
Abstract
Background
Four of the five species of Telopea (Proteaceae) are distributed in a latitudinal replacement pattern on the south-eastern Australian mainland. In similar circumstances, a simple allopatric speciation model that identifies the origins of genetic isolation within temporal geographic separation is considered as the default model. However, secondary contact between differentiated lineages can result in similar distributional patterns to those arising from a process of parapatric speciation (where gene flow between lineages remains uninterrupted during differentiation). Our aim was to use the characteristic distributional patterns in Telopea to test whether it reflected the evolutionary models of allopatric or parapatric speciation. Using a combination of genetic evidence and environmental niche modelling, we focused on three main questions: do currently described geographic borders coincide with genetic and environmental boundaries; are there hybrid zones in areas of secondary contact between closely related species; did species distributions contract during the last glacial maximum resulting in distributional gaps even where overlap and hybridisation currently occur?
Results
Total genomic DNA was extracted from 619 individuals sampled from 36 populations representing the four species. Seven nuclear microsatellites (nSSR) and six chloroplast microsatellites (cpSSR) were amplified across all populations. Genetic structure and the signature of admixture in overlap zones was described using the Bayesian clustering methods implemented in STUCTURE and NewHybrids respectively. Relationships between chlorotypes were reconstructed as a median-joining network. Environmental niche models were produced for all species using environmental parameters from both the present day and the last glacial maximum (LGM).
The nSSR loci amplified a total of 154 alleles, while data for the cpSSR loci produced a network of six chlorotypes. STRUCTURE revealed an optimum number of five clusters corresponding to the four recognised species with the additional division of T. speciosissima into populations north and south of the Shoalhaven River valley. Unexpectedly, the northern disjunct population of T. oreades grouped with T. mongaensis and was identified as a hybrid swarm by the Bayesian assignment test implemented in NewHybrids. Present day and LGM environmental niche models differed dramatically, suggesting that distributions of all species had repeatedly expanded and contracted in response to Pleistocene climatic oscillations and confirming strongly marked historical distributional gaps among taxes.
Conclusions
Genetic structure and bio-climatic modeling results are more consistent with a history of allopatric speciation followed by repeated episodes of secondary contact and localised hybridisation, rather than with parapatric speciation. This study on Telopea shows that the evidence for temporal exclusion of gene flow can be found even outside obvious geographical contexts, and that it is possible to make significant progress towards excluding parapatric speciation as a contributing evolutionary process.
Similar content being viewed by others


Background
Models of speciation are generally categorised on the role and nature of geographic isolation during the origination of reproductive barriers between differentiating populations. Usually, allopatric speciation is considered as the default model since it specifies a simpler, more biologically plausible process than other models [1, 2]. In an allopatric model of speciation, genetic isolation resulting from temporal geographic separation can lead to the evolution of reproductive barriers and speciation. Consequently, biogeographic patterns are of particular interest when attempting to test allopatric models [3]. In contrast, the parapatric and sympatric speciation models involve the differentiation of lineages despite the possibility for ongoing exchange of genetic material. In these models, selective barriers must have a strong impact in order to preserve between-lineage divergence [4, 5].
The spatial arrangement of genetically divergent populations is important in allopatric speciation, as it involves the geographic separation of a previously continuous metapopulation by geological or climatic causes, or a founder event resulting from long distance dispersal [6]. Lack of gene flow, drift and secondary adaptation to local environmental conditions among geographically separated populations can eventually result in morphologically and genetically distinct lineages. However, current isolating mechanisms are not necessarily the same as those that initiated speciation and, as a result, secondary contact and hybridisation can ensue [3]. Although the presence of hybrid fronts between closely related lineages can confirm allopatric speciation with secondary contact, such fronts are difficult to differentiate from zones of primary parapatric speciation. In fact, any mechanism that can cause divergence in allopatry can theoretically also act in parapatry, as long as the selective gradient acting on differentiation is sufficiently strong to counterbalance continuing gene flow [1].
Here, we aim to identify the main speciation mechanism involved in the diversification of the plant genus Telopea (the waratahs; Proteaceae). Telopea is a genus comprising five species of long-lived, lignotuberous, sclerophyllous, bird-pollinated, wind-dispersed shrubs and small trees distributed in mesic environments of south eastern Australia, including Tasmania. They live in eucalypt-dominated forests and woodlands, growing on acidic, nutrient-poor, well drained soils derived from sandstone, granite, chert or quartzite from sea level in the Sydney region to 1200 m altitude in subalpine habitats in Tasmania. Weston & Crisp [7] reconstructed Telopea phylogeny in a cladistic analysis of morphological characters and the species limits of the four mainland Australian species were tested morphometrically by Crisp & Weston [8].
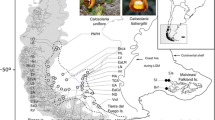
Phylogenetically, the four recognised mainland taxa are grouped into two morphologically distinct clades comprising T. speciosissima and T. aspera to the north, and T. oreades and T. mongaensis to the south. Their distribution follows a latitudinal pattern ranging from southern Victoria to northern New South Wales (Figure 1). The species are mostly separated by large distributional gaps. The exceptions are a site at Monga National Park (NP), where a geographically disjunct population of T. oreades is found in sympatry with T. mongaensis, and an area also south of the Sydney Basin where a potential distributional overlap between the latter species and T. speciosissima occurs (recent fieldwork has located a single morphologically intermediate individual).

Distribution map for the four Telopea species investigated in this study. Map showing all verified distributional records (used as training data for the Maxent models) for the four study species (purple T. aspera; green T. speciosissima; red T. mongaensis; blue T. oreades). The major geographic points of interest discussed in the text are presented.
Although terminally-winged, the seeds of Telopea are not easily dispersed and tend to germinate in proximity to the maternal parent [9]. As a result, the latitudinal distribution of the genus suggests that interruptions of connectivity between once continuously distributed lineages could have contributed to allopatric speciation with the development of morphological and adaptive differentiation likely to have been secondary events. In a previous landscape genetic study on T. speciosissima, we suggested that substrate-mediated allopatry and temperature-dependent reproductive barriers were likely to affect temporal changes in connectivity among populations [10]. While genetic differentiation in the south corresponded to a distributional gap caused by changes in edaphic conditions, altitudinal constraints in gene flow were the consequence of a strong association between temperature and flowering time.
In south-eastern Australia, the glacial cycles of the Quaternary shaped the distribution of plants and plant communities by increasing aridity, fire frequency and fire intensity [11]. The limited fossil record suggests that dynamic successional processes affected local vegetation types, and that localised moist refugia along the Great Dividing Range provided some protection against localised extinctions [12, 13]. Plant distributional changes during the Quaternary have been particularly well documented in northern latitudes [14, 15] and more recently in eastern Australia [16]. Similar temporal changes can also be expected for Telopea. The extent to which a species’ distribution can respond to changing climatic conditions is affected by niche breadth, whilst the ability of a species to establish in new climatically suitable areas is dependant upon numerous ecological factors, including dispersal ability and competitive advantage. The use of environmental niche modeling (ENM) to investigate historical environmental suitability in combination with molecular data can enhance our understanding of temporal changes in population dynamics [17]. Here we integrate genetic and bio-climatic methods to investigate speciation across all mainland species of Telopea and test the alternative scenarios of parapatric or allopatric speciation. In particular we focus on three main questions:
-
1.
Do currently described geographic borders coincide with genetic and environmental boundaries? Distributional and morphological data mostly support this [8]. Here we aim to validate between-species distinctiveness by combining present-day genetic (nSSRs, cpSSRs) and bio-climatic (ENM) analyses.
-
2.
Are there hybrid zones in areas of secondary contact between closely related species? Here we use targeted population genetic data in overlap zones to investigate hybridisation between species. A previous study on T. speciosissima suggested that admixture between previously differentiated populations can take place as a consequence of the post-LGM re-establishment of gene flow caused by temperature-mediated phenological shifts along the altitudinal gradients [10].
-
3.
Did species distribution contract during the last glacial maximum, resulting in distributional gaps even where overlap and hybridisation currently occur? We use ENM to predict historical ranges for the four species and gather evidence for distributional discontinuities. The existence of such patterns would provide support for secondary rather than primary contact zones and consequently favour a scenario of allopatric over parapatric speciation [3].
Methods
Study species
Five species of Telopea are distributed from the Gibraltar Range in south-eastern mainland Australia (29° 28’ S) to southern Tasmania (43° 33’ S). Of the four mainland species, T. aspera in the north is the only one completely separated from its nearest congeners by about 400 km, while the other three have potential contact points at the extremes of their distributions (Figure 1). All species have naturally fragmented distributions, of which the most intriguing is that of T. oreades, the most northern population of which overlaps with the most southerly population of T. mongaensis but is located over 160 km from the next most northerly population of T. oreades[8]. Telopea mongaensis also shows a significant internal gap of over 45 km imposed by the Shoalhaven River valley, with its most north-easterly populations occurring within 20 km of the nearest populations of T. speciosissima. Telopea speciosissima is the most widespread species with populations south of the Shoalhaven River valley being isolated from those on the northern side by a over 30 km [10].
According to the morphology-based cladistic analysis of Proteaceae subtribe Embothriinae published by Weston & Crisp [7], a clade of T. speciosissima and T. aspera is sister to a clade including T. truncata (Tasmania), T. oreades and T. mongaensis. The speciosissima-aspera clade is characterised by three macromorphological synapomorphies: presence of marginal leaf teeth (versus entire or lobed margins), and enlarged, bright red involucral bracts (versus small and green, to dusky pink involucral bracts). The truncata-oreades-mongaensis clade is characterised by basitonic inflorescences (flowers open from the tip of the inflorescence towards the base, in contrast to the acrotonic inflorescences of the speciosissima-aspera clade and outgroups, in which flowers open from the base towards the tip). Telopea oreades and T. mongaensis form a clade characterised by markedly bicolorous perianths in which the adaxial surfaces are a much brighter shade of red than the abaxial surfaces, in contrast to the concolorous perianths in other Telopea species.
Sampling, DNA extractions and PCRs
Sampling was aimed at obtaining an account of the genetic diversity across the entire distribution of mainland species (Table 1). Leaf material was collected from a total of 619 individuals, and total genomic DNA was extracted using DNeasy® 96 plant kits (QIAGEN®, Hilden, Germany).
Seven nuclear microsatellites (nSSR) specifically developed for T. speciosissima (TS03bgt, TS04bgt, TS12bgt, TS13bgt, TS18bgt, TS23bgt and TS27bgt) were amplified across all individuals using the PCR protocols of Porter et al. [18]. Genotyping results were checked with Microchecker [19] for evidence of scoring errors due to stuttering, large allele dropout and null alleles with ranked-based and binomial-based estimates showing no evidence of scoring error.
Chloroplast microsatellites (cpSSR) were amplified from three individuals per population, using ccmp10[20], psbA [21], trnG (UCC)ex1 – atpA [22], Lomcp1 Lomcp2 and Lomcp3 (M Milner pers. com.). Reaction conditions were as described in Ebert & Peakall [22] using Schuelke’s [23] method of fluorescently-labelled M13 forward primers. Amplification products were visualized using a 3130xl Genetic Analyzer (Applied Biosystems) and scored for size using GeneMapper 3.7 (Applied Biosystems).
To test genotyping accuracy approximately 20% of all PCR reactions were repeated for each primer pair across random individuals. Less than 5% of those PCR repeats identified errors that needed confirmation through replication of PCR and genotyping. Finally, to verify that the amplified loci indeed contained the expected nSSR repeats across all Telopea species and to verify the accuracy of allelic sizes, representatives of species/locus combinations were sequenced [GenBank accession no. JF931649-JF931672]. Unique cpSSR allele sizes were also sequenced across species to ascertain that the variation was due to mononucleotide repeat regions [GenBank accession no. JQ778988-JQ779019].
Defining genetic boundaries
Species-level averages of nSSR allelic diversity (A), expected (He) and observed (Ho) heterozygosities as well as number of private alleles (Ap) were calculated using GDA 1.1 [24]. In order to avoid bias caused by uneven sampling [25], a standardized estimate of allelic richness (R63) independent of sample size [26] was calculated using the program FSTAT 2.9.3 [27]. Pariwise Fst[28] values were also obtained through FSTAT and used to provide within and between species averages.
We used the Bayesian clustering method described by Pritchard et al. [29] and implemented in STUCTURE 2.3.3 on the nSSR data to identify genetically differentiated groups of individuals in the absence of preliminary information on group boundaries. The model assumes the existence of K clusters (the real number being unknown) and uses the allelic frequencies at each locus to assign individuals to these clusters through a Markov chain Monte Carlo (MCMC) probabilistic approach in which individuals are assigned to clusters so as to maximize Hardy-Weinberg equilibrium within populations. All analyses were based on 10 independent runs for each K value, with each individual run being based on 2.5x105 MCMC iterations following a burn-in period of 5x104 iterations without prior information on the taxonomy or the locality of origin of the individuals sampled. The admixture frequency model was run under the assumption of correlated allele frequencies to improve clustering of related lineages and identify possible hybridisation patterns [30]. The optimal number of clusters was verified using the ΔK statistical approach suggested by Evanno et al. [31].
After a preliminary test aimed at finding a suitable range for K and the optimal burn-in period, we tested K from 1 to 20 for the full dataset including the four species under study. We also tested K from 1 to 12 for the dataset including only T. oreades and T. mongaensis to specifically investigate assignment patterns for the only sympatric individuals located at the Monga NP site.
Analysis of molecular variance (AMOVA; [32]) was then used to quantify variance components and the significance of the genetic subdivisions identified by the Bayesian tests, as well as a range of other relevant groupings. Permutation tests for two-level AMOVAs were implemented to test whether levels of differentiation were significantly greater than zero. AMOVA results were obtained using GenAlEx6.3 [33].
Assuming that cpDNA does not recombine and can be treated as a single locus, we combined the sizes of all six cpSSR fragments to give the one chloroplast haplotype (chlorotype) per individual. A median-joining network [34] was constructed using NETWORK v4.5.1.6 [35], a program that constructs networks based on size differences among haplotypes.
Detecting hybridisation
Since STUCTURE (and the other analytical approaches used) is not explicitly designed to assign individuals to admixed classes, we used the Bayesian model implemented in NewHybrids [36] to detect admixed individuals resulting from the interbreeding between distinct species. The original tests and simulations by Anderson & Thompson [36] show that admixture can be detected without the need for diagnostic alleles. Although a high number of informative loci produces much better posterior probabilities (PP) for assigning to hybrid categories, when Fst > 0.2 (as is the case in this study; Table 2) a smaller number of loci is sufficient for detecting hybrids [37].
Our objective was not to identify and quantify specific admixture categories but rather to ascertain whether differentiated genomes, particularly those of T. oreades and T. mongaensis, have been mixed where the species overlap. To this end, we conducted a series of preliminary analyses that culminated in a run using all 11 populations from the two species. Jeffreys priors were used with a burn-in of 1x105 sweeps followed by 3.5x105 sweeps. Posterior probability of assignment as pure, F1s, F2s, and backcrosses were initially measured, and proportions of admixture vs. pure were then calculated from these results. An individual was assigned as a pure or hybrid individual when its posterior probability of belonging to that genotypic class exceeded PP = 90% [38, 39].
Factorial correspondence analysis (FCA) of diploid genetic data [40] was carried out using GENETIX 4.05 [41] to produce comparative graphical representations of genotypic relationships between all T. oreades and T. mongaensis individuals sampled.
NewHybrids was also used to analyse the T. speciosissima and T. mongaensis populations that were geographically closest to the only known morphological intermediate between these two species. This was to serve as a test for the analytical approach and to investigate the possible presence of introgression.
Current and historical environmental niche modelling
We used the machine-learning automated statistical algorithm Maxent 3.3.3 (Maximum entropy modeling application; [42]) to identify the multivariate correlations between the 19 available ~920 m x 770 m pixel (30 arc-second) WorldClim [43] climate axes (environmental data) and climate conditions present at geographic localities matching the known native distributions of each of the Telopea species. We employed this environmental niche modeling (ENM) technique to determine whether present day habitat characteristics correspond to genetic divergence between taxa. Maxent was chosen as it has been shown to outperform other modeling methods when generating predictions of species’ ranges [44].
Occurrence records were compiled from all Australian Herbaria (voucher - high taxonomic integrity) and taxonomically corrected records from the (non-voucher - high geographic integrity) databases: YETI plots [45]; and ATLAS incidental observations [46]. We ran multiple model sets for Telopea as a whole and for each Telopea species to determine consistent geographic extents between models and consistently dominant climatic factors [47]. Model run 1) only included locality records with accuracies better than 100 m (Table 3). These high precision sites were also of high accuracy (giving ±0.1 climatic pixel accuracy) and those sites that fell within 100 m of the climatic pixel boundary were excluded. This gave a single climatic pixel that best represents the sites sampled. This model represents a minimum estimate of the realised climatic space (climatic conditions in which the species is specifically recorded) of each species but could result in over-fitting of the models [47]. Model 2) included Herbaria records of geographic accuracies better than 1 km, (±1 climatic pixel accuracy or a potential of 9 climatic pixel values per site); and model 3) the above plus non-voucher records better than 1 km accuracy (Table 3). Whilst the climatic data submitted to the model is less precise, substantially more points are submitted providing more robust correlations. Model runs 2) and particularly 3) provide better estimates of the fundamental climatic space (the climatic conditions under which the species is capable of reproducing and persisting) and minimise the effect of false omissions from the training localities which would usually result in models underestimating suitable climatic ranges [47]. The three sets of model runs were compared for between-model consistencies. The larger numbers of localities submitted to the training data better capture the realised climate space and avoid having (climatic) variable numbers approaching the number of sample locations which leads to over-fitting of the models [47].
Model outputs were projected onto to the eastern states of Australia using the same 19 WorldClim variables for 0 ka (pre-industrial) and 21 ka (LGM) time frames. Models were run in the default settings but with 10 replicates under the cross-validation option, and with response curves and jackknife settings enabled. However, this resulted in over-fitted models (as shown geographically by over fragmentation and underestimation of suitable climatic habitat within expected available soil/plant community types) so the models were rerun with hinge features disabled.
The models were run initially with all 19 variables to determine the relative contributions of each environmental variable to the Maxent model. Then, to reduce the effects of overfitting [47], only variables that were not auto-correlated and contributed more than 5% to the Maxent model were included in a subsequent model run.
Telopea species are highly constrained by soil type, and non-climatic abiotic factors like this can constrain realised distributions to portions of the fundamental climatic space. If obviously dominant factors of this nature are excluded from the assessment of suitable geographic extents to submit to the training of the models, then this has the effect of submitting more false omissions to the model, producing an underestimation of suitable habitat [47]. To better determine the available realised habitat and estimate the (true) pseudo-absences within the fundamental space, the geographic extents of the training data were clipped to suitable soil types. To make soil type allocation relevant to current and historic time frames, surface geological units were used rather than soil type and were buffered to 1 km to capture likely surface soil types. Soil types for each species were assessed by overlaying occurrence records with soils mapping and resulted in the identification of mostly sedimentary siliciclastic, quartz-rich arenite to rudite or igneous felsic intrusive lithologies.
In order to maintain the integrity of the pseudo-absence reference that is integral to the software and to reduce the effects of over fitting [47], model runs were repeated for the 19 training environmental variables from 0 ka (pre-industrial time) and the geographic extents of training data were constrained to within 200 km of: 1) the genus point locations; 2) the species point locations; 3) the species point locations and to suitable soil type. These three sets of model runs represent increasing removal of false omissions from the training data. Each of these model runs was projected onto the following two geographic extents: a) the eastern Australian states; and b) the eastern Australian states constrained by available suitable soil type. The latter output represented a reduction in the false commissions in the projection.
To determine the extent of overlap between species pairs, the outputs of the last model run (3b) were intersected for each species pair in ArcGIS 9.2 to produce a map layer of the probability of the two species to overlap (Table 4).
Results
Defining genetic boundaries among lineages
The seven SSR loci amplified a total of 154 alleles across the 36 populations representing four recognised Telopea species. Species-level allelic richness ranged from R63 = 6.3 to 17.2, and heterozygosity measures from He = 0.492 to 0.794 and from Ho = 0.380 to 0.686 (Table 2). Telopea speciosissima was consistently the most, and T. mongaensis consistently the least diverse species. When analysed on their own, the three most southerly populations of T. speciosissima still contained high levels of diversity, while shifting the putative T. oreades population at Monga NP to T. mongaensis made little difference to their respective diversity measures (Table 2).
Average pairwise Fst values suggest strongest genetic differentiation within T. oreades (particularly when the Monga NP population is included) and weakest within T. aspera. Telopea mongaensis was the most genetically differentiated from all other taxa (Table 2).
Using the seven nSSR loci, STRUCTURE’s Bayesian clustering produced the most substantial increases in LnP(D) values at K = 5, with the ΔK statistic preferentially supporting K = 5 (ΔK5 = 358.7) as the most robust division. Here we visualise data from the next most robust division (ΔK6 = 198.5) as it better represents previously-described sub-structure within T. speciosissima[10]. Assignment values of Q ≥ 95% were usually obtained at the species level (Table 2). However, two major taxonomic inconsistencies emerged: a) the assignment of the T. oreades population at Monga NP to T. mongaensis; b) the separation of the southern populations of T. speciosissima from the rest of the species.
AMOVA supported significant segregation among Telopea species. High levels of genetic variance were partitioned to between-species differentiation (42.7% of variation; P < 0.001), although similar levels of genetic differentiation were partitioned to within-species (43.6% of variation; P < 0.001). A total of six distinct chlorotypes were obtained across the four mainland species and none of the sampled populations amplified more than one chlorotype each (Table 2). A single, shared chlorotype (chlorotype 4) was amplified for all T. oreades (Figure 2g), all but one population of T. mongaensis (Figure 2e) as well as the Brogers Creek population of T. speciosissima (Figure 2c). Multiple chlorotypes were obtained for T. speciosissima, with chlorotype 6 being found only among the southern populations as well as the Gunrock Creek population of T. mongaensis (Figure 2e).

Modelled bio-climatic distribution of four Telopea species (LGM and current) and measured population-level diversity (chloroplast and nuclear). Maxent modeled bio-climatic distribution for each Telopea species for the LGM (21 ka) and current (pre-industrial) time frames (showing only model run 3b being all records projected only onto soils suitable for Telopea), showing population-level nSSR and cpSSR (colour-coded haplotypes) data for each of the sampled populations. The nSSR pie charts partition mean individual-level assignment value to a species (based on the Bayesian clustering approach implemented in STRUCTURE, at K = 6). The chloroplast network is shown in (a), and the model probability (likelihood occurrence of suitable habitat) is in a gray scale from 0 (absent) to1 (certain presence) in (b). a) T. aspera, LGM habitat and cpSSR; b) T. aspera, current habitat and nSSR; c) T. speciosissima, LGM habitat and cpSSR; d) T. speciosissima, current habitat and nSSR; e) T. mongaensis, LGM habitat and cpSSR; f) T. mongaensis, current habitat and nSSR; g) T. oreades, LGM habitat and cpSSR; h) T. oreades, current habitat and nSSR.
Detecting hybridisation
Further STRUCTURE analyses were conducted to refine and clarify the interpretations of the assignment among the two southern taxa. Bayesian clustering across 199 individuals from the two southern Telopea species, produced substantial increases in LnP(D) values at K = 2 and K = 3, with the ΔK statistic preferentially supporting K = 3 (ΔK2 = 417.4; ΔK3 = 1009.7). This result identified the southern-most population at Monga NP (including both putative T. oreades and T. mongaensis individuals) as differentiated from the two species. K = 2 differentiated between the two species but assigned the T. oreades population at Monga NP to T. mongaensis. The FCA plot provided further insight in the K = 3 result by placing the individuals at the Monga NP site (morphologically identified as either of the parental species or hybrids) as a third intermediate group suggesting either a differentiated lineage or a hybrid population (Figure 3a).

Testing hybridisation patterns at a distributional overlap zone ( T. oreades and T. mongaensis at Monga NP). a) Factorial analysis comparing all individuals from all tested populations from T. oreades (white squares) and T. mongaensis (black triangles). The individuals from the Monga NP site morphologically assigned to either of the two species are represented in grey. b) NewHybrids results comparing the individuals (single bars) from the Monga NP site to the geographically closest populations of T. mongaensis (Dasyurus PA, River Forest Road) and T. oreades (Waratah Creek). T.m. DPA: T. mongaensis – Dasyurus PA; T.m. RFrd: T. mongaensis – River Forest Road; T.m. MNP: T. mongaensis – Monga NP (sympatric); T.o. MNP: T. oreades – Monga NP (sympatric); T.o. WC: T. oreades – Waratah Creek. Black represents% assignment to T. mongaensis genotype, white represents% assignment to T. oreades, grey represents% assignment to hybrid.
The Bayesian assignment test implemented in NewHybrids correctly identified the only morphologically intermediate individual between T. speciosissima and T. mongaensis (collected at Brogers Creek) as a hybrid (PP = 92%) when no priors were given, and as an F1 hybrid (PP = 98%) if an F1 prior was used in the analysis. No further sign of admixture was detected between the two species, even among the other individuals at Brogers Creek (average assignment to T. speciosissima for the other Brogers Creek individuals; PP = 98%).
NewHybrids analysis of the T. mongaensis and T. oreades populations identified extensive hybridisation at the Monga NP site (Figure 3b), with 13% of individuals assigned to pure T. mongaensis (PP > 90%), and the remainder assigned to a potential hybrid category (PP < 90% of being pure) with 60% having PP > 90% of being either F2 or back-cross to T. mongaensis. None of the Monga NP individuals (identified based on morphology to either T. mongaensis or T. oreades) were assigned to pure T. oreades (only four individuals had a PP > 0% of being pure T. oreades). At the other sites, all individuals (with the exception of three T. mongaensis individuals at River Forest Rd, the site closest to the Monga NP site) were assigned with PP > 90% to either pure T. oreades or pure T. mongaensis.
Current and historical environmental niche modelling
The average test AUC for the 10 replicate runs indicates excellent model fit for all species tested across all model sets indicating an excellent fit to estimated realised climatic spaces for Telopea and for each species tested [47]. For the model sets 1-3b (trained on extents to within 200 km of the species point locations and to suitable soil type, and projected onto only suitable soil types in the eastern Australian states) the average test AUC was: T. aspera 0.998 (std dev 0.001); T. speciosissima 0.943 (std dev 0.018); T. mongaensis 0.990 (std dev 0.006); and T. oreades 0.983 (std dev 0.011). However, this does not mean that the models sufficiently estimated the fundamental climatic distributions [47]. The model run for the Herbarium records of T. aspera with 100 m accuracy (with lower number of training localities) was the most fragmented, indicating overfitting of the model. Fragmentation of the remaining model sets within the expected distribution was minimal, particularly for the soils runs, indicating good representation of the fundamental climatic space for each species. Interestingly, the distribution of soil types suitable to Telopea closely matches the modelled climatic distribution, consequently modelled climatic probabilities and areas did not differ appreciably between model runs, at species or genus levels, with or without soils included. Telopea aspera responded most to higher precipitation in the warmest quarter. Telopea speciosissima responded to higher mean temperatures for the warmest and wettest quarters and to a higher precipitation in the driest quarter. Telopea mongaensis responded to higher temperatures in the wettest quarter and lower temperatures in the driest quarter (low seasonal temperature and precipitation variation). Telopea oreades responded to low maximum temperatures (for the warmest month and quarter).
The modelled available bio-climatic habitats for T. aspera and T. speciosissima expanded from the LGM to present (Figure 2a-d). In contrast, the distribution of habitats suitable for T. mongaensis and T. oreades declined from the LGM to present (Figure 2e-h). As Telopea generally occurs in topographically variable habitats it is probable that there was greater in-situ preservation than the LGM modeling indicates, particularly in microhabitats too fine to be observed in the model pixel size of 30 arc seconds (approximately 850 m).
The LGM bio-climatic models for the various lineages support appreciable overlap only for T. speciosissima and T. mongaensis, and T. mongaensis and T. oreades. Due to the modelled current expansion of T. aspera and T. speciosissima, overlap of distributions increased between all species pairs excluding between T. mongaensis and T. oreades (Table 4). Although some low-level potential overlaps are modelled between these two species, a 160 km wide distributional gap currently exists. Similarly, despite a large potential habitat overlap between T. speciosissima and T. oreades they are significantly separated geographically. Similarly T. aspera is currently too geographically disjunct (400 km) from all species for any interaction to occur.
Discussion
Concordance between geographic range and the distribution of genes and suitable habitat
The species boundaries described for Telopea[7] follow a pattern of latitudinal differentiation along the Great Dividing Range (GDR). The GDR, running north–south along the length of eastern Australia, is a dominant topographic feature. Despite having relatively low elevation, it is sufficiently close to the coast to provide precipitation gradients. These gradients support a range of species and communities that differ considerably from those found further west across the prevalently dry continent. The GDR supported refugial habitats during the climatic oscillations of the Quaternary, as revealed by regional species turnover [48, 49] and deep between-population genetic divergences [50, 51]. Based on palaeoecological evidence [12, 13], phylogeographic divergences are interpreted as the genetic signatures of distributional contractions caused by climate-induced loss of suitable habitat. Habitat contractions were followed by re-expansions at the return of more favourable conditions [52–54]. These recurring Quaternary patterns have the potential to isolate populations and initiate allopatric speciation, even in the absence of strong intrinsic reproductive barriers [55, 56].
The nSSR data provided fine-scale resolution and broad support for latitudinal species-level genetic divergences (Figure 2). The highest diversity was measured in the most widespread species (T. speciosissima) and, as expected in an allopatric speciation model [57], the more diverse species also showed signs of significant structure particularly in relation to latitudinal landscape barriers and elevation. Current environment niche models are also in agreement with species being differentiated along the latitudinal gradient, suggesting relatively small bio-climatic overlaps between species (Table 4). Genetic and bio-climatic boundaries correspond to breaks in the GDR. T. aspera and T. speciosissima are separated by the Hunter River Corridor (an important floristic boundary; [58, 59]). With the exception of the T. oreades population at Monga NP, T. mongaensis and T. oreades are separated by an elevation decline of the GDR (previously reported as affecting genetic structure in other plants; [60]) and the fragmented nature of suitable edaphic conditions.
Chloroplast SSRs could not fully resolve species boundaries (only six chlorotypes were discovered across the entire distribution of the four continental species), however the geographic distribution of the chlorotypes confirmed a latitudinal partitioning of genetic variation, even confirming within-species latitudinal divergences in T. speciosissima (Figure 2). Thus, genetic and bio-climatic analyses largely corroborate an allopatric speciation model where the latitudinal gradient is a major factor influencing lineage divergence. Nevertheless, present-day distributions include potential contact zones between species, and localised chlorotype-sharing suggests that between-species admixture is possible. If confirmed, hybridisation events could provide supporting evidence either for secondary contact between once-disjunct taxa or for parapatric differentiation along the borders separating sister taxa. Consequently, we explored in greater detail the possible occurrence of hybridisation at known contact zones.
Supporting evidence for hybridisation at contact zones
Artificial hybrids among Telopea species are common in the horticultural trade and all species are known to be potentially inter-fertile in cultivation [61]. A single and isolated individual that is morphologically intermediate between T. speciosissima and T. mongaensis was confirmed as a hybrid (potentially an F1) by the nSSR analyses. No T. mongaensis individuals were found in the vicinity, and the closest Telopea population was T. speciosissima at Brogers Creek. Interestingly, the Brogers Creek population shared chlorotype 4 with T. mongaensis despite bearing no morphological or nuclear signature of admixture. Furthermore, while the nSSR data preferentially assigned this same population to the southern T. speciosissima group, the geographically closest T. mongaensis population shared chlorotype 6 with the southern T. speciosissima, while being unequivocally assigned to T. mongaensis by nSSRs (Figure 2). These patterns suggest temporal variations in species overlap in response to changing climatic conditions, leaving genetic signatures similar to those observed in Tasmanian eucalypts [62, 63].
Telopea oreades and T. mongaensis are sister taxa and the only qualitative morphological character that differentiates them is the absence of foliar sclereids in T. mongaensis. They also differ in average leaf dimensions, bract width and prominence of leaf venation but variation in these characters overlaps between the species [8]. Consequently, morphological intermediates are difficult to characterise. These two species are generally geographically disjunct, but variation in leaf morphology, including the presence of foliar sclereids in large-leaved plants, suggested that T. oreades and T. mongaensis may occur in sympatry at Monga NP (the southern distributional end of T. mongaensis). While both species share chlorotype 4 (Figure 2), which could be interpreted as a result of incomplete lineage sorting in view of limited chloroplast variation, the nSSR-based Bayesian analyses implemented in NewHybrids presented an unambiguous admixture scenario at Monga NP (Figure 3) suggesting repeated backcrossing to T. mongaensis but no evidence of the persistence of pure T. oreades. Interestingly, when Crisp & Weston [8] excluded sclereids from their morphometric analysis, individuals sampled from Monga NP spanned the full range of morphological variation between pure T. mongaensis and pure T. oreades. Their results were consistent with our genetic evidence but when viewed through the lens of a categorical distinction between plants possessing or lacking sclereids, morphological variation at Monga NP was interpreted as circumscribing two sympatric, albeit sporadically hybridising species rather than a single, morphologically variable population [8].
Rather than a scenario of parapatric speciation, where hybrid zones continuously occur along adaptive clines at the distributional extremes of species [3], our findings are more supportive of a scenario similar to the one proposed for T. speciosissima and T. mongaensis. Temporal climatic fluctuations caused distributional changes that resulted in repeated cycles of secondary contact. As suitable habitat contracted, the T. oreades population at Monga NP remained isolated and was integrated by T. mongaensis. A history of allopatric speciation is unlikely to result in rapid and strong postzygotic reproductive isolation and consequently, complex hybridisation events can be expected in secondary contact zones [1]. However, further evidence for temporal changes in connectivity was needed in order to exclude a scenario of parapatric speciation.
Bio-climatic evidence for temporal changes in connectivity
A previous study on T. speciosissima suggested that the genetic differentiation measured among population groups representing coastal, upland and southern distributions was the consequence of cyclical distributional adjustments during the Quaternary [10]. An exploration of temporal changes in the availability of suitable habitat supports contractions into refugia during the LGM (Figure 2). In fact, Hesse et al. [64] suggested that local environmental conditions were so extreme during the LGM that upland areas were largely devoid of woody vegetation.
Unexpectedly, whilst the temporal distributional variation that provides evidence for intra-specific allopatric differentiation in T. speciosissima is characterised by habitat contractions during the LGM (also evident for its northern sister species, T. aspera), the circumstances are reversed for the two southern taxa (Figure 2). Current interglacial bio-climatic conditions confirm the existence of an inter-specific distributional gap, while modelled LGM ENMs provide support for expansions of suitable habitat. The LGM models support the scenario of a T. oreades x T. mongaensis hybrid population at Monga NP, currently disjunct from other T. oreades populations and likely to have originated from historical habitat expansions resulting in a now-vanished overlap zone.
Overall, the bio-climatic models show that cyclical contractions/expansions of suitable habitat for all Telopea are likely to have generated temporally variable inter-specific distribution gaps consistent with allopatric differentiation. Contraction to small population size and the selection for even small adaptive differences would have enhanced the chance of faster stochastic allopatric differentiation [6]. Conversely, there is no evidence for the persistence of the continuous contact zones essential to a parapatric differentiation model [1].
Conclusions
Although the traditional separation of speciation processes into allopatric and parapatric categories might not capture the full complexity of spatial relationships among taxa [65], our study shows that the evidence for temporal exclusion of gene flow can be found even outside obvious geographical contexts (such as continental drift and island radiations for example). In Telopea, between-species differentiation has been regulated mostly by temporal changes in bio-climatic conditions that have repeatedly disrupted the continuum in distribution of the various evolving lineages.
We also show that it is possible to make significant progress towards excluding parapatric speciation as a contributing evolutionary process in a selected study system. Telopea includes allopatric taxa that are at different stages of speciation involving some ecological adaptation but no complete reproductive isolation. Niche differentiation does not imply the establishment of fixed reproductive barriers [3], and the combination of species-specific bio-climatic envelopes and hybrid zones suggests that selective filters shaped local adaptive distinctiveness as a secondary process rather than being the driver of speciation via the establishment of selection-based reproductive barriers.
Current studies involving fine-scale coalescent analyses and transcriptome-based investigations of differential adaptive potential among Telopea populations will bring further insights on speciation mechanisms operating in Telopea. Comparing genome-level patterns of diversity between populations can facilitate the identification of genomic regions that do not conform to the expectation of neutral demographic models. However, strong divergent selection at a small number of loci would not be expected in an allopatric speciation scenario. Instead, analogous coalescent patterns across different loci could identify the signature of past periods of allopatry. For instance, targeting multiple low recombination regions has been shown to be particularly useful in describing the contribution of allopatric processes to sympatric divergences [66].
References
Turelli M, Barton NH, Coyne JA: Theory and speciation. Trends Ecol Evol. 2001, 16: 330-343.
Mayr E: Systematics and the Origin of Species. 1942, New York: Columbia University Press
Coyne JA, Orr HA: Speciation. 2004, Massachusetts: Sinauer Associates, Sunderland
Slatkin M: Gene flow in natural populations. Annu Rev Ecol Syst. 1985, 16: 393-430.
Smith JM: Sympatric speciation. Am Nat. 1966, 100: 637-650.
Gavrilets S: Perspective: Models of speciation: What have we learned in 40 years?. Evolution. 2003, 57: 2197-2215.
Weston PH, Crisp MD: Cladistic biogeography of waratahs (Proteaceae, Embothrieae) and their allies across the pacific. Aust Syst Bot. 1994, 7: 225-249.
Crisp MD, Weston PH: Geographic and ontogenic variation in morphology of Australian Waratahs (Telopea, Proteaceae). Syst Biol. 1993, 42: 49-76.
Denham AJ, Auld TD: Flowering, seed dispersal, seed predation and seedling recruitment in two pyrogenic flowering resprouters. Aust J Bot. 2002, 50: 545-557.
Rossetto M, Thurlby KAG, Offord CA, Allen CB, Weston PH: The impact of distance and a shifting temperature gradient on genetic connectivity across a heterogeneous landscape. BMC Evol Biol. 2011, 11: 126-
Bowman DMJS: Australian rainforest: islands of green in a land of fire. 2000, Cambridge: Cambridge University Press
Black MP, Mooney SD, Martin HA: A > 43,000-year vegetation and fire history from Lake Baraba, New South Wales, Australia. Quaternary Sci Rev. 2006, 25: 3003-3016.
Kershaw AP, Bretherton SC, van der Kaars S: A complete pollen record of the last 230 ka from Lynch's Crater, north-eastem Australia. Palaeogeogr Palaeoclimatol Palaeoecol. 2007, 251: 23-45.
Petit RJ, Csaikl UM, Bordacs S, Burg K, Coart E, Cottrell J, van Dam B, Deans JD, Dumolin-Lapegue S, Fineschi S, Finkeldey R, Gillies A, Glaz I, Goicoechea PG, Jensen JS, Konig AO, Lowe AJ, Madsen SF, Matyas G, Munro RC, Olalde M, Pemonge MH, Popescu F, Slade D, Tabbener H, Taurchini D, de Vries SGM, Ziegenhagen B, Kremer A: Chloroplast DNA variation in European white oaks - Phylogeography and patterns of diversity based on data from over 2600 populations. For Ecol Manag. 2002, 156: 5-26.
Brewer S, Cheddadi R, de Beaulieu JL, Reille M: The spread of deciduous Quercus throughout Europe since the last glacial period. For Ecol Manag. 2002, 156: 27-48.
Mellick R, Lowe A, Allen CB, Hill RS, Rossetto M: Palaeodistribution modelling and genetic evidence highlight differential post-glacial range shifts of a rain forest conifer distributed across a broad latitudinal gradient. J Biogeogr. 2012, 10.1111/j.1365-2699.2012.02747.x.
Scoble J, Lowe AJ: A case for incorporating phylogeography and landscape genetics into species distribution modelling approaches to improve climate adaptation and conservation planning. Divers Distrib. 2010, 16: 343-353.
Porter C, Rymer PD, Rossetto M: Isolation and characterization of microsatellite markers for the waratah, Telopea speciosissima (Proteaceae). Mol Ecol Notes. 2006, 6: 446-448.
van Oosterhout C, Weetman D, Hutchinson WF: Estimation and adjustment of microsatellite null alleles in nonequilibrium populations. Mol Ecol Notes. 2006, 6: 255-256.
Weising K, Gardner RC: A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome. 1999, 42: 9-19.
Sang T, Crawford DJ, Stuessy TF: Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am J Bot. 1997, 84: 1120-1136.
Ebert D, Peakall R: A new set of universal de novo sequencing primers for extensive coverage of noncoding chloroplast DNA: new opportunities for phylogenetic studies and cpSSR discovery. Mol Ecol Res. 2009, 9: 777-783.
Schuelke M: An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol. 2000, 18: 233-234.
Lewis PO, Zaykin D: Genetic Data Analyser: Computer Program for the Analysis of Allelic Data v1.1. 2002, http://www.eeb.uconn.edu/people/plewis/software.php,
Leberg PL: Estimating allelic richness: Effects of sample size and bottlenecks. Mol Ecol. 2002, 11: 2445-2449.
El Mousadik A, Petit RJ: High level of genetic differentiation for allelic richness among populations of the argan tree Argania spinosa (L) Skeels endemic to Morocco. Theor Appl Genet. 1996, 92: 832-839.
Goudet J: FSTAT (Version 1.2): A computer program to calculate F-statistics. J Hered. 1995, 86: 485-486.
Weir BS, Cockerham CC: Estimating F-statistics for the analysis of population structure. Evolution. 1984, 38: 1358-1370.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959.
Falush D, Stephens M, Pritchard JK: Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003, 164: 1567-1587.
Evanno G, Regnaut S, Goudet J: Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005, 14: 2611-2620.
Excoffier L, Smouse PE, Quattro JM: Analysis of molecular variance inferred from metric distances among DNA haplotypes - application to human mitochondrial DNA restriction data. Genetics. 1992, 131: 479-491.
Peakall R, Smouse PE: Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006, 6: 288-295.
Bandelt HJ, Forster P, Rohl A: Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16: 37-48.
fluxus-engineering.com: http://www.fluxus-engineering.com,
Anderson EC, Thompson EA: A model-based method for identifying species hybrids using multilocus genetic data. Genetics. 2002, 160: 1217-1229.
Vähä JP, Primmer CR: Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol. 2006, 15: 63-72.
Beaumont M, Barratt EM, Gottelli D, Kitchener AC, Daniels MJ, Pritchard JK, Bruford MW: Genetic diversity and introgression in the Scottish wildcat. Mol Ecol. 2001, 10: 319-336.
Viscosi V, Lepais O, Gerber S, Fortini P: Leaf morphological analyses in four European oak species (Quercus) and their hybrids: A comparison of traditional and geometric morphometric methods. Plant Biosyst. 2009, 143: 564-574.
She JX, Autem M, Kotulas G, Pasteur N, Bonhomme F: Multivariate analysis of genetic exchanges between Solea aegyptiaca and Solea senegalensis (Teleosts, Soleidae). Biol J Linn Soc. 1987, 32: 357-371.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F: GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5171. 2004, Montpellier (France): Université de Montpellier II
Phillips SJ, Anderson RP, Schapire RE: Maximum entropy modeling of species geographic distributions. Ecol Model. 2006, 190: 231-259.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A: Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 2005, 25: 1965-1978.
Elith J, Graham CH, Anderson RP, Dudik M, Ferrier S, Guisan A, Hijmans RJ, Huettmann F, Leathwick JR, Lehmann A, Li J, Lohmann LG, Loiselle BA, Manion G, Moritz C, Nakamura M, Nakazawa Y, Overton JM, Peterson AT, Phillips SJ, Richardson K, Scachetti-Pereira R, Schapire RE, Soberon J, Williams S, Wisz MS, Zimmermann NE: Novel methods improve prediction of species' distributions from occurrence data. Ecography. 2006, 29: 129-151.
Vegetation Information System: http://www.environment.nsw.gov.au/research/VISplot.htm,
Atlas of NSW Wildlife: http://wildlifeatlas.nationalparks.nsw.gov.au/wildlifeatlas/watlas.jsp,
Rodda GH, Jarnevich CS, Reed RN: Challenges in Identifying Sites Climatically Matched to the Native Ranges of Animal Invaders. PLoS ONE. 2011, 6 (6): e14670-
Kooyman R, Rossetto M, Allen C, Cornwell W: Australian tropical and sub-tropical rain forest community assembly: phylogeny, functional biogeography, and environmental gradients. Biotropica. 2012, 10.1111/j.1744-7429.2012.00861.x.
Kooyman R, Rossetto M, Cornwell W, Westoby M: Phylogenetic tests of community assembly across regional to continental scales in tropical and subtropical rain forests. Glob Ecol Biogeogr. 2011, 20: 707-716.
Chapple DG, Hoskin CJ, Chapple SNJ, Thompson MB: Phylogeographic divergence in the widespread delicate skink (Lampropholis delicata) corresponds to dry habitat barriers in eastern Australia. BMC Evol Biol. 2011, 11: 191-
Schneider CJ, Cunningham M, Moritz C: Comparative phylogeography and the history of endemic vertebrates in the Wet Tropics rainforests of Australia. Mol Ecol. 1998, 7: 487-498.
Nicholls JA, Austin JJ: Phylogeography of an east Australian wet-forest bird, the satin bowerbird (Ptilonorhynchus violaceus), derived from mtDNA, and its relationship to morphology. Mol Ecol. 2005, 14: 1485-1496.
Rossetto M, Crayn D, Ford A, Mellick R, Sommerville K: The influence of environment and life-history traits on the distribution of genes and individuals: a comparative study of 11 rainforest trees. Mol Ecol. 2009, 18: 1422-1438.
Rossetto M, Crayn D, Ford A, Ridgeway P, Rymer P: The comparative study of range-wide genetic structure across related, co-distributed rainforest trees reveals contrasting evolutionary histories. Aust J Bot. 2007, 55: 416-424.
Hewitt G: The genetic legacy of the Quaternary ice ages. Nature. 2000, 405: 907-913.
Hewitt GM: Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc. 1996, 58: 247-276.
Lorenz-Lemke AP, Togni PD, Mäder G, Kriedt RA, Stehmann JR, Salzano FM, Bonatto SL, Freitas LB: Diversification of plant species in a subtropical region of eastern South American highlands: a phylogeographic perspective on native Petunia (Solanaceae). Mol Ecol. 2010, 19: 5240-5251.
Cracraft J: Patterns of diversification within continental biotas: Hierarchical congruence among the areas of endemism of Australian vertebrates. Aust Syst Bot. 1991, 4: 211-227.
Playford J, Bell JC, Moran GF: A Major Disjunction in Genetic Diversity Over the Geographic Range of Acacia melanoxylon R.Br. Aust J Bot. 1993, 41: 355-368.
Worth JRP, Marthick JR, Jordan GJ, Vaillancourt RE: Low but structured chloroplast diversity in Atherosperma moschatum (Atherospermataceae) suggests bottlenecks in response to the Pleistocene glacials. Ann Bot. 2011, 108: 1247-1256.
Offord CA: Analysis of characters and germplasm of significance to improvement of australian native waratahs (Telopea spp., family Proteaceae) for cut flower production. Genet Resour Crop Evol. 2006, 53: 1263-1272.
McKinnon GE, Jordan GJ, Vaillancourt RE, Steane DA, Potts BM: Glacial refugia and reticulate evolution: the case of the Tasmanian eucalypts. Philos Trans R Soc B. 2004, 359: 275-284.
McKinnon GE, Vaillancourt RE, Jackson HD, Potts BM: Chloroplast sharing in the Tasmanian eucalypts. Evolution. 2001, 55: 703-711.
Hesse PP, Humphreys GS, Selkirk PM, Adamson DA, Gore DB, Nobes DC, Price DM, Schwenninger JL, Smith B, Tulau M, Hemmings F: Late Quaternary aeolian dunes on the presently humid Blue Mountains, eastern Australia. Quatern Int. 2003, 108: 13-32.
Butlin R, Debelle A, Kerth C, Snook RR, Beukeboom LW, Castillo Cajas RF, Diao W, Maan ME, Paolucci S, Weissing FJ, van de Zande L, Hoikkala A, Geuverink E, Jennings J, Kankare M, Knott KE, Tyukmaeva VI, Zoumadakis C, Ritchie MG, Barker D, Immonen E, Kirkpatrick M, Noor M, Garcia CM, Schmitt T, Schilthuizen M: What do we need to know about speciation?. Trends Ecol Evol. 2012, 27: 27-39.
Feder JL, Berlocher SH, Roethele JB, Dambroski H, Smith JJ, Perry WL, Gavrilovic V, Filchak KE, Rull J, Aluja M: Allopatric genetic origins for sympatric host-plant shifts and race formation in Rhagoletis. Proc Natl Acad Sci U S A. 2003, 100: 10314-10319.
Acknowledgements
This project was funded by the Hermon Slade Foundation (HSF 07–1). The authors would like to thank D. Benson, D. Burge and P. Rymer for useful discussions; and R Milner and J Baum for collection support and N. Weston and J. Siow for checking occurrence records.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing Interests
The authors have no financial or other competing interest to declare.
Authors' contributions
MR, MLM and PHW conceived and designed the study and collected samples; KAGT and CBA contributed laboratory and modeling data, MLM collected samples and contributed cpDNA data and analyses; MR analysed the nDNA molecular data and prepared the manuscript with all authors editing and approving the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rossetto, M., Allen, C.B., Thurlby, K.A. et al. Genetic structure and bio-climatic modeling support allopatric over parapatric speciation along a latitudinal gradient. BMC Evol Biol 12, 149 (2012). https://doi.org/10.1186/1471-2148-12-149
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-12-149