearance was obtained from provincial ethical review committees of Guangxi and Anhui, and Kunming Medical University Institutional Review Board, respectively. The samples used for sequencing have been identified as single-clone infections by genotyping the merozoite surface protein (msp) 3α and msp3β genes [20, 25]. The highly polymorphic nature of these two genes should allow us to exclude most of the mixed-clone infections. Parasite DNA was extracted from blood filters using the QIAamp DNA Mini Kit (Qiagen, USA) with minor modifications. Primers were designed to amplify the complete P. vivax mt genome based on the sequence of the reference strain Sal-I (GenBank: AY598140). Long-range, high-fidelity PCR amplification was performed using Advantage HD DNA Polymerase Mix (Clontech, Mountain View, CA), which has efficient 3' → 5' exonuclease proof-reading activity. PCR reactions in 50 μl contained DNA template, 1 μM of each oligonucleotide primer, 10 × Advantage HD PCR Buffer, 1 mM deoxynucleosides (dNTPs), and 5 units of polymerase mix. PCR was performed at 94°C for 1 min, followed by 30 cycles of 94°C for 1 min, 60°C for 2 min, and 72°C for 3 min. A final extension was done at 72°C for 10 min. PCR products were purified with the PrepEase Gel Extraction Kit (USB, Affymetrix, USA) and sequenced using the BigDye version 1.1 on an ABI 3730XL sequencer. Overlapping sequences were obtained with the primers listed in Additional file 1. Sequences were base-called and assembled with PHRED and PHRAP [27, 28], and heterozygous bases were called with the Polyphred program [29] and confirmed by visual inspection of the corresponding trace files using the CONSED trace file viewer [30]. To remove potential nucleotide ambiguities, certain sequences were confirmed by two independent PCR reactions from the same DNA template. The sequenced fragments were assembled into complete mt sequences and deposited in GenBank (JQ240331-JQ240429).
Sequence alignment and haplotype network
For phylogenetic analysis, we retrieved additional 291 complete P. vivax mt genome sequences from GenBank (AY791517-AY791692, AY598035-AY598140, and AB550270-AB550280). Nucleotide diversity (π) - the average number of nucleotide differences per site between two sequences, and haplotype diversity (h) - the number and frequency of different haplotypes in the samples were estimated using DnaSP version 5.10 [31]. The three protein-coding genes were identified through alignment with annotated genes from the published P. vivax mt genomes. Sequence alignments were performed using inGAP v 2.2 [32] and ClustalW [33], and were manually checked using BioEdit http://www.mbio.ncsu.edu/BioEdit/bioedit.html. To detect selection acting on these coding sequences, the number of synonymous nucleotide substitutions per synonymous site (d S ) and number of nonsynonymous nucleotide substitutions per nonsynonymous site (d N ) were calculated using the SNAP program http://www.hiv.lanl.gov/content/sequence/SNAP/SNAP.html. Phylogenetic trees were constructed by using the maximum likelihood method implemented in BioNumerics v5.10 (Applied-Maths, Sint Maartens-Latem, Belgium). In order to define the relationship among the isolates at a micro-evolutionary level, we performed allelic profile-based comparison using a minimum spanning tree analysis with the TCS1.21 [34] as well as the BioNumerics v5.10 software. In TCS, inferences depend on the chosen probability of parsimony, for which we tried a value of 91%-95%. The number of mutational differences associated with the probability just before the 91% cutoff is the maximum number of connections between sequences. Ambiguities were treated according to the previous criteria [35].
Population structure
Analysis of population structure was performed using STRUCTURE [36], which uses a Bayesian approach to calculate the posterior probability of the number of populations in the sample single nucleotide polymorphism (SNP) set. Replicate runs of STRUCTURE used a burn-in period of 50,000 iterations followed by a Markov chain Monte Carlo (MCMC) 100,000 iterations from which estimates were obtained. All runs were based on the admixture model without prior population information [36]. Twenty replicate unsupervised runs were performed for each value of the number of clusters K from 1 to 14. The number of population clusters was inferred according to Evanno et al. [37] and the ad hoc statistic ΔK was calculated for the presence of individuals with ancestry in two or more of the K populations. Graphs of the STRUCTURE results were made using DISTRUCT. Sequences were analyzed for maximum sequence diversity and visually inspected with highlighter tools http://www.hiv.lanl.gov.
ARLEQUIN software package version 3.11 was used to estimate genetic diversity indices and to assess population differentiation [38]. Pairwise comparisons FST and ΦST values were estimated by permutation analyses using 1,000 permutations with an assumption of no difference between populations. The P-value was considered as the proportion of permutations resulting in the higher FST or ΦST value or equal to the observed one. Analysis of molecular variance (AMOVA) was used to evaluate the extent to which sequence variation was partitioned among populations and areas.
Neutrality tests and historical expansions
Neutrality statistics and pairwise nucleotide differences were calculated to examine the historical demographic expansions of P. vivax. Tajima's D test [39] and Fu's Fs test [40] were used to test whether the mt genome data conform to the expectations of neutrality. Tajima's D test compares two estimators of the mutation parameter θ, Watterson's estimator θs and Tajima's estimator θπ, and significant D values can be due to factors such as population expansion, bottlenecks and selection [39]. Fu's Fs test compares the number of haplotypes observed with the number of haplotypes expected in a random sample under the assumption of an infinite-sites model without recombination, and Fs was sensitive to population demographic expansion [40]. In addition, historic demographic expansions were also investigated by examining frequency distributions of pairwise differences between sequences (mismatch distribution), which is based on three parameters: θ0, θ1 (θ before and after the population growth) and τ (time since expansion expressed in units of mutational time) [41, 42]. The expected mismatch distribution under a constant population size was estimated by the equation F i = θi/(θ+1)i+1, where F i is the expected frequency of pairwise comparisons showing i differences, and θ is estimated by the observed mean of pairwise differences. The distribution usually has a ragged profile at demographic equilibrium due to stochastic lineage loss, but it is usually unimodal in populations following a recent population demographic expansion and population range expansion [41, 43]. This reflects an underlying star-like genealogy in which all of the coalescent events occurred in a narrow time window. Both mismatch analysis and neutrality tests were performed in ARLEQUIN.
Estimating the most recent common ancestor (TMRCA)
TMRCA of P. vivax was estimated using the strict-clock Bayesian MCMC method as implemented in BEAST v1.5.4 [44] with non-coding region of the mitochondrial genome. We assumed a general time-reversible model of nucleotide substitution with gamma-distributed rate heterogeneity among sites and a proportion of invariant sites. In addition, we assumed an uncorrected log normal distribution molecular clock model of rate variation among branches in the tree. As previously described, TMRCA was calculated using the divergence between Plasmodium fragile and Plasmodium knowlesi, assuming that they diverged as part of the radiation of their hosts (the silenus group of macaques and the divergence of the sinica and fascicularis groups) between 3.5 and 4.7 million years ago [14]. Analyses were performed for 1,000,000 generations with the model of Yule Process and Bayesian skyline analysis. The initial 20% of each run was discarded. The effective sample size for parameter estimates and convergence was checked using Tracer http://beast.bio.ed.ac.uk/Tracer.
Results
Genetic variation in P. vivaxmt genomes from Myanmar, China and Korea
We sequenced the complete mt genomes from 99 P. vivax samples representing four populations collected in temperate and warm temperate regions of East Asia and Myanmar (Figure 1 and Table 1). Alignment of these complete mt genomes identified 26 SNPs (indels excluded), including 15 transversions and 11 transitions (Additional file 2). Sixteen of 26 SNPs are located in the three coding regions (COX3, COX1 and CYTB). Nucleotide diversity ranged from 0.00020 for China's Anhui population to 0.00096 for the combined Korean population. The dN/dS ratios of coding regions from all geographical sample sets were well below one, confirming that purifying selection is the predominant force in mtDNA evolution (Table 1). The 26 SNPs defined a total of 30 mt genome haplotypes. Compared with the number of haplotypes/population (8-38) identified in earlier studies, the numbers of haplotypes in our studied populations were modest, being the lowest (5) in China's Anhui population and highest (16) in the Myanmar population (Table 1). Likewise, haplotype diversity also varied, ranging from 0.584 in the Anhui population to 0.850 in the Myanmar population. The two temperate populations from China had much lower haplotype diversity and were comparable with the New World parasite population (0.58-0.68).
Phylogeography
Analysis of P. vivax mt genomes from 99 newly collected Asian samples as well as 291 previously described isolates from 11 populations identified a total of 141 haplotypes [12, 13, 45]. Most geographical regions had one predominant haplotype, whereas Melanesia, China, and Southeast Asia had two predominant haplotypes. To characterize the frequencies and relationships of different haplotypes, a minimum spanning tree was constructed. This haplotype network clearly showed geographical clustering of the haplotypes (Figure 2). Inclusion of the temperate P. vivax samples did not dramatically change the overall topology of the network. Consistently, significant sharing of the haplotypes was detected between South/West Asian and African populations [12]. For the four temperate populations, most haplotypes were connected to the South/West Asian populations, which is consistent with the fact that these geographic regions are physically connected and past genetic exchanges might have occurred between these populations. Interestingly, the two temperate P. vivax populations from China's Anhui and Guizhou provinces formed a local network consisting of two predominant, related haplotypes. In contrast, the Chinese samples analyzed earlier (some from Guangxi Province in southern China, Figure 1) [12, 13] formed two clusters, which might indicate discrete origins: some haplotypes were clustered in the East Asian branch, whereas others were shared and linked with haplotypes from Southeast Asia (Thailand, Vietnam and Indonesia). As reported previously [45], most Korean haplotypes were shared with those from southern China [12, 13], but diverged from the central China populations.

A haplotype network based on P. vivax mt genome sequences. Each circle represents a haplotype with size of the circle equaling to the frequency of the haplotype in the samples. Lines separating haplotypes represent mutational steps. Each color shows the countries of origin of the samples.
To further illustrate the relationship of the mt genome haplotypes, a maximum likelihood phylogenetic tree based on the complete genome sequences was constructed (Additional file 3). All mt genomes from P. vivax were closely related to one another; no branches within the P. vivax cluster received strong bootstrap support (data not shown). In general, the topology of this tree roughly mirrored that of the minimum spanning tree. Both analyses showed that East Asian populations exhibited relatively large divergence from the remaining populations, suggesting that the temperate parasite populations in East Asia had a different demographic history.
Population Structure
Earlier studies using both mt genome and microsatellites have found significant differentiation of the extant P. vivax populations [7, 12]. To further define genetic structure of the P. vivax populations, we used a Bayesian admixture procedure implemented in STRUCTURE to calculate the potential number of populations. Using a series of K values from 1 to 14, we detected a large incremental increase of the likelihood value as the number of genetic clusters in the model increased from 1 to 4 (data not shown). Significant genetic structure was found between parasite populations, and most parasites formed geographical clusters. At K = 2, only the East Asian cluster was separated from the rest of the populations, although this division was also supported from analysis of the distribution of ΔK using the method of Evanno et al. [37]. Posterior probability of the entire sample set at 4 populations was most consistent with a clear separation of populations according to large geographical regions of Melanesia, East Asia, Southeast Asia and the rest of the samples (Figure 3). It is noteworthy that South American and Myanmar populations appeared to be composed of a mixture of haplotypes from South/West Asia as well as Southeast Asia, suggestive of population boundaries or shared demographic histories of the parasite populations in these regions.

Graphical display of estimated population structure at K = 4. Each vertical line represents one individual. At K = 4, the samples are partitioned into four populations, which are consistent with large geographical regions of Melanesia, East Asia, Southeast Asia and the rest of the world. Populations are separated by black lines and occurred in the same order as given in Table 2.
To further determine whether population differentiation exists, we estimated FST (only based on haplotype frequency) and ΦST (based on both haplotype frequency and genetic distance) for each population using the mtDNA sequences. The majority of pairwise FST values were greater than 0.25 (ranging from 0.25 to 0.85), suggesting that strong population differentiation exists among the populations. Notable exceptions included the following pairwise comparisons: South/West Asia and Africa, South/West Asia and South America, South/West Asia and Southeast Asia, as well as Korea and southern China (Table 2). In Southeast Asia, parasite populations from the Greater Mekong subregion, Indonesia and Melanesia were also distinct with FST values of 0.32 - 0.62 and ΦST values of 1.58 - 4.84 (P = 0.01). For the temperate parasite populations from Asia, which include northeast Myanmar, Korean, and three Chinese P. vivax populations, between-population genetic differences were strong (FST = 0.32-0.75, ΦST = 0.97-5.13) and statistically significant after sequential Bonferroni correction (P < 0.0001). The only exception was between the Korean and southern Chinese population (exact origin of the samples unknown), which showed little genetic differentiation (FST = 0.05, P = 0.08; ΦST = 0.20, P = 0.08) (Table 2). We further grouped all studied populations into eight geographical groups consisting of Africa, America, South/West Asia, Southeast Asia, Myanmar, East Asia, Indonesia, and Melanesia, and performed a hierarchical AMOVA analysis. The three covariance components (within population, among population/within group, and among groups) could explain 22.7%, 32.3% and 45.0% of the variance, respectively. Apparently, considerable variation was preserved at the population level.
Demographic history
The star-like shape of the haplotype network is consistent with significant population expansion (Figure 2). To test this hypothesis, we performed mismatch distribution and skyline analyses. Mismatch distribution was unimodal for the entire P. vivax sample set (Figure 4A), a pattern characteristic of populations that have undergone large-scale expansion. To obtain more precise estimates for the temperate parasite populations, we divided our recent samples into two large groups represented by the Myanmar and East Asian populations (Chinese and Korean samples combined), and performed neutrality tests and mismatch distribution analysis. Although mismatch distributions for these two populations did not perfectly fit the expected pattern under the sudden expansion model (Figure 4B and 4C), they did not differ significantly (P > 0.05) from this model and therefore were suitable for the analysis of demographic patterns. The Fs test and Tajima's D test agreed well with the mismatch analyses. The Fs values of the entire P. vivax populations as well as the East Asian and Myanmar populations were negative and significant (P = 0.025 for East Asian populations and P = 0.005 for Myanmar population). Tajima's D test compares two mutation estimators θs and θπ, which are affected by the size of the present-day and original population, respectively [39]. Consequently, a history of population growth would inflate S relative to π and generate a negative value of D [46, 47]. Consistent with a past population expansion, Tajima's D test was negative (-2.05) and statistically significant (P = 0.001) for the entire P. vivax populations analyzed. For the Myanmar and East Asian populations, the D statistic was negative and only significant for the Myanmar population (P = 0.021). The τ value, which reflects the location of the mismatch distribution crest, provides a rough estimate of the time when rapid population expansion started. The observed value of the age expansion parameter (τ) was 4.18 units (95% CI: 2.56-10.41) of mutational time in P. vivax. In comparison, the τ value of the Myanmar population was smaller (4.10, 95% CI: 1.36- 6.59). Yet, the East Asian populations had a τ value of 5.13 (95% CI: 2.01- 13.3), which was even greater than that for all vivax populations, suggesting that the East Asian parasite populations may be at demographic equilibrium in recent evolutionary history. The results of the skyline analysis further supported P. vivax population expansion showing a rapid increase in effective population size starting ~ 200,000 years before present (ybp), followed by a more recent increase in population numbers starting at ~50,000 ybp (Figure 4D).

Historical demographic history of P. vivax inferred from whole mt genome. A. Pairwise mismatch distributions for worldwide P. vivax. Observed distributions (columns) were compared with the expected values for populations that have undergone historical demographic expansion (lines and circles). B. Pairwise mismatch distributions for P. vivax from Myanmar. C. Pairwise mismatch distributions for vivax species from East Asia. D. Bayesian skyline plots for the same regional groups with A. Black lines represent median population estimates and grey lines denote upper and lower confidence limits (95% highest posterior density).
We estimated the divergence time of Myanmar P. vivax population to be 385,000 (± 3,800) years ago, whereas the East Asian P. vivax population diverged at 383,000 (± 7,300) years ago. Our estimate also indicated that the TMRCA for the whole vivax populations was around 346,000 to 452,400 years ago (95% highest posterior density).
Discussion
The demographic history and genetic diversity of P. vivax provide a foundation for the development of effective malaria control measures. Phylogenetic analysis of P. vivax using sequences from the mt genome and nuclear genes has placed this parasite in a clade that includes the Asian monkey parasites P. cynomolgi, P. simiovale and P. knowlesi [11–13]. This phylogeny is consistent with an Asian origin of P. vivax, suggesting this parasite is probably the descendent of an ancestor that switched from Asian monkeys to humans. Regardless of what calibration points were used, TMRCA estimates support an ancient demographic history of the extant P. vivax parasites [12–14, 48], which concurs with the genetic diversity of global P. vivax strains. P. vivax parasites also display significant phenotypic diversity such as different relapse patterns, which serve to distinguish tropical from temperate strains. Temperate strains with primary infection occurring eight months or more following inoculation by an infected mosquito were proposed to be a subspecies of P. vivax [49]. Similarly, New World P. vivax parasites were postulated to represent a separate subspecies based on molecular polymorphism and difference in vector preference [50], albeit such a hypothesis was not supported by the mt haplotype analysis [12]. Therefore, more detailed sampling of P. vivax populations from its entire geographical range is necessary to better understand the evolutionary history of P. vivax.
Extant P. vivax populations may have been influenced by historical population expansions and more recent migrations. In addition, the evolutionary process, often estimated from the geographical pattern of genetic variation, can be influenced by colonization events such as range expansion or reduction [51, 52]. Several lines of evidence support the hypothesis of ancient population expansion of P. vivax. First, the existence of a phylogeny consisting of several distinct, but closely related lineages suggests a rapid population expansion in the recent past (Figure 2). Second, we found an extremely close fit between the observed pairwise mismatch distribution and the expected distribution based on a model of rapid population expansion (Figure 4). Finally, the skyline plot shows a period of a more recent increase in population size. This period is concordant with a time of human migration. Homo sapiens had occupied Africa about 150,000 Mya. They moved out of Africa 70,000 years ago and spread into Asia, Europe and Australia 40,000 years ago. It is also possible that the population expansion of P. vivax was linked to the expansion not only in the human host but also in adaptation of the mosquito vectors. However, it needs to be cautioned that mtDNA has a smaller effective population size and provides only part of a species' history; thus more robust data from nuclear genes are needed to corroborate this conclusion [53, 54].
Despite generally low endemicity, the global P. vivax populations display high genetic diversity at microsatellite, SNP, and antigenic loci [5, 7, 15, 55–57]. Previous studies of P. vivax parasites from Myanmar and central China demonstrated high genetic diversity and multiple-clone infections [20, 55, 58]. Our analysis of the mt genome also detected comparable, high-level genetic diversity among these temperate P. vivax populations. Consistent with high malaria endemicity in Myanmar, haplotype diversity was also high (0.85 ± 0.057) and comparable to other highly endemic areas of the world [12]. However, haplotype diversity of the two temperate populations from China was lower and at similar levels to those of the New World parasite populations. This result appeared to be consistent with the recent history of malaria epidemiology in central China. Historically, temperate P. vivax malaria was highly prevalent in central China, but it was considerably curtailed during the global malaria eradication campaign in the 1950s and 1960s [59]. However, in the last two decades P. vivax malaria has resurged and outbreaks occurred in several central provinces [18, 60]. We thus speculate that past strenuous control efforts might have caused a population bottleneck in the parasite population and as a result the diversity of resurging parasites was reduced. This bottleneck effect on P. vivax population was also found in southern Thailand, where P. vivax population displayed a high level of clonality [61].
For a finite population, unless there is complete panmixia and random sampling, a pattern of genetic isolation by geographic distance is generally expected [62]. This principle applies well to the P. vivax populations. Within its geographic range, P. vivax exhibits substantial population differentiation, especially between different continents [7, 12]. The clear differentiation between parasites from Melanesia and those from Southeast Asian countries is much surprising, since previous microsatellite-based analyses of both P. vivax [8] and P. falciparum [63] failed to show such a clear pattern. FST and ΦST statistics revealed significant population differentiation between Myanmar and East Asian P. vivax populations. Even within a short distance, genetic differentiation may be significant due to possible migration or ecological constraints [64]. Substantial genetic structure existed between the Chinese Guizhou and Anhui P. vivax populations despite their geographic proximity. Interestingly, two major genotypes in China were also observed in the South Korean population [45, 65]. Population genetic structure can result from both species-specific biological traits and abiotic factors. The temperate populations of P. vivax have developed a trait of long relapsing liver hypnozoites, an adaptation to the long winter period of temperate climate when transmission is interrupted [66]. Because of the obligatory role of mosquito vectors in malaria transmission, reciprocal selection between malaria parasites and mosquito vectors can lead to local adaptation of the parasite [16]. It is unknown whether vector adaptation plays any role in the population structure of these temperate parasites. Also, genetic drift acting on small populations (from areas of low endemicity) may be a force driving population differentiation [7, 9, 67].
Since genetic diversity of the global P. vivax populations has been suggested to be the result of ancient hominid geographical expansion [13], the relationships among the extant parasite populations might reflect past demographic histories of the parasites and the routes by which parasite populations have expanded. Most mtDNA haplotypes from the four temperate and warm temperate populations were unique but related, suggesting that they might be descendents from the same lineage(s). Haplotype network analysis suggested South/West Asia as the root or origin of the parasite populations (Figure 2), but this conjecture does not exclude a possible African origin of P. vivax, as African parasites shared the major mtDNA haplotype with the South/West Asian samples. Culleton and collegues proposed that the present-day African and American populations may be the closest extant relatives of the African ancestor [68]. Since clustering in the network is often affected by the methodologies used, the exact origin of the vivax ancestor is still not clear. Haplotype network analysis also showed that samples collected in China formed two divergent lineages: one (possibly from subtropical southern China) was closely related to the Southeast Asian samples (Indonesia, Thailand, and Vietnam), whereas the other (mostly temperate strains) was directly diverged from the northeast Myanmar population. Myanmar is connected to East Asia, Southeast Asia, and South/West Asia and such a geographical location may be critical for elucidating the population expansion and evolutionary history of P. vivax. The relationship of the temperate Chinese and northeast Myanmar P. vivax populations points to a possibility of population expansion from South/West Asia to temperate China via northeast Myanmar, which seems to make sense from a geographic point of view. It is noteworthy that the P. vivax parasites from northeast Myanmar, China and Korea all have similar, long relapsing patterns characteristic of temperate P. vivax strains [69]. Furthermore, our results are consistent with the notion that temperate and warm temperate P. vivax parasites may represent a unique lineage, which is important to elucidate the genetic structure and history of expansion of P. vivax.
Conclusion
The present study readdressed the issue of the extant P. vivax population structure by focusing on temperate zone parasite populations in East and Southeast Asia. Analysis of the complete mt genomes from 99 clinical samples confirmed that P. vivax displays extensive genetic diversity and natural populations are clearly structured. While most mtDNA haplotypes from the four temperate populations were related, suggestive of descendent from the same lineage(s), local population subdivision was also apparent. Multiple tests further confirm the ancient expansion of the P. vivax population.
Abbreviations
- Mt:
-
mitochondrial
- d S :
-
the number of synonymous nucleotide substitutions per synonymous site
- d N :
-
the number of nonsynonymous nucleotide substitutions per nonsynonymous site
- Markov chain Monte Carlo (MCMC):
-
TMRCA: the most recent common ancestor
- SNP:
-
single nucleotide polymorphism
- ybp:
-
years before present.
References
Hay SI, Guerra CA, Tatem AJ, Noor AM, Snow RW: The global distribution and population at risk of malaria: past, present, and future. The Lancet infectious diseases. 2004, 4 (6): 327-336. 10.1016/S1473-3099(04)01043-6.
Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM: Vivax malaria: neglected and not benign. Am J Trop Med Hyg. 2007, 77 (6 Suppl): 79-87.
Anstey NM, Russell B, Yeo TW, Price RN: The pathophysiology of vivax malaria. Trends in parasitology. 2009, 25 (5): 220-227. 10.1016/j.pt.2009.02.003.
Menard D, Barnadas C, Bouchier C, Henry-Halldin C, Gray LR, Ratsimbasoa A, Thonier V, Carod JF, Domarle O, Colin Y, Bertrand O, Picot J, King CL, Grimberg BT, Mercereau-Puijalon O, Zimmerman PA: Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proceedings of the National Academy of Sciences of the United States of America. 2010, 107 (13): 5967-5971. 10.1073/pnas.0912496107.
Cui L, Escalante AA, Imwong M, Snounou G: The genetic diversity of Plasmodium vivax populations. Trends in parasitology. 2003, 19 (5): 220-226. 10.1016/S1471-4922(03)00085-0.
Leclerc MC, Durand P, Gauthier C, Patot S, Billotte N, Menegon M, Severini C, Ayala FJ, Renaud F: Meager genetic variability of the human malaria agent Plasmodium vivax. Proc Natl Acad Sci USA. 2004, 101 (40): 14455-14460. 10.1073/pnas.0405186101.
Imwong M, Nair S, Pukrittayakamee S, Sudimack D, Williams JT, Mayxay M, Newton PN, Kim JR, Nandy A, Osorio L, Carlton JM, White NJ, Day NP, Anderson TJ: Contrasting genetic structure in Plasmodium vivax populations from Asia and South America. Int J Parasitol. 2007, 37 (8-9): 1013-1022. 10.1016/j.ijpara.2007.02.010.
Karunaweera ND, Ferreira MU, Munasinghe A, Barnwell JW, Collins WE, King CL, Kawamoto F, Hartl DL, Wirth DF: Extensive microsatellite diversity in the human malaria parasite Plasmodium vivax. Gene. 2008, 410 (1): 105-112. 10.1016/j.gene.2007.11.022.
Ferreira MU, Karunaweera ND, da Silva-Nunes M, da Silva NS, Wirth DF, Hartl DL: Population structure and transmission dynamics of Plasmodium vivax in rural Amazonia. J Infect Dis. 2007, 195 (8): 1218-1226. 10.1086/512685.
Orjuela-Sanchez P, Da Silva-Nunes M, Da Silva NS, Scopel KK, Goncalves RM, Malafronte RS, Ferreira MU: Population dynamics of genetically diverse Plasmodium falciparum lineages: community-based prospective study in rural Amazonia. Parasitology. 2009, 136 (10): 1097-1105. 10.1017/S0031182009990539.
Escalante AA, Cornejo OE, Freeland DE, Poe AC, Durrego E, Collins WE, Lal AA: A monkey's tale: the origin of Plasmodium vivax as a human malaria parasite. Proceedings of the National Academy of Sciences of the United States of America. 2005, 102 (6): 1980-1985. 10.1073/pnas.0409652102.
Mu J, Joy DA, Duan J, Huang Y, Carlton J, Walker J, Barnwell J, Beerli P, Charleston MA, Pybus OG, Su XZ: Host switch leads to emergence of Plasmodium vivax malaria in humans. Mol Biol Evol. 2005, 22 (8): 1686-1693. 10.1093/molbev/msi160.
Jongwutiwes S, Putaporntip C, Iwasaki T, Ferreira MU, Kanbara H, Hughes AL: Mitochondrial genome sequences support ancient population expansion in Plasmodium vivax. Mol Biol Evol. 2005, 22 (8): 1733-1739. 10.1093/molbev/msi168.
Cornejo OE, Escalante AA: The origin and age of Plasmodium vivax. Trends Parasitol. 2006, 22 (12): 558-563. 10.1016/j.pt.2006.09.007.
Feng X, Carlton JM, Joy DA, Mu J, Furuya T, Suh BB, Wang Y, Barnwell JW, Su XZ: Single-nucleotide polymorphisms and genome diversity in Plasmodium vivax. Proceedings of the National Academy of Sciences of the United States of America. 2003, 100 (14): 8502-8507. 10.1073/pnas.1232502100.
Joy DA, Gonzalez-Ceron L, Carlton JM, Gueye A, Fay M, McCutchan TF, Su XZ: Local adaptation and vector-mediated population structure in Plasmodium vivax malaria. Molecular biology and evolution. 2008, 25 (6): 1245-1252. 10.1093/molbev/msn073.
Garnham PCC: Malaria parasites of man: life-cycles and morphology. Malaria:Principles and Practice of Malariology. Edited by: Wernsdorfer WH, Sir McGregor I. 1988, London: Churchill Livingstone, 61-96.
Xu BL, Su YP, Shang LY, Zhang HW: Malaria control in Henan Province, People's Republic of China. The American journal of tropical medicine and hygiene. 2006, 74 (4): 564-567.
Park JW, Jun G, Yeom JS: Plasmodium vivax malaria: status in the Republic of Korea following reemergence. Korean J Parasitol. 2009, S39-50. 47 Suppl
Yang Z, Miao J, Huang Y, Li X, Putaporntip C, Jongwutiwes S, Gao Q, Udomsangpetch R, Sattabongkot J, Cui L: Genetic structures of geographically distinct Plasmodium vivax populations assessed by PCR/RFLP analysis of the merozoite surface protein 3beta gene. Acta tropica. 2006, 100 (3): 205-212. 10.1016/j.actatropica.2006.10.011.
Conway DJ: Tracing the dawn of Plasmodium falciparum with mitochondrial genome sequences. Trends Genet. 2003, 19 (12): 671-674. 10.1016/j.tig.2003.10.007.
Perkins SL: Molecular systematics of the three mitochondrial protein-coding genes of malaria parasites: corroborative and new evidence for the origins of human malaria. Mitochondrial DNA. 2008, 19 (6): 471-478.
Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, Krettli AU, Ho M, Wang A, White NJ, Suh E, Beerli P, Su XZ: Early origin and recent expansion of Plasmodium falciparum. Science (New York, NY. 2003, 300 (5617): 318-321. 10.1126/science.1081449.
Miao M, Yang Z, Cui L, Ahlum J, Huang Y, Cui L: Different allele prevalence in the dihydrofolate reductase and dihydropteroate synthase genes in Plasmodium vivax populations from China. The American journal of tropical medicine and hygiene. 2010, 83 (6): 1206-1211. 10.4269/ajtmh.2010.10-0259.
Zhong D, Bonizzoni M, Zhou G, Wang G, Chen B, Vardo-Zalik A, Cui L, Yan G, Zheng B: Genetic diversity of Plasmodium vivax malaria in China and Myanmar. Infect Genet Evol. 2011, 11 (6): 1419-1425. 10.1016/j.meegid.2011.05.009.
Cui L, Yan G, Sattabongkot J, Cao Y, Chen B, Chen X, Fan Q, Fang Q, Jongwutiwes S, Parker D, Sirichaisinthop J, Kyaw MP, Su XZ, Yang H, Yang Z, Wang B, Xu J, Zheng B, Zhong D, Zhou G: Malaria in the Greater Mekong Subregion: Heterogeneity and complexity. Acta tropica. 2011
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8 (3): 186-194.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8 (3): 175-185.
Nickerson DA, Tobe VO, Taylor SL: PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997, 25 (14): 2745-2751. 10.1093/nar/25.14.2745.
Gordon D, Abajian C, Green P: Consed: a graphical tool for sequence finishing. Genome Res. 1998, 8 (3): 195-202.
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25 (11): 1451-1452. 10.1093/bioinformatics/btp187.
Qi J, Zhao F, Buboltz A, Schuster SC: inGAP: an integrated next-generation genome analysis pipeline. Bioinformatics. 2009, 26 (1): 127-129.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22 (22): 4673-4680. 10.1093/nar/22.22.4673.
Clement M, Posada D, Crandall KA: TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000, 9 (10): 1657-1659. 10.1046/j.1365-294x.2000.01020.x.
Pfenninger M, Posada D: Phylogeographic history of the land snail Candidula unifasciata (Helicellinae, Stylommatophora): fragmentation, corridor migration, and secondary contact. Evolution. 2002, 56 (9): 1776-1788.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155 (2): 945-959.
Evanno G, Regnaut S, Goudet J: Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005, 14 (8): 2611-2620. 10.1111/j.1365-294X.2005.02553.x.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform Online. 2005, 1: 47-50.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123 (3): 585-595.
Fu YX: Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997, 147 (2): 915-925.
Rogers AR, Harpending H: Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992, 9 (3): 552-569.
Walker MJ, Stockman AK, Marek PE, Bond JE: Pleistocene glacial refugia across the Appalachian Mountains and coastal plain in the millipede genus Narceus: evidence from population genetic, phylogeographic, and paleoclimatic data. BMC Evol Biol. 2009, 9: 25-10.1186/1471-2148-9-25.
Slatkin M, Hudson RR: Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991, 129 (2): 555-562.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7: 214-10.1186/1471-2148-7-214.
Iwagami M, Hwang SY, Fukumoto M, Hayakawa T, Tanabe K, Kim SH, Kho WG, Kano S: Geographical origin of Plasmodium vivax in the Republic of Korea: haplotype network analysis based on the parasite's mitochondrial genome. Malaria journal. 2010, 9: 184-10.1186/1475-2875-9-184.
Fay JC, Wu CI: A human population bottleneck can account for the discordance between patterns of mitochondrial versus nuclear DNA variation. Mol Biol Evol. 1999, 16 (7): 1003-1005.
Tajima F: The effect of change in population size on DNA polymorphism. Genetics. 1989, 123 (3): 597-601.
Ricklefs RE, Outlaw DC: A molecular clock for malaria parasites. Science (New York, NY. 2010, 329 (5988): 226-229. 10.1126/science.1188954.
Nicolaiev BP: Subspecies of the parasite of tertian malaria (Plasmodium vivax). Dokl Akad Nauk (USSR). 1949, 67: 201-210.
Li J, Collins WE, Wirtz RA, Rathore D, Lal A, McCutchan TF: Geographic subdivision of the range of the malaria parasite Plasmodium vivax. Emerging infectious diseases. 2001, 7 (1): 35-42. 10.3201/eid0701.010105.
Walton C, Handley JM, Tun-Lin W, Collins FH, Harbach RE, Baimai V, Butlin RK: Population structure and population history of Anopheles dirus mosquitoes in Southeast Asia. Mol Biol Evol. 2000, 17 (6): 962-974.
Lee HJ, Boulding EG: Mitochondrial DNA variation in space and time in the northeastern Pacific gastropod, Littorina keenae. Mol Ecol. 2007, 16 (15): 3084-3103. 10.1111/j.1365-294X.2007.03364.x.
Zhang DX, Hewitt GM: Nuclear DNA analyses in genetic studies of populations: practice, problems and prospects. Mol Ecol. 2003, 12 (3): 563-584. 10.1046/j.1365-294X.2003.01773.x.
Ballard JW, Whitlock MC: The incomplete natural history of mitochondria. Mol Ecol. 2004, 13 (4): 729-744. 10.1046/j.1365-294X.2003.02063.x.
Gunawardena S, Karunaweera ND, Ferreira MU, Phone-Kyaw M, Pollack RJ, Alifrangis M, Rajakaruna RS, Konradsen F, Amerasinghe PH, Schousboe ML, Galappaththy GN, Abeyasinghe RR, Hartl DL, Wirth DF: Geographic structure of Plasmodium vivax: microsatellite analysis of parasite populations from Sri Lanka, Myanmar, and Ethiopia. Am J Trop Med Hyg. 2010, 82 (2): 235-242. 10.4269/ajtmh.2010.09-0588.
Gunasekera AM, Wickramarachchi T, Neafsey DE, Ganguli I, Perera L, Premaratne PH, Hartl D, Handunnetti SM, Udagama-Randeniya PV, Wirth DF: Genetic diversity and selection at the Plasmodium vivax apical membrane antigen-1 (PvAMA-1) locus in a Sri Lankan population. Molecular biology and evolution. 2007, 24 (4): 939-947. 10.1093/molbev/msm013.
Putaporntip C, Jongwutiwes S, Sakihama N, Ferreira MU, Kho WG, Kaneko A, Kanbara H, Hattori T, Tanabe K: Mosaic organization and heterogeneity in frequency of allelic recombination of the Plasmodium vivax merozoite surface protein-1 locus. Proceedings of the National Academy of Sciences of the United States of America. 2002, 99 (25): 16348-16353. 10.1073/pnas.252348999.
Moon SU, Lee HW, Kim JY, Na BK, Cho SH, Lin K, Sohn WM, Kim TS: High frequency of genetic diversity of Plasmodium vivax field isolates in Myanmar. Acta Trop. 2009, 109 (1): 30-36. 10.1016/j.actatropica.2008.09.006.
Yip K: Antimalarial work in China: a historical perspective. Parassitologia. 1998, 40 (1-2): 29-38.
Sheng HF, Zhou SS, Gu ZC, Zheng X: [Malaria situation in the People's Republic of China in 2002]. Zhongguo ji sheng chong xue yu ji sheng chong bing za zhi = Chinese journal of parasitology & parasitic diseases. 2003, 21 (4): 193-196.
Jongwutiwes S, Putaporntip C, Hughes AL: Bottleneck effects on vaccine-candidate antigen diversity of malaria parasites in Thailand. Vaccine. 2010, 28 (18): 3112-3117. 10.1016/j.vaccine.2010.02.062.
Wright S: Isolation by Distance. Genetics. 1943, 28 (2): 114-138.
Anderson TJ, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, Bockarie M, Mokili J, Mharakurwa S, French N, Whitworth J, Velez ID, Brockman AH, Nosten F, Ferreira MU, Day KP: Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000, 17 (10): 1467-1482.
Hernandez-Vera G, Mitrovic M, Jovic J, Tosevski I, Caldara R, Gassmann A, Emerson BC: Host-associated genetic differentiation in a seed parasitic weevil Rhinusa antirrhini (Coleptera: Curculionidae) revealed by mitochondrial and nuclear sequence data. Mol Ecol. 19 (11): 2286-2300.
Hwang SY, Kim SH, Kho WG: Genetic characteristics of polymorphic antigenic markers among Korean isolates of Plasmodium vivax. Korean J Parasitol. 2009, S51-58. 47 Suppl
White NJ: Determinants of relapse periodicity in Plasmodium vivax malaria. Malaria journal. 2011, 10 (1): 297-10.1186/1475-2875-10-297.
Orjuela-Sanchez P, Karunaweera ND, da Silva-Nunes M, da Silva NS, Scopel KK, Goncalves RM, Amaratunga C, Sa JM, Socheat D, Fairhust RM, Gunawardena S, Thavakodirasah T, Galapaththy GL, Abeysinghe R, Kawamoto F, Wirth DF, Ferreira MU: Single-nucleotide polymorphism, linkage disequilibrium and geographic structure in the malaria parasite Plasmodium vivax: prospects for genome-wide association studies. BMC Genet. 2010, 11: 65-
Culleton R, Coban C, Zeyrek FY, Cravo P, Kaneko A, Randrianarivelojosia M, Andrianaranjaka V, Kano S, Farnert A, Arez AP, Sharp PM, Carter R, Tanabe K: The Origins of African Plasmodium vivax; Insights from Mitochondrial Genome Sequencing. PLoS One. 2011, 6 (12): e29137-10.1371/journal.pone.0029137.
Yang BL, Wan WJ, Wang WR, Hu HX, Li HX, Li XL, Li CZ, Huang GZ: [Experimental studies on the biological characteristics of Plasmodium vivax in south Yunnan]. Ji sheng chong xue yu ji sheng chong bing za zhi = Journal of parasitology & parasitic diseases. 1986, 4 (2): 101-105.
Acknowledgements
We are grateful to Yi Wu for help with structure analysis. This work was supported by NIH grants GM080586 and U19AI089672.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
LC designed experiments, guided the work and helped to draft the manuscript. MM carried out the molecular studies, analyzed data and drafted the manuscript. ZY provided the samples and carried some molecular studies. YH provided the samples. PH and AE revised the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2011_1993_MOESM1_ESM.DOC
Additional file 1: Primer sequences. Primers used for amplifying and sequencing the mitochondrial genome of Plasmodium vivax. (DOC 31 KB)
12862_2011_1993_MOESM2_ESM.DOC
Additional file 2: Distribution of SNPs in the Plasmodium vivax mitochondrial genomes isolated from Myanmar, China and South Korea. The graph shows the positions of the SNPs in the 5900 bp mt genome for the 30 haplotypes (h1 - h30) of P. vivax populations. (DOC 50 KB)
12862_2011_1993_MOESM3_ESM.DOC
Additional file 3: Phylogeny of Plasmodium vivax parasites. A maximum likelihood tree based on mitochondrial genomes of 390 Plasmodium vivax samples. (DOC 150 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Miao, M., Yang, Z., Patch, H. et al. Plasmodium vivax populations revisited: mitochondrial genomes of temperate strains in Asia suggest ancient population expansion. BMC Evol Biol 12, 22 (2012). https://doi.org/10.1186/1471-2148-12-22
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-12-22