
Abstract
Background
Atlantolacerta andreanskyi is an enigmatic lacertid lizard that, according to the most recent molecular analyses, belongs to the tribe Eremiadini, family Lacertidae. It is a mountain specialist, restricted to areas above 2400 m of the High Atlas Mountains of Morocco with apparently no connection between the different populations. In order to investigate its phylogeography, 92 specimens of A. andreanskyi were analyzed from eight different populations across the distribution range of the species for up to 1108 base pairs of mitochondrial DNA (12S, ND4 and flanking tRNA-His) and 2585 base pairs of nuclear DNA including five loci (PDC, ACM4, C-MOS, RAG1, MC1R).
Results
The results obtained with both concatenated and coalescent approaches and clustering methods, clearly show that all the populations analyzed present a very high level of genetic differentiation for the mitochondrial markers used and are also generally differentiated at the nuclear level.
Conclusions
These results indicate that A. andreanskyi is an additional example of a montane species complex.
Similar content being viewed by others


Background
An emerging pattern among European biotas is that the accentuated environmental instability that occurred during the Pleistocene did not lead to increased speciation rates, with many species and populations originating during the Miocene and proceeding through the Quaternary [1, 2]. In many species, population fragmentation was triggered by the beginning of the Messinian Salinity Crisis, a short (600 000 years) but crucial period that occurred between 5.9 and 5.3 Mya during which the Mediterranean Sea desiccated almost completely, producing a general and drastic increase in aridity around the Mediterranean Basin [3, 4]. As a result of this increased aridity, forests continued to be replaced by more open and arid landscapes forcing the mesic species to retreat to the moister Atlantic-influenced areas and to the mountainous regions, leading to high speciation in some groups [5, 6].
Various studies have attempted to unravel the different roles that the global aridification at the end of the Miocene and the Pleistocene glacial cycles have played in the diversity and distribution of European faunas [7]. However, little is known about the effects that these climatic changes had on species living further South, in the African continent. Recent assessments of central African chameleons have uncovered evidence of long-isolated evolutionary histories, with the survival of palaeoendemics leading to considerable diversity [8]. In general, reptiles are excellent model organisms to assess the relative role that the Pre-Quaternary and Quaternary major climatic events have played in the origin, evolution and distribution of species [9]. Available data from some herpetofauna indicate that a similar pattern to the neighboring Iberian Peninsula exists in North Africa, with deep lineages originating at the end of the Miocene (Chalcides[10], Acanthodactylus[11–13], Podarcis[2, 14, 15], Saurodactylus[16], Ptyodactylus[17], Salamandra[18], Pleurodeles[19]). However, the lack of informative nuclear markers in most of these studies may prevent the recovery of the true evolutionary history of the group [eg. [20, 21], and makes it difficult to ascertain if these lineages correspond to species complexes or not. Since there is a strong likelihood of discordance between gene trees and species trees [22–24], information from different genetic markers (mitochondrial and nuclear) is thus necessary for delimiting evolutionary lineages, as well as for establishing phylogenetic relationships.
Despite being key concepts in the fields of systematic and evolutionary biology, recognizing and delimiting species are highly controversial issues ([e.g. [25, 26]). Recognizing species is not only a taxonomic challenge, but is also essential for other biological disciplines such as biogeography, ecology and evolutionary biology [27], and has serious consequences for conservation biology and the design of effective conservation plans [28, 29]. Delimiting species is also the first step towards discussing broader questions on evolution, biogeography, ecology or conservation. Recently, thanks to intellectual progress made in the field with the aim of identifying a common element among all the different species concepts, a single, more general, concept of species known as General Lineage Species Concept has been suggested [30]. This unified species concept emphasizes the common element found in many species concepts, which is that species are separately evolving lineages. Therefore, properties like reciprocal monophyly at one or multiple loci, phenotypic diagnosability, ecological distinctiveness, etc. are not part of the species concept but are used to assess the separation of lineages and to species delimitation [31]. This separation between species conceptualization and species delimitation and the proposal of a unified species concept has concentrated efforts in the development of new approaches for species delimitation, as for example with “integrative taxonomy” [32, 33], among others]. Under this new approach, species delineation is regarded as an objective scientific process that results in a taxonomic hypothesis. Therefore, the level of confidence in the taxonomic hypothesis supported by several independent character sets is much higher than for species supported by only one character [34]. Such an integrative view is especially useful in the case of taxonomic groups that are morphologically conservative, where cryptic species have probably been overlooked [17, 35, 36].
Normally, high altitude species carry signatures of the expansion and contraction cycles occurred during glacial and interglacial periods [37–39]. Because of this, they are of particular interest to study historical responses to climate change, since they are adapted to a small window of environmental changes, and usually present low tolerance to high temperatures [40]. In Europe, high altitude species often seem to have persisted through glacial periods by short movements to lower altitudes rather than to the classic "southern refugia" of lowland species. In this way current ranges may primarily reflect postglacial expansions [41]. However, it is not clear if the same phenomenon occurs in African montane taxa.
Atlantolacerta andreanskyi (Werner, 1929) is a lacertid lizard endemic to the western and central parts of the High Atlas Mountains of Morocco. It is restricted to areas above 2400 m [42, 43], where it is frequently found in the vicinity of small watercourses or plateaus in the top of the mountains that retain some water from rain or snowmelt. Habitat is normally screes and areas with boulders, meadows and, in particular, the base of cushion-like thorny plants in these places [42]; personal observation]. Although A. andreanskyi had initially been placed in several different genera within the subtribe Lacertini [44–48], recent phylogenetic analyses based on mitochondrial DNA and a combination of mitochondrial and nuclear markers [49, 50] suggest that A. andreanskyi is a member of the subtribe Eremiadini, and apparently sister to the remaining Eremiadini. This position would conform to this species lacking the synapomorphies that characterize most other Eremiadini, namely a derived condition of the ulnar nerve and the presence of a fully developed armature in the hemipenis, which has folded lobes when retracted. It is also distinctive within the Eremiadini regarding the presence of enlarged masseteric scale [49]. Because of its phylogenetic position, without close relationship to any other genus of Eremiadini and its distinctive morphology it was recently placed in a new monotypic genus, Atlantolacerta[49]. Atlantolacerta andreanskyi is distributed across 440 Km (straight line) of mountainous terrain, with the different populations presenting an apparently disjunct distribution ([42, 43]; see Figure 1). As with many montane species, the situation observed in A. andreanskyi is similar to an archipelago, with the different “islands” being represented by mountaintops disconnected due to areas of unsuitable habitat below 2400 m. As a result of this scenario, minimal gene flow is currently expected between the different populations; however, it is not known how the different climatic events occurred during the Miocene and Pleistocene have affected this species. Even though some aspects of the biology of A. andreanskyi are already well known [e.g. [51, 52], the genetic structure of the different populations, as well as the relationships between the different populations have never been assessed before.

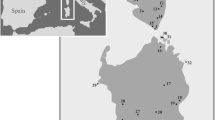
Atlantolacerta andreanskyi distribution map. The color dots represent the localities of the populations sampled for this work, J. Awlime (yellow), J. Sirwa (pink), Oukaimeden (red), Toubkal (orange), Tizin Tichka (dark blue), J. Azourki (light blue), Outabati (light green), and J. Ayache (dark green). The white dots represent the distributions of the species by Bons and Geniez [42].
Therefore, in order to shed some light on the previous questions and attempt to assess the evolutionary history of the species and identify the number of lineages, we sampled the distribution area of the species and performed several combined phylogenetic reconstructions and clustering analyses, using both mtDNA and nuclear markers.
Results
Mitochondrial genealogies
A total of 1108 base pairs (bp) of concatenated mtDNA (12S rRNA 330 bp, ND4 592 bp and tRNA-His 186 bp) were obtained for 89 A. andreanskyi. The concatenated alignment of the ingroup sequences revealed 30 haplotypes (3 from Tizin Tichka, 7 from J. Ayache, 5 from J. Sirwa, 2 from Oukaimeden, 7 from J. Azourki, 2 from Outabati, 2 from Toubkal and 2 from J. Awlime) and contained 241 variable sites, of which 232 were parsimony informative.
Analyses of the concatenated mtDNA data were mostly congruent (Figure 2A). Seven well-supported lineages were recovered from these analyses (pp > 0.95 and BP > 70%), corresponding to the populations from J. Awlime, J. Sirwa, Tizin Tichka, J. Azourki, Outabati, J. Ayache, and Oukaimeden and nearby Toubkal that were grouped together. Regarding the relationships among these clades, we could distinguish three main groups, Oukaimeden and Toubkal with J. Sirwa from the southern end of the distribution range; J. Ayache with Outabati from the northern distribution, and Tizin Tichka with J. Azourki from the central distribution range. The population from J. Awlime, from the extreme South of the range, is a genetically distinct lineage related to the northern group, although, both ML and BI analysis weakly support this topology (see Figure 2A).

Trees resulting from partitioned Bayesian analysis. (A) mitochondrial DNA tree (12S, ND4 and flanking tRNA-His), (B) nuclear concatenated tree (RAG1, ACM4, MC1R, PDC and C-MOS), (C) Concatenated tree from the combined mitochondrial and nuclear DNA data. The partitions used the models described in the text. Bayesian posterior probabilities (0–1) and bootstrap values (> 50%) for ML (1–100) are indicated near the branches, (D) Species tree from mitochondrial and nuclear DNA data from the Bayesian Inference of Species Trees (STARBEAST). Clade posterior probabilities are shown to the left of the nodes, and divergence times and 95% intervals (calculated in BEAST using only ND4 + tRNA-His), to the right of the nodes. The trees were rooted using Podarcis bocagei, P. hispanica and P. carbonelli. The colors represent the different populations.
All the populations present a low level of diversity in the mitochondrial DNA (uncorrected genetic distances 0–0.5% for the ND4 + tRNA-His and 0 – 0.2% for the 12S; see Table 1), and a very high level of genetic divergence between populations (5.5 – 16.5% in the ND4 + tRNA-His and 2.5 – 6.6% in the 12S).
Nuclear genealogies
A total of 77 specimens of A. andreanskyi were sequenced for five nuclear genes. The ACM4 was 447 bp long, presenting 47 haplotypes and 34 polymorphic sites, 33 of them parsimony informative; C-MOS was 534 bp long, with 32 haplotypes and 21 polymorphic sites, all of them parsimony informative; MC1R was 635 bp long, with 57 haplotypes and 36 variable sites, 35 of them parsimony informative; PDC was 441 bp long, with 60 haplotypes and 29 variable sites, 26 of them parsimony informative; RAG1 was 528 bp long, with 38 haplotypes and 19 variable sites, 18 of them parsimony informative.
The differences in the genetic distances between the lineages are congruent with the geographic distance between them, supporting the grouping of the lineages in three main groups as seen in the analysis of mitochondrial sequences.
The concatenated analyses of the 5 unphased nuclear markers are congruent with the results obtained in the mitochondrial DNA tree, although with some differences (Figure 2B). Despite recovering the three main groups observed in the mtDNA analysis, according to the nuclear markers the J. Awlime population is not sister to the northernmost populations but branches off inside a polytomy with the westernmost lineages at the base of the tree. It is possible to distinguish some of the lineages, although in some cases they are not monophyletic. The J. Ayache population is monophyletic but makes Outabati paraphyletic. The same happens with Tizin Tichka, which makes the population from J. Azourki paraphyletic. The population from Oukaimeden is polyphyletic.
Concatenated analysis (mtDNA and nDNA)
The results of the ML and BI analyses of the mtDNA and nDNA (Figure 2C) support the same seven lineages as recovered in the mitochondrial analysis, although in this case J. Awlime is sister to the central and northern lineages (Tizin Tichka, J. Azourki, Outabati, and J. Ayache) instead of being sister to only the northernmost lineages (Figure 2A). As in the mtDNA analysis (Figure 2A), the relationship of J. Awlime with the central and northern lineages is very poorly supported. This result was expected, given the higher resolving power of the mtDNA that contributed with 241 variable sites versus the 150 from the nDNA.
Nuclear networks
As show in Figure 3 and Table 2, there is a moderate degree of haplotype sharing between populations, with most of them lacking private alleles for the nuclear genes analyzed.

Parsimony networks corresponding to MC1R (A), RAG1 (B), C-MOS (C), ACM4 (D) and PDC (E) nDNA sequence variation from all the populations. The colors used were the same as the used in the map (Figure 1) and trees (Figure 2), J. Awlime (yellow), Toubkal (orange), Oukaimeden (red), J. Sirwa (pink), Tizin Tichka (dark blue), J. Azourki (light blue), Outabati (light green), and J. Ayache (dark green). Lines represent a mutation step, circles represent haplotypes and dots missing haplotypes. The size of the circles is proportional to the number of alleles.
Clustering analysis and individual assignment
In our study, the obtained K differs with the combination between the ancestry model and the allele frequency model. When combined the No Admixture Model (ancestry model) with the Allele Frequencies Independent Model (allele frequency model) the best resulting K values where for K = 3: South (Oukaimeden, J. Sirwa, Toubkal and J. Awlime), center (Tizin Tichka and J. Azourki) and North (Outabati and J. Ayache) groups. With the other three combinations between the models the best result were for K = 6: J. Sirwa, Tizin Tichka, J. Azourki, Outabati, J. Ayache, and a group formed by Oukaimeden, Toubkal and J. Awlime (Figure 4).

Population structure estimation. Each individual is represented by a thin vertical line, which is partitioned into K colored segments that represent the individual’s estimated membership fractions in K clusters. The bigger vertical divisions separate individuals from different populations. Populations are labeled below the figure. The colors used are the same used in Figure 1 and Figure 2.
Species tree and divergence time estimates
The results of the clustering analysis with K = 6 were used to define the species for the species tree analysis in STARBEAST. The tree inferred with information from mitochondrial and nuclear markers (phased) (figure 2D) recovered the same topology as in Figure 2C, with all the relationships between the lineages supported by previous analyses.
The divergence time estimates were calculated for the six populations (Table 1). High effective sample sizes were observed for all parameters in all BEAST analysis (posterior ESS values > 1000 for all four analyses) and assessment of convergence statistics in Tracer indicated that all analyses had converged. Maximum clade credibility tree for ND4 + tRNA-His was identical in topology to those produced by Bayesian and ML analyses. According to the inferred dates resulted from BEAST (Figure 2D), the two main mitochondrial lineages of A. andreanskyi (South versus central and North) split approximately 7.6 Ma (95% high posterior density (HPD) interval 4.3-11.9 Ma). The populations that are grouped in the three main clades (South, central and North) split approximately at the same time, being Tizin Tichka and J. Azourki the first to split at about 4.3 Ma (1.4-7.8), followed by Oukaimeden and J. Sirwa 2.9 Ma (1–5.6), and Outabati and J. Ayache 2.4 Ma (0.8-4.4). Tizin Tichka and J. Azourki diverged from Outabati and J. Ayache approximately 6.4 Ma (3.1-10.2).
Discussion
Extreme mtDNA diversity in A. andreanskyi
Several recently published analyses of North African herpetofauna have revealed high levels of endemism and cryptic species [12, 14, 15, 17]. In this analysis, the surprising result was the extreme diversity of mitochondrial DNA found between almost all the populations analyzed The genetic differentiation observed between populations (2.8% - 6.6% in 12S and 5.5% - 16.5% in ND4 + tRNA-His) is similar and, in some cases, higher than the divergence found between Iberolacerta species (between 7.4% and 8.2% in the cytochrome b gene, [53]), a lacertid genus with most of its species occurring in the mountains of the Iberian Peninsula [41, 54]. Initially considered one species, there are now seven recognized species of Iberolacerta in the Iberian Peninsula. Genetic differentiation between these species is lower than between the different populations of A. andreanskyi.
Although the mitochondrial phylogeny supports the existence of seven distinct groups, the clustering analysis only supports the existence of six lineages (J. Sirwa, Tizin Tichka, J. Azourki, Outabati, J. Ayache and a lineage formed by Oukaimeden, Toubkal and J. Awlime). Toubkal samples were always part of the same lineage as Oukaimeden, although, they show some divergence at least at the mitochondrial DNA level (1.7% in ND4 + tRNA-His and 0.3% in 12S). This is not unexpected, as these populations are geographically very close and are part of the High Atlas Mountains, where interconnectivity between populations could occur. The mitochondrial phylogenetic analyses supported the existence of a seventh isolated lineage, J. Awlime, however clustering analysis and the nuclear phylogeny did not support the distinctiveness of this population, possibly because of the small sampling size. Unfortunately, despite multiple attempts to sample in this remote region, only three individuals were captured. The analyses also could not recover the genetic relationship between J. Awlime and the other populations, because its position in the trees fluctuated between the two main groups (North and South), without support in any of the trees.
Non-reciprocal monophyly in nuclear markers and species delimitation
In the phylogenetic analyses of the concatenated nuclear loci, some of the lineages supported by mtDNA data were not monophyletic. This was observed only between the geographically closest lineages, as in the case of Oukaimeden and J. Sirwa; Tizin Tichka and J. Azourki; and Outabati and J. Ayache, that presumably were in contact more recently than the others. This may be due to the larger effective population size of the nuclear DNA compared to the mitochondrial DNA and the consequent stronger effect of the incomplete lineage sorting at each single nuclear loci [55]. Additionally, the slow evolutionary rate of some of these markers may be a factor. The conjugation of these two effects probably explains the absence of concordance in the single nuclear gene trees (not show), although the same general topology was recovered in the concatenated nuclear phylogeny. Reciprocal monophyly is one of the primary criteria to delimit species [31, 56]. Although it is possible to delimit species without observing monophyly in gene trees, since a considerable amount of time must pass after the beginning of divergence of species until they show reciprocal monophyly at a sample of multiple loci [57, 58]. Pinho et al.[59] have shown that Podarcis from the Iberian Peninsula and North Africa have a similar pattern (between mtDNA and nuclear) but in a smaller time window and using faster evolving nuclear loci and, in contrast to our case, some populations are in contact.
Although we are aware that the determination of K, in STRUCTURE, is only an ad hoc guide to describe consistence between models and the data [60], the program has been commonly used for this end [61]. Several methods based on Bayesian clustering have been developed [62–64], however, STRUCTURE is the most widely used, and various studies show its efficiency in assigning individuals to their population of origin [65–68] and its ability to construct an appropriate clustering hypothesis [61]. However, in the present example the analysis was limited because it was based only in haplotype information. The obtained K differ with the combination model used, but in most of the combinations the analysis supports a K = 6 corresponding to the geographical populations and to the results recovered by the other analyses. This analysis also placed the samples from the J. Awlime population together with the Oukaimeden lineage, possibly due to the limited haplotype sampling. Similarly, the concatenated phylogenetic tree, based on all the genes, supports the existence of 7 lineages giving once more a low support to the relationship between J. Awlime and the other lineages.
The networks of the individual nuclear loci show high percentage of private alleles in some of the lineages, which fluctuate depending on the gene.
Dating the trees
All the lineages are grouped in two main clusters, the northern group composed by J. Ayache, Outabati, J. Azourki and Tizin Tichka; and the southern group that includes Oukaimeden and J. Sirwa. The divergence obtained for these two lineages was around 7.6 Mya, (4.3-11.9), which coincides approximately with the time of the final closing of the Rifian Strait (7.2 Mya; [3]). During the Miocene, tectonic activity in the region was intense and included the uplift of the Atlas Mountains that occurred around 9.0 Mya [69, 70]. It was more or less at the same time that Podarcis invaded North Africa (7.5 ± 1.2 Mya, [2]) and the Iberian clade of Iberolacerta started to fragment (6.1 Mya, [1]). The split of the six lineages must have occurred later, probably during the Quaternary Glaciations (4.3 ± 3; 2.4 ± 2; 2.9 ± 2 Mya). However, the confidence intervals obtained were very large, increasing the time window for the events and the associated error. Determination of the time of the speciation events is important to understand the evolutionary biogeography of species [71]. However, it is difficult to estimate ages in phylogenies without several sources of error. Clearly the lineages of A. andreanskyi are pre-Pleistocenic and, as found in Central African chameleons [8] can be considered paleoendemics. However, without better calibration points it is difficult to date the split of the lineages more precisely than this.
Conclusions
Phylogeographic assessments of several taxa in northwest Africa have indicated the presence of cryptic diversity in organisms ranging from scorpions [72] to mammals [73], and reptiles are not an exception [e.g. [11, 17, 74]. What is exceptional in the case of A. andreanskyi are the high levels of mitochondrial divergence between almost every sampled populations, ranging from 5.5 up to 16.5% (ND4 + tRNA-His) between populations separated by low geographic distances (for example just 60 Km between Oukaimeden and J. Sirwa and 45 Km between Oukaimeden and Tizin Tichka). Six of the eight analyzed populations are highly distinct based on both mtDNA and multiple nuclear markers. This raises the issue not of whether A. andreanskyi is a species complex, but just how many species may occur within the group. Presumably, far more than the six possible species identified in this study, since, probably, many populations remain unsampled. However, preliminary morphological analyses suggest that all the different populations included in the present study are very homogeneous (unpublished data). This may imply the presence of cryptic diversity, but definitive conclusions should wait until a complete morphological study is carried out (work in progress).
Current models of reptiles species accessed for the region indicate low levels of diversity across much of the High Atlas Mountains [75]. Indeed only a few species are recorded at altitudes above 2000 m; typically A. andreanskyi, Quedenfeldtia species (Q. trachyblepharus and Q. moerens), Chalcides montanus and Vipera monticola [e.g. [42, 76]. However, the finding of high genetic diversity in A. andreanskyi indicates that unidentified lineages occur, and that the other high mountain species should also be assessed as possible cryptic species candidates. Our results are also essential from a conservation point of view, as many forms may actually have smaller ranges than currently thought, and small isolated populations on high mountains have been identified as those of high concern under typical global warming scenarios [77]. Given these results it is necessary to increase the sampling in order to understand the relationship of J. Awlime with the other populations and try to find new populations. Furthermore it is very important to conduct a through morphological study to determine if there is phenotypic variation, and then to revise the taxonomy of the genus Atlantolacerta.
Methods
Species concept and integrative approach
Although the present study does not include a taxonomic revision of the genus Atlantolacerta, like many other works in which some of the authors of the present manuscript have participated [35, 78, 79], we advocate for the use of the General Lineage Species Concept proposed by de Queiroz [30]. Two lines of evidence have been defined on the basis of alleged independence of their respective datasets: mitochondrial DNA and nuclear DNA. In the present study, we have decided to retain as “putative species” only these lineages that were recovered as monophyletic in the phylogenetic analysis of the mtDNA data and that were supported by the analysis of the nDNA using STRUCTURE v.2.3.2 [60]. Within the framework of an integrative approach, and pending the inclusion of morphological data, this would correspond to Integration by total congruence (ITC). However, it is important to take into account that in the absence of a thorough morphological analysis we do not consider the molecular data presented here enough to revise the taxonomy of the genus Atlantolacerta.
DNA extraction, amplification, and sequencing
A total of 92 individuals from eight different populations distributed across the entire range of Atlantolacerta andreanskyi were sampled for this study: 14 from Oukaimeden, 15 from Tizin Tichka, 14 from Jebel Ayache, 15 from Jebel Azourki, 14 from Outabati, 15 from Jebel Sirwa and 2 from Toubkal and 3 from J. Awlime (Figure 1 and Table 3). Specimens were caught by hand, identified on the basis of external features, measured and photographed for later morphological studies. Tail tips where collected and stored in 96% ethanol, after which individuals were released in the same place where they were caught.
Genomic DNA was extracted from ethanol-preserved tissue samples using standard high-salt protocols [80]. A total of 89 specimens of Atlantolacerta andreanskyi plus three outgroups (Podarcis hispanica, Podarcis carbonelli and Podarcis bocagei) were sequenced for two mitochondrial regions: partial 12S rRNA (12S) and partial NADH dehydrogenase 4 (ND4) and flanking tRNA (tRNA-His) and 77 specimens for five nuclear gene fragments, recombination-activating gene 1 (RAG1), acetylcholinergic receptor M4 (ACM4), melanocortin receptor 1 (MC1R), oocyte maturation factor Mos (C-MOS) and phosducin (PDC). Primers used for both amplification and sequencing were: 12Sa and 12Sb [81] for the 12S following the PCR conditions described in Harris and Arnold [82], ND4 and Leu for ND4 + tRNA-His, PCR conditions described in Arévalo et al.[83]; L2408 and H2920 for RAG1 following the PCR conditions from Vidal and Hedges [84]; tg-F and tg-R [85] for ACM4 with PCR conditions following Gamble et al.[86]; MC1RF and MC1RR for MC1R following PCR conditions described in Pinho et al.[87]; Lsc1 and Lsc2 for C-MOS following the PCR conditions from Godinho et al.[88]; and PHOF2 and PHOF1 for PDC, following PCR conditions described in Bauer et al.[89]. PCRs were carried out in 25 μl volumes, containing 5.0 μl of 10 reaction Buffer, 2.0 mM of MgCl2, 0.5 mM each dNTP, 0.2 μM each primer, 1 U of Taq DNA polymerase (Invitrogen), and approximately 100 ng of template DNA. Finally, PCR products were purified using exosap IT and the resulting amplified fragments were sequenced on an Applied Biosystem DNA Sequencing Apparatus. Chromatographs were checked manually, assembled and edited using Bioedit 7.0.1 [90]. Sequences were aligned for each gene independently using the online version of MAFFT v.6 [91] with default parameters (gap opening penalty = 1.53, gap extension = 0.0) and FFT-NS-1 algorithm. Coding gene fragments (ND4, C-MOS, ACM4, RAG1, PDC and MC1R) were translated into amino acids and no stop codons were observed, suggesting that the sequences were all functional. Heterozygous individuals were identified based on the presence of two peaks of approximately equal height at a single nucleotide site. SEQPHASE [92] was used to convert the input files, and the software PHASE v2.1.1 to resolve phased haplotypes [93]. Default settings of PHASE were used except for phase probabilities that were set as ≥ 0.7 [94]. All polymorphic sites with a probability of < 0.7 were coded in both alleles with the appropriate IUPAC ambiguity code. Phased nuclear sequences were used for the structure analysis; networks and species tree analysis, and the unphased sequences for the phylogenetic analyses (see below). DnaSP [95] was used to calculate the number of haplotypes (h) and mutations (η). Mega v.3.0 [96] was used to estimate uncorrected p-distances and to obtain the number of variable and parsimony informative sites.
Phylogenetic analyses
Phylogenetic analyses were performed using maximum likelihood (ML) and Bayesian (BI) methods. JModelTest [97] was used to select the most appropriate model of sequence evolution under the Akaike Information Criterion [98]. ML analyses were performed with RAxML v.7.0.4 [99] with 100 random addition replicates. A GTR + I + G model was used and parameters were estimated independently for each partition (by gene). Reliability of the ML tree was assessed by bootstrap analysis [100] including 1000 replications. Bayesian analyses were performed with MrBayes v.3.1.2 [101] with best fitting models applied to each partition by gene and all parameters unlinked across partitions. The models selected for the different partitions were: 12S, GTR + I + G; ND4, GTR + G; tRNA-His, GTR + I + G; ACM4, HKY + I; C-MOS, GTR + I + G; MC1R, HKY + I + G; PDC, GTR + I + G; and RAG1, GTR + I. Two independent runs of 5x106 generations were carried out, sampling at intervals of 1000 generations producing 5000 trees. Convergence and appropriate sampling were confirmed examining the standard deviation of the split frequencies between the two simultaneous runs and the Potential Scale Reduction Factor (PSRF) diagnostic. Burn-in was performed discarding the first 1250 trees of each run (25%) and a majority-rule consensus tree was generated from the remaining trees. In both ML and BI alignment gaps were treated as missing data and the nuclear gene sequences were not phased.
Nuclear Networks
The genealogical relationships between the populations were assessed with haplotype networks for all the individual nuclear genes, constructed using statistical parsimony [102] implemented in the program TCS v 1.21 [103] with a connection limit of 95%. This analysis was made with the phased sequences. Haplotypes were colored taking into account the population of origin.
Population structure – Clustering analyses
A model-based Bayesian clustering method was applied to all haplotypes using STRUCTURE v.2.3.2 [60, 104, 105]. In this analysis, individuals are probabilistically assigned to either a single cluster (the population of origin), or more than one cluster (if there is admixture). STRUCTURE was run with haplotype information from the nuclear fragments independently. We ran our data with the all parameters combinations between the Ancestry Model and the Allele Frequency Model to compare the results. The genetic structure was forced to vary from K = 2 to K = 10 clusters, the latter corresponding to the number of geographic populations sampled plus two. STRUCTURE ran for 550 000 steps, of which the first 50 000 were discarded as burn-in. For each value of K ten independent replicates of the Markov Chain Monte Carlo (MCMC) were conducted. To detect the true number of clusters (K) we followed the graphical methods and algorithms outlined in Evanno et al.[61], with the comparison of the average posterior probability values for K (log likelihood; ln L) using the online version, STRUCTURE HARVESTER v0.6.5 (available at: http://taylor0.biology.ucla.edu/struct_ harvest/, April 2011).
Species tree, and divergence time estimates
Here we applied the coalescent-based species-tree approach implemented in STARBEAST [106] an extension of BEAST v1.6.1 [107] to test the origin and diversification patterns in Atlantolacerta, and to compare these results to those obtained from the ML and BI analyses of the concatenated dataset. This analysis needs a priori information regarding the species/populations delimitation and the species/populations assignation of the individuals in order to reconstruct the topology of the species tree. For this approach, we used the results obtained from previous clustering analyses to define the groups of individuals to be used as “species” (populations) in STARBEAST [106]. The clustering analysis supported the existence of six lineages, as Oukaimeden, Toubkal and J. Awlime were included in the same lineage.
All five nuclear gene fragments, 12S and the fragment consistent of the ND4 and flanking tRNA-His were included in the analyses as 7 independent partitions. The phased dataset was used for the nuclear loci.
The input file was formatted with the BEAUti utility included in the software package. We performed two independent runs of 1.5 x 108 generations, sampling every 15 000 generations, from which 10% were discarded as burn-in. Models and prior specifications applied were as follows (otherwise by default): 12S - GTR + G; ND4 and tRNA-His - HKY + G; MC1R - HKY + I; ACM4 - HKY + I; C-MOS - GTR + I + G; RAG1 - HKY + I; PDC - GTR + I; Relaxed Uncorrelated Lognormal Clock (estimate); Yule process of speciation; random starting tree; alpha Uniform (0, 10).
For all analyses implemented in BEAST, convergence for all model parameters was assessed by examining trace plots and histograms in Tracer v1.5 [108] after obtaining an effective sample size (ESS) > 200. The initial 10% of samples were discarded as burn-in. Runs were combined using LogCombiner, and maximum credibility trees with divergence time means and 95% highest probability densities (HPDs) were produced using Tree Annotator (both part of the BEAST package). Trees were visualized using the software FigTree v1.3.1 [109].
Several studies have already calculated divergence rates for reptiles, and particularly for lacertids [2, 15, 49]. Pinho et al. [15] used well-known and dated independent geological events in the Aegean [110] to estimate a maximum and minimum mutation rate for the ND4 mitochondrial fragment (and flanking tRNA-His) for the lacertid lizards of the genus Podarcis (0.0278 and 0.0174 mutation/site/million years, respectively). However, this was the only information available for our data, since we did not have any fossils or calibrations for nuclear markers. It is important to bear in mind that, in the absence of accurate calibration points in the phylogeny from external and independent data (fossil records, known biogeographic events, or paleoclimatic reconstructions) or as a result of the heterogeneity in the evolutionary rate between the calibrated and uncalibrated taxa, temporal estimates by means of molecular data could be a potential source of inference error, and, therefore, they should be treated with caution [111]. Despite the limitations of molecular clocks [111, 112], divergence time estimates can still provide a proxy for the temporal window of evolutionary diversification in species groups of interest. Therefore and taking into account our data limitations and availability, we used BEAST v.1.6.1 [107] to estimate dates of the cladogenetic events using only ND4 and flanking tRNA-His. We used a phylogeny pruned arbitrarily to include one representative from each of the major lineages uncovered with the concatenated analysis (6 specimens in total, we excluded J. Awlime population, because of the lack of support of the branch in previous analyses). This method excludes closely related terminal taxa because the Yule tree prior (see below) does not include a model of coalescence, which can complicate rate estimation for closely related sequences [113]. Analyses were run four times for 5x107 generations with a sampling frequency of 10 000. Models and prior specifications applied were as follows (otherwise by default): GTR + G for 12S; HKY + G for ND4 and tRNA-His; HKY + I for MC1R; HKY + I for ACM4; GTR + G + I for C-MOS; HKY + I for RAG1; GTR + I for PDC; Relaxed Uncorrelated Lognormal Clock (estimate); Yule process of speciation; random starting tree; alpha Uniform (0, 10); ucld.mean of ND4 Normal (initial value: 0.0226, mean: 0.0226, Stdev: 0.0031).
References
Arribas O, Carranza S: Morphological and genetic evidence of the full species status of Iberolacerta cyreni martinezricai (Arribas, 1996). Zootaxa. 2004, 634: 1-24.
Carretero MA: An integrated assessment of a group with complex systematics: the Iberomaghrebian lizard genus Podarcis (Squamata, Lacertidae). Integr Zool. 2008, 3 (4): 247-266.
Duggen S, Hoernle K, van den Bogaard P, Rupke L, Morgan JP: Deep roots of the Messinian salinity crisis. Nature. 2003, 422 (6932): 602-606.
Krijgsman W, Hilgen FJ, Raffi I, Sierro FJ, Wilson DS: Chronology, causes and progression of the Messinian salinity crisis. Nature. 1999, 400 (6745): 652-655.
Pinho C, Ferrand N, Harris DJ: Reexamination of the Iberian and North African Podarcis (Squamata: Lacertidae) phylogeny based on increased mitochondrial DNA sequencing. Molecular Phylogenetics and Evolution. 2006, 38 (1): 266-273.
Santos X, Roca J, Pleguezuelos JM, Donaire D, Carranza S: Biogeography and evolution of the Smooth snake Coronella austriaca (Serpentes: Colubridae) in the Iberian Peninsula: evidence for Messinian refuges and Pleistocenic range expansions. Amphibia-Reptilia. 2008, 29 (1): 35-47.
Schmitt T: Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Frontiers in Zoology. 2007, 4 (1): 11-
Tolley KA, Chase BM, Forest F: Speciation and radiations track climate transitions since the Miocene Climatic Optimum: a case study of southern African chameleons. J Biogeogr. 2008, 35 (8): 1402-1414.
Camargo A, Sinervo B, Sites JW: Lizards as model organisms for linking phylogeographic and speciation studies. Mol Ecol. 2010, 19 (16): 3250-3270.
Carranza S, Arnold EN, Geniez P, Roca J, Mateo JA: Radiation, multiple dispersal and parallelism in the skinks, Chalcides and Sphenops (Squamata: Scincidae), with comments on Scincus and Scincopus and the age of the Sahara Desert. Molecular Phylogenetics and Evolution. 2008, 46 (3): 1071-1094.
Fonseca MM, Brito JC, Paulo OS, Carretero MA, Harris DJ: Systematic and phylogeographical assessment of the Acanthodactylus erythrurus group (Reptilia: Lacertidae) based on phylogenetic analyses of mitochondrial and nuclear DNA. Molecular Phylogenetics and Evolution. 2009, 51 (2): 131-142.
Fonseca MM, Brito JC, Rebelo H, Kalboussi M, Larbes S, Carretero MA, Harris DJ: Genetic variation among spiny-footed lizards in the Acanthodactylus pardalis group from North Africa. African Zoology. 2008, 43 (1): 8-15.
Harris DJ, Batista V, Carretero MA: Assessment of genetic diversity within Acanthodactylus erythrurus (Reptilia: Lacertidae) in Morocco and the Iberian Peninsula using mitochondrial DNA sequence data. Amphibia-Reptilia. 2004, 25 (2): 227-232.
Kaliontzopoulou A, Pinho C, Harris DJ, Carretero MA: When cryptic diversity blurs the picture: a cautionary tale from Iberian and North African Podarcis wall lizards. Biol J Linn Soc. 2011, 103 (4): 779-800.
Pinho C, Harris DJ, Ferrand N: Contrasting patterns of population subdivision and historical demography in three western Mediterranean lizard species inferred from mitochondrial DNA variation. Mol Ecol. 2007, 16 (6): 1191-1205.
Rato C, Harris DJ: Genetic variation within Saurodactylus and its phylogenetic relationships within the Gekkonoidea estimated from mitochondrial and nuclear DNA sequences. Amphibia-Reptilia. 2008, 29 (1): 25-34.
Perera A, Harris DJ: Genetic variability within the Oudri’s fan-footed gecko Ptyodactylus oudrii in North Africa assessed using mitochondrial and nuclear DNA sequences. Molecular Phylogenetics and Evolution. 2010, 54: 634-639.
Carranza S, Romano A, Arnold EN, Sotgiu G: Biogeography and evolution of European cave salamanders, Hydromantes (Urodela: Plethodontidae), inferred from mtDNA sequences. J Biogeogr. 2008, 35: 724-738.
Carranza S, Arnold EN: History of West Mediterranean newts, Pleurodeles (Amphibia: Salamandridae), inferred from old and recent DNA sequences. Syst Biodivers. 2004, 1 (3): 327-337.
Rato C, Carranza S, Harris DJ: When selection deceives phylogeographic interpretation: The case of the Mediterranean house gecko, Hemidactylus turcicus (Linnaeus, 1758). Molecular Phylogenetics and Evolution. 2011, 58: 365-373.
Rato C, Carranza S, Perera A, Carretero MA, Harris DJ: Conflicting patterns of nucleotide diversity between mtDNA and nDNA in the Moorish gecko, Tarentola mauritanica. Molecular Phylogenetics and Evolution. 2010, 56 (3): 962-971.
Degnan JH, Rosenberg NA: Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends in ecology & evolution. 2009, 24 (6): 332-340.
Edwards SV: Is a New and General Theory of Molecular Systematics Emerging?. Evolution. 2009, 63 (1): 1-19.
Maddison WP: Gene trees in species trees. Syst Biol. 1997, 46 (3): 523-536.
Agapow PM, Bininda-Emonds ORP, Crandall KA, Gittleman JL, Mace GM, Marshall JC, Purvis A: The impact of species concept on biodiversity studies. Q Rev Biol. 2004, 79 (2): 161-179.
Mayden RL: A hierarchy of species concepts: the denouement in the saga of the species problem. Species: The units of diversity. Edited by: Claridge MF, Dawah HA, Wilson MR. 1997, London: Chapman and Hall, 381-423.
Sites JW, Marshall JC: Delimiting species: a Renaissance issue in systematic biology. Trends in Ecology & Evolution. 2003, 18 (9): 462-470.
Agapow M: Species: demarcation and diversity. Phylogeny and Conservation. Edited by: Purvis A, Gittleman JL, Brooks T. 2005, Cambridge, UK: Cambridge University, 57-75.
Sattler T, Bontadina F, Hirzel AH, Arlettaz R: Ecological niche modelling of two cryptic bat species calls for a reassessment of their conservation status. J Appl Ecol. 2007, 44 (6): 1188-1199.
de Queiroz K, Donoghue MJ: Phylogenetic Systematics and the Species Problem. Cladistics. 1988, 4 (4): 317-338.
de Queiroz K: Species Concepts and Species Delimitation. Syst Biol. 2007, 56 (6): 879-886.
Dayrat B: Towards integrative taxonomy. Biol J Linn Soc. 2005, 85: 407-415.
Padial JM, Miralles A, De la Riva I, Vences M: The integrative future of taxonomy. Front Zool. 2010, 7: 16-
Schlick-Steiner BC, Steiner FM, Seifert B, Stauffer C, Christian E, Crozier RH: Integrative taxonomy: a multisource approach to exploring biodiversity. Annu Rev Entomol. 2010, 55: 421-438.
Miralles A, Vasconcelos R, Perera A, Harris DJ, Carranza S: An integrative taxonomic revision of the Cape Verdean skinks (Squamata, Scincidae). Zoologica Scripta. 2010, 40: 16-44.
Vasconcelos R, Carranza S, Harris DJ: Insight into an island radiation: the Tarentola geckos of the Cape Verde archipelago. J Biogeogr. 2010, 37 (6): 1047-1060.
Galbreath KE, Hafner DJ, Zamudio KR: When cold is better: Climate-driven elevation shifts yield complex patterns of diversification and demography in an Alpine Specialist (American Pika, Ochotona Princeps). Evolution. 2009, 63 (11): 2848-2863.
Hewitt GM: Genetic consequences of climatic oscilations in the quaternary. Philos Trans R Soc Lond. 2004, 359: 183-195.
Knowles LL: Did the Pleistocene glaciations promote divergence? Tests of explicit refugial models in montane grasshopprers. Mol Ecol. 2001, 10 (3): 691-701.
Hughes L: Climate change and Australia: Trends, projections and impacts. Austral Ecol. 2003, 28 (4): 423-443.
Mouret V, Guillaumet A, Cheylan M, Pottier G, Ferchaud AL, Crochet PA: The legacy of ice ages in mountain species: post-glacial colonization of mountain tops rather than current range fragmentation determines mitochondrial genetic diversity in an endemic Pyrenean rock lizard. J Biogeogr. 2011, 38 (9): 1717-1731.
Bons J, Geniez P: Amphibiens et reptiles du Maroc (Sahara Occidental compris) Atlas Biogéographique. 1996, Barcelone: Asociación Herpetologica Espanola
Schleich HH, Kastle W, Kabisch K: Amphibians and Reptiles from North Africa. 1996, Königstein, Germany: Koeltz Scientific Publications
Arnold EN: Relationships of the Palaearctic lizards assigned to the genera Lacerta, Algyroides and Psammodromus (Reptila, Lacertidae). 1973, London: British Museum (Natural History)
Arnold EN: Towards a phylogeny and biogeography of the Lacertidae: relationships within an Old-World family of lizards derived from morphology. 1989, London: British Museum (Natural History)
Harris DJ: Molecular systematics and evolution of lacertid lizards. Natura Croatica. 1999, 83 (3): 161-180.
Mayer W, Bischoff W: Beiträge zur taxonomischen Revision der Gattung Lacerta (Reptilia: Lacertidae) Teil 1: Zootoca, Omanosaura, TimonundTeira als eigenstandige Gattungen. Salamandra. 1996, 32 (3): 163-170.
Oliverio M, Bologna MA, Mariottin P: Molecular biogeography of the Mediterranean lizards Podarcis Wagler, 1830 and Teira Gray, 1838 (Reptilia, Lacertidae). J Biogeogr. 2000, 27: 1403-1420.
Arnold EN, Arribas O, Carranza S: Systematics of the Palaearctic and Oriental lizard tribe Lacertini (Squamata: Lacertidae: Lacertinae), with descriptions of eight new genera. Zootaxa. 2007, 1430: 1-86.
Pavlicev M, Mayer W: Fast radiation of the subfamily Lacertinae (Reptilia: Lacertidae): History or methodical artefact?. Molecular Phylogenetics and Evolution. 2009, 52 (3): 727-734.
Busack SD: Notes on the biology of Lacerta andreanszkyi (Reptilia: Lacertidae). Amphibia-Reptilia. 1987, 8: 231-236.
Carretero MA, Perera A, Harris DJ, Batista V, Pinho C: Spring diet and trophic partitioning in an alpine lizard community from Morocco. African Zoology. 2006, 41 (1): 113-122.
Crochet PA, Chaline O, Surget-Groba Y, Debain C, Cheylan M: Speciation in mountains: phylogeography and phylogeny of the rock lizards genus Iberolacerta (Reptilia: Lacertidae). Molecular Phylogenetics and Evolution. 2004, 30 (3): 860-866.
Carranza S, Arnold EN, Amat F: DNA phylogeny of Lacerta (Iberolacerta) and other lacertine lizards (Reptilia: Lacertidae): did competition cause long-term mountain restriction?. Syst Biodivers. 2004, 2 (01): 57-77.
Funk DJ, Omland KE: Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu Rev Ecol Evol S. 2003, 34: 397-423.
Knowles LL, Carstens BC: Delimiting species without monophyletic gene trees. Syst Biol. 2007, 56 (6): 887-895.
Hudson RR, Coyne JA: Mathematical consequences of the genealogical species concept. Evolution. 2002, 56 (8): 1557-1565.
Hudson RR, Turelli M: Stochasticity overrules the "three-times rule": Genetic drift, genetic draft, and coalescence times for nuclear loci versus mitochondrial DNA. Evolution. 2003, 57 (1): 182-190.
Pinho C, Harris DJ, Ferrand N: Non-equilibrium estimates of gene flow inferred from nuclear genealogies suggest that Iberian and North African wall lizards (Podarcis spp.) are an assemblage of incipient species. BMC Evol Biol. 2008, 8: 63-(http://www.biomedcentral.com/1471-2148/8/63)
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959.
Evanno G, Regnaut S, Goudet J: Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study Molecular ecology. 2005, 14: 2611-2620.
Banks MA, Eichert W: WHICHRUN (version 3.2): A computer program for population assignment of individuals based on multilocus genotype data. J Hered. 2000, 91 (1): 87-89.
Corander J, Waldmann P, Sillanpaa MJ: Bayesian analysis of genetic differentiation between populations. Genetics. 2003, 163 (1): 367-374.
Dawson KJ, Belkhir K: A Bayesian approach to the identification of panmictic populations and the assignment of individuals. Genet Res. 2001, 78 (1): 59-77.
Manel S, Berthier P, Luikart G: Detecting wildlife poaching: Identifying the origin of individuals with Bayesian assignment tests and multilocus genotypes. Conserv Biol. 2002, 16 (3): 650-659.
Pritchard JK, Donnelly P: Case–control studies of association in structured or admixed populations. Theor Popul Biol. 2001, 60 (3): 227-237.
Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW: Genetic structure of human populations. Science. 2002, 298 (5602): 2381-2385.
Turakulov R, Easteal S: Number of SNPS loci needed to detect population structure. Hum Hered. 2003, 55 (1): 37-45.
Babault J, Teixell A, Arboleya ML, Charroud M: A late cenozoic age for long-wavelength surface uplift of the atlas mountains of Morocco. Terra Nova. 2008, 20 (2): 102-107.
Gomez F, Beauchamp W, Barazangi M: Role of the Atlas Mountains (northwest Africa) within the African-Eurasian plate-boundary zone. Geology. 2000, 28 (9): 775-778.
Brown JW, Rest JS, Garcia-Moreno J, Sorenson MD, Mindell DP: Strong mitochondrial DNA support for a Cretaceous origin of modern avian lineages. BMC Biol. 2008, 6: 6-(http://www.biomedcentral.com/1741-7007/6/6/)
Sousa P, Froufe E, Harris DJ, Alves PC, van der Meijden A: Genetic diversity of Maghrebian Hottentotta (Scorpiones: Buthidae) scorpions based on CO1: new insights on the genus phylogeny and distribution. Afr Invertebr. 2011, 52 (1): 135-143.
Masembe C, Muwanika VB, Nyakaana S, Arctander P, Siegismund HR: Three genetically divergent lineages of the Oryx in eastern Africa: Evidence for an ancient introgressive hybridization. Conserv Genet. 2006, 7 (4): 551-562.
Perera A, Vasconcelos R, Harris DJ, Brown RP, Carretero MA, Perez-Mellado V: Complex patterns of morphological and mtDNA variation in Lacerta perspicillata (Reptilia; Lacertidae). Biol J Linn Soc. 2007, 90 (3): 479-490.
de Pous P, Beukema W, Weterings M, Dummer I, Geniez P: Area prioritization and performance evaluation of the conservation area network for the Moroccan herpetofauna: a preliminary assessment. Biodivers Conserv. 2011, 20 (1): 89-118.
Barata M, Perera A, Harris DJ, Van Der Meijden A, Carranza S, Ceacero F, García-Muñoz E, Gonçalves D, Henriques S, Jorge F, et al: New observations of amphibians and reptiles in Morocco, with a special emphasis on the eastern region. Herpetological Bulletin. 2011, 116: 4-14.
Pounds JA, Fogden MPL, Campbell JH: Biological responses to climate change on a tropical mountain. Nature. 1999, 398: 611-615.
Vasconcelos R, Perera A, Geniez P, Harris DJ, Carranza S: An integrative taxonomic revision of the Tarentola geckos (Squamata, Phyllodactylidae) of the Cape Verde Islands. Zool J Linn Soc-Lond. 2012, 164: 328-360.
Carranza S, Arnold EN: A review of the geckos of the genus Hemidactylus (Squamata: Gekkonidae) from Oman based on morphology, mitochondrial and nuclear data, with descriptions of eight new species. Zootaxa. 2012, 3378: 1-95.
Sambrook J, Fritsch EF, Maniatis T: Molecular cloning: a laboratory manual, 3nd edt edn. 1989, New York: Cold Sring Harbor Laboratory Press
Kocher TD, Thomas WK, Meyer A, Edwards SV, Pääbo S, Villablanca FX, Wilson AC: Dynamics of mitochondrial-DNA evolution in animals - Amplification and sequencing with conserved primers. Proc Natl Acad Sci U S A. 1989, 86 (16): 6196-6200.
Harris DJ, Arnold EN: Relationships of wall lizards, Podarcis (Reptilia: Lacertidae) based on mitochondrial DNA sequences. Copeia. 1999, 3: 749-754.
Arévalo E, Davis SK, Sites JW: Mitochondrial-DNA Sequence Divergence and Phylogenetic-Relationships among 8 Chromosome Races of the Sceloporus-Grammicus Complex (Phrynosomatidae) in Central Mexico. Syst Biol. 1994, 43 (3): 387-418.
Vidal N, Hedges SB: Molecular evidence for a terrestrial origin of snakes. P R Soc B. 2004, 271: S226-S229.
Gamble T, Bauer AM, Greenbaum E, Jackman TR: Evidence for Gondwanan vicariance in an ancient clade of gecko lizards. J Biogeogr. 2008, 35 (1): 88-104.
Gamble T, Bauer AM, Greenbaum W, Jackman TR: Out of the blue: a novel, trans-Atlantic clade of geckos (Gekkota, Squamata). Zoologica Scripta. 2008, 37 (4): 355-366.
Pinho C, Rocha S, Carvalho BM, Lopes S, Mourao S, Vallinoto M, Brunes TO, Haddad CFB, Goncalves H, Sequeira F, et al: New primers for the amplification and sequencing of nuclear loci in a taxonomically wide set of reptiles and amphibians. Conserv Genet Resour. 2010, 2: 181-185.
Godinho R, Crespo EG, Ferrand N, Harris DJ: Phylogeny and evolution of the green lizards, Lacerta spp. (Squamata: Lacertidae) based on mitochondrial and nuclear DNA sequences. Amphibia-Reptilia. 2005, 26 (3): 271-285.
Bauer AM, de Silva A, Greenbaum E, Jackman T: A new species of day gecko from high elevation in Sri Lanka, with a preliminary phylogeny of Sri Lankan Cnemaspis (Reptilia, Squamata, Gekkonidae). Zoosystematics and Evolution. 2007, 83 (S1): 22-32.
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series. 1999, 41: 95-98.
Katoh K, Misawa K, Kuma K, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30 (14): 3059-3066.
Flot J-F: SeqPHASE: a web tool for interconverting PHASE input/output files and FASTA sequence alignments. Mol Ecol Resour. 2010, 372: 372-(http://www.biomedcentral.com/1471-2148/10/372/)
Stephens M, Donnelly P: A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003, 73 (5): 1162-1169.
Harrigan RJ, Mazza ME, Sorenson MD: Computation vs. cloning: evaluation of two methods for haplotype determination. Mol Ecol Resour. 2008, 8 (6): 1239-1248.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19 (18): 2496-2497.
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004, 5 (2): 150-163.
Posada D: jModelTest: Phylogenetic model averaging. Mol Biol Evol. 2008, 25 (7): 1253-1256.
Akaike H: A new look at the statistical model identification. IEEE Trans Autom Control. 1974, 19 (6): 716-723.
Stamatakis A: RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006, 22 (21): 2688-2690.
Felsenstein J: Confidence-Limits on Phylogenies - an Approach Using the Bootstrap. Evolution. 1985, 39 (4): 783-791.
Huelsenbeck JP, Ronquist F: MrBayes: Bayesian inference of phylogeny. Bioinformatics. 2001, 17: 754-755.
Templeton AR, Crandall KA, Sing CF: A Cladistic-Analysis of Phenotypic Associations with Haplotypes Inferred from Restriction Endonuclease Mapping and DNA-Sequence Data.3. Cladogram Estimation. Genetics. 1992, 132 (2): 619-633.
Clement M, Posada D, Crandall KA: TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000, 9: 1657-1659.
Falush D, Stephens M, Pritchard JK: Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003, 164: 1567-1587.
Documentation for STRUCTURE software. http://pritch.bsd.uchicago.edu, 2,
Heled J, Drummond AJ: Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution. 2010, 27 (3): 570-580.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7 (1): 214-
Rambaut A, Drummond AJ: Tracer v1.4. 2007, United Kingdom: University of Edinburgh
Rambaut A: FigTree v1.3.1. 2008, University of Edinburgh, UK: Institute of Evolutionary Biology
Poulakakis N, Lymberakis P, Valakos E, Pafilis P, Zouros E, Mylonas M: Phylogeography of Balkan wall lizard (Podarcis taurica) and its relatives inferred from mitochondrial DNA sequences. Mol Ecol. 2005, 14: 2433-2443.
Heads M: Dating nodes on molecular phylogenies: a critique of molecular biogeography. Cladistics. 2005, 21 (1): 62-78.
Benton MJ, Ayala FJ: Dating the Tree of Life. Science. 2003, 300 (5626): 1698-1700.
Ho SYW, Phillips MJ: Accounting for calibration uncertainty in phylogenetic estimation of evolutionary divergence times. Syst Biol. 2009, 58 (3): 367-380.
Acknowledgements
MB is supported by the FCT grant SFRH/BD/41488/2007. This work was funded by FCT grant PTDC/BIA-BDE/74349/2006 and by grant CGL2009-11663 from the Ministerio de Educación y Ciencia, Spain to SC. Fieldwork in Morocco in 2008 and 2009 was conducted under permit decision 84° issued by Haut Commissariat aux Eaux et Forêts et à la Lutte Contre la Désertification, issued to David Donaire plus other permits issued to the latter along a 10 year period.
Thanks to all colleagues from CIBIO who assisted during the hard fieldwork, especially to Anna Perera, Daniele Salvi, Fatima Jorge and Fernando Martinez-Freiria. We also want to thank to the anonymous reviewers that helped to improve this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors’ contributions
MB carried out the molecular laboratory work, analyzed the data and drafted a preliminary version of the manuscript. All authors participated in the conception and design of the study, collection of samples, writing and approval of the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Barata, M., Carranza, S. & Harris, D.J. Extreme genetic diversity in the lizard Atlantolacerta andreanskyi(Werner, 1929): A montane cryptic species complex. BMC Evol Biol 12, 167 (2012). https://doi.org/10.1186/1471-2148-12-167
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-12-167