
Abstract
Background
Plant leucine-rich repeat receptor-like kinases (LRR-RLKs) are receptor kinases that contain LRRs in their extracellular domain. In the last 15 years, many research groups have demonstrated major roles played by LRR-RLKs in plants during almost all developmental processes throughout the life of the plant and in defense/resistance against a large range of pathogens. Recently, a breakthrough has been made in this field that challenges the dogma of the specificity of plant LRR-RLKs.
Results
We analyzed ~1000 complete genomes and show that LRR-RK genes have now been identified in 8 non-plant genomes. We performed an exhaustive phylogenetic analysis of all of these receptors, revealing that all of the LRR-containing receptor subfamilies form lineage-specific clades. Our results suggest that the association of LRRs with RKs appeared independently at least four times in eukaryotic evolutionary history. Moreover, the molecular evolutionary history of the LRR-RKs found in oomycetes is reminiscent of the pattern observed in plants: expansion with amplification/deletion and evolution of the domain organization leading to the functional diversification of members of the gene family. Finally, the expression data suggest that oomycete LRR-RKs may play a role in several stages of the oomycete life cycle.
Conclusions
In view of the key roles that LRR-RLKs play throughout the entire lifetime of plants and plant-environment interactions, the emergence and expansion of this type of receptor in several phyla along the evolution of eukaryotes, and particularly in oomycete genomes, questions their intrinsic functions in mimicry and/or in the coevolution of receptors between hosts and pathogens.
Similar content being viewed by others

Background
Receptor-like kinases (RLKs) are plant-specific transmembrane (TM) receptor kinases (RKs) that are closely related to the Pelle proteins, a family of animal cytoplasmic kinases. These RLK/Pelle proteins are involved in host defense against a range of pathogens and are also key regulators of many developmental processes in both plants and animals [1–4]. In an extended phylogenetic analysis of eukaryotic receptor kinases, Shiu and Bleecker (2001) have shown that, with respect to the kinase domain (KD), the RLK/Pelle, receptor tyrosine kinase (RTK), receptor serine/threonine kinase (RSK) and Raf protein subfamilies form a monophyletic group, the receptor kinase group (RKG), that is distinct from all other eukaryotic kinases [5, 6]. All plant RLKs possess a single-pass TM domain and an intracytoplasmic KD but differ in their extracellular domain (ECD) [5]. Members of the largest RLK subfamily, the leucine-rich repeat receptor-like kinases (LRR-RLKs), contain 1 to 30 leucine-rich repeats (LRRs) in their ECDs (Figure 1A). Plant LRR-RLKs are involved in many developmental processes and in host responses to biotic and abiotic stresses [7, 8]. Animals also possess LRR-containing receptors (the Toll and Toll-like receptors) that play a role in development and immunity [9]. Interestingly, these animal receptors contain LRRs in their ECD but do not possess a KD, and several transduce their signal by activating kinase-containing co-receptors through the binding to adaptor proteins. Because the structural organization (LRRs exclusively in the ECD, followed by a TM and a KD with serine/threonine kinase activity) was previously found only in plants, a plant-specific dogma for LRR-RLKs was expounded for many years. However, LRR and KD domains are highly versatile with respect to their associations with other protein domains and are predicted to be present in all genomes from bacteria to humans [10].

Features and number of LRR-containing receptors in the analyzed genomes. (A) Schematic representation of LRR-RLK (land plants) and LRR-RK (other genomes) receptors. Each of these proteins contains a signal peptide (SP, empty box), 1 to 30 LRRs (black boxes) in their extracellular domain (ECD), a transmembrane domain (TM, dark gray box) and an intracytoplasmic kinase domain (KD, pale gray box). (B) Schematic phylogenetic representation of all of the genomes analyzed. The tree is based on [34] and on [35] for the green algae lineage. The number of genomes in which LRR-RKs or LRR-RLKs (land plants) were found is followed in parentheses by the total number of genomes analyzed for each kingdom. On the right, the number of LRR-RLKs or LRR-RKs is given following the name of each species. Among the green algae, 6 genomes were studied; only one (Chlorella variabilis NC64A) contains LRR-RK proteins. Among the oomycetes, 1 genome of Saprolegniales (Saprolegnia parasitica) and 4 genomes of Peronosporales (Pythium ultimum, Phytophthora infestans, Phytophthora ramorum and Phytophthora sojae) were analyzed. Only proteins with an LRR-TM-KD organization are considered to be LRR-RKs. 1Soanes et al. 2010, 2Cock et al. 2010, 3this study, N.D. Not Determined.
In this report, we show that LRR-KD subfamilies have been reinvented in several eukaryotic genomes outside plants. Moreover, the evolutionary history of these LRR-RKs is comparable to the one described for the LRR-RLK plant subfamilies.
Results and discussion
LRR-containing receptor kinases are not plant-specific
As LRRs and KDs are present in all genomes, we searched for the presence of structurally related LRR-RLKs in non-plant lineages. We analyzed 884 bacterial, 50 archaeal and 77 eukaryotic genomes to identify LRR-containing RKs that were structurally related to plant LRR-RLKs (Figure 1B). Additional file 1 details the references and links for all of the genomes analyzed. Our study reveals that, among all of the genomes outside of land plants that were analyzed, LRR receptor kinase (LRR-RK) subfamilies are present in the genomes of Monosiga brevicollis (a choanoflagellate), Chlorella variabilis NC64A (a green alga) and several stramenopiles (Ectocarpus siliculosus [a brown alga] and all of the oomycetes analyzed) (Figure 1B). Some of these findings have recently been reported, albeit unobtrusively, mentioned as only a side discovery notice in two articles [11, 12]. Our detailed analysis shows that the Ectocarpus, Chlorella and Monosiga genomes contain 2, 5 and 7 LRR-RK genes, respectively, whereas the oomycete species (Saprolegnia parasitica, Pythium ultimum, Phytophthora infestans, Phytophthora ramorum and Phytophthora sojae) contain up to 34 LRR-RK genes per genome. The step-by-step procedure used to detect the LRR-RK genes is described in Additional file 2. These LRR-RKs possess up to 26 LRRs in their ECDs (Additional file 3). Interestingly, although we searched for other domains (known to be present in plant ECDs, including lectins, duf26, EGF, lysM, Slocus, thaumatin and PAN) associated with RKs in oomycetes, we did not find any.
LRR-RK subfamilies have been reinvented several times in eukaryotic genomes and evolved independently from each other
A phylogenetic analysis of a representative subset of animal and plant eukaryotic kinases and all of the LRR-RKs, including receptors that are closely related to LRR-RKs but lack TM domains and/or LRRs, has been performed (Figure 2; see Additional file 4 for the sequence alignment and Additional file 5 for the details of Figure 2). This analysis reveals that the Ectocarpus, Chlorella, Monosiga and oomycete LRR-RK genes form four separate lineage-specific clades within the eukaryotic RKG. Because we did not observe incongruence between the phylogenetic and the species trees, we excluded the hypothesis of lateral transfer of the LRR-RK genes between species. However, because the tree is not fully resolved, we cannot rule out the possibility that the KDs of the LRR-RKs are more closely related to one of the RKG subfamilies. Nevertheless, with regard to the combination of the three modules (LRRs, TM and KD) to form new genes that are structurally related to LRR-RLKs, the most parsimonious hypothesis, based primarily on the species tree (Figure 1B), is that the association of LRRs with RKs appeared independently at least four times in eukaryotic evolutionary history. This hypothesis is supported by previous work on plants that showed that the KD of the RLK families has been recruited several times to form all of the LRR-RLK subfamilies [5]. Moreover, LRR domains are known to be highly versatile with respect to their associations with other protein domains [10]. Thus, our study expands what has been observed in plant lineages to several eukaryotic genomes. Alternatively, even if much less parsimonious, we cannot completely rule out a third hypothesis, which states that the LRR-RK and LRR-RLK gene families evolved from a common ancestor and have been subjected to massive gene losses.

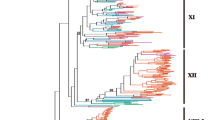
Topology of the maximum likelihood tree representing the phylogenetic relationships among the LRR-RK KDs and other eukaryotic kinases. The phylogenetic tree was generated from an alignment of the KDs (Additional file 4) of representative Arabidopsis and animal protein kinases, LRR-containing RKs and closely related receptors lacking TM domains and/or LRRs from Monosiga brevicollis (Monosiga, brown), Chlorella variabilis NC64A (Chlorella, greenish blue), Ectocarpus siliculosus (Ectocarpus, gray) and oomycetes (oomycetes, blue). Oomycete LRR-RKs, Monosiga LRR-RKs, Chlorella LRR-RKs and Ectocarpus LRR-RKs are included in the monophyletic receptor kinase group (RKG, red box), consisting of the plant receptor-like kinase (RLK) and animal cytoplasmic Pelle (forming the RLK/Pelle subfamily, green), receptor serine/threonine kinase (RSK, light blue), receptor tyrosine kinase (RTK, khaki) and Raf (Raf, pink) proteins. The RKG members are distinct from the other eukaryotic kinases. See Additional file 5 for the detailed phylogenetic tree.
LRR-RK genes are in expansion in certain oomycete genomes
In plants, LRR-RLKs show a pattern of expansion with amplification/deletion and evolution of domain organization leading to the functional diversification of the members of the gene family [1]. The same evolutionary history can be observed in the oomycete LRR-RK subfamily. Indeed, we were able to identify gains and losses of LRR-RKs in each oomycete genome analyzed (Additional file 6). Moreover, some of the gene subgroups are Saprolegnia- or Phytophthora-specific, suggesting that several duplication events occurred independently in the Saprolegnia and Phytophthora genomes to give rise to ~25 copies in each lineage (Additional file 5B). In the Phytophthora-specific subgroups, most of the duplications are present in all of the Phytophthora genomes, indicating that these genetic changes occurred in the last common ancestor of the Phytophthora species. However, a few duplications are species specific, implying the possibility for the acquisition of new functions of these genes in these species. Notably, the absence of amplification in the Pythium genome and the independent amplifications in the Saprolegnia and Phytophthora genomes suggest that these genes may be involved in signaling pathways and, therefore, in functions that have diverged between Saprolegnia, Phytophthora and Pythium species. Indeed, similar to what is known in plants, these oomycete receptors could be involved in the perception of diverse signals leading to multiple cellular responses [8, 13, 14].
Oomycete LRR-RKs may play a role in several stages of the oomycete life cycle
To determine whether these oomycete LRR-RK genes are expressed, we first performed an in silico gene expression analysis by identifying expressed sequenced tags (ESTs) in the public databases (Additional file 7). Among the 85 oomycete LRR-RK genes assessed, 22 are represented by one to eight ESTs. These ESTs were obtained from libraries of various developmental stages, indicating that the oomycete LRR-RK genes are expressed during vegetative growth, mating, dissemination and host infection. To analyze the expression of these LRR-RK genes further, we used the plant pathosystem available in our laboratory: the Phytophthora parasitica/Arabidopsis thaliana interaction [15]. First, we searched for ESTs of Phytophthora parasitica and retrieved 6 ESTs (Additional file 8). Next, we verified and analyzed the expression patterns of these LRR-RK genes using quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) at several key stages during the course of the pathogen/host interaction (Figure 3): penetration (At-2.5 hpi (hours post inoculation)), biotrophic invasive growth (At-6 hpi and At-10.5 hpi), switch to necrotrophy (At-30 hpi) and necrotrophy (At-96 hpi). The data reveal that these genes are actually differentially expressed in the course of Arabidopsis thaliana infection. Moreover, the divergences in the expression profiles among the LRR-RK genes analyzed indicate that functional diversification may have occurred in the oomycete LRR-RK family. Taken together, these results suggest that these receptors could be involved in the perception of environmental cues, in the adaptation to specific conditions encountered in the host and/or in various developmental processes.

Expression analysis of 5 Phytophthora parasitica LRR-RK genes during their interaction with Arabidopsis thaliana. The relative mRNA levels have been quantified by qRT-PCR at different development stages: samples corresponding to Phytophthora parasitica mycelium grown in V8 medium (M), Phytophthora parasitica motile zoospores (Z), and Arabidopsis thaliana roots collected 2.5 (At-2.5 hpi), 6 (At-6 hpi), 10.5 (At-10.5 hpi), 30 (At-30 hpi) and 96 (At-96 hpi) hours after inoculation with Phytophthora parasitica zoospores [30]. The data are presented as expression ratios relative to the mean expression values of three reference genes (2-DCT). Two independent RNA extractions corresponding to a pool of more than 5 biological replicates each were used. • and ×, biological replicates; bars, mean values.
Conclusions
In conclusion, we have shown that the Monosiga, Chlorella, Ectocarpus and oomycete LRR-RK receptors belong to the RKG and are likely to have acquired the LRRs in their ECDs independently. The evolutionary history of the oomycete LRR-RK receptor subfamily is consistent with the molecular evolution of plant LRR-RLKs [16, 17]. Saprolegnia and Phytophthora species have developed, expanded and functionally diversified a subfamily of receptors that are structurally, but not phylogenetically, related to plant LRR-RLKs. Considering the key roles that plant LRR-RLKs play throughout the plant life cycle and in plant-environment interactions, it is tempting to propose that oomycete LRR-RKs may be important regulators of the oomycete life cycle. Future work should focus on deciphering the functions of oomycete LRR-RKs in host-oomycete interactions to reveal new targets to help combat these pathogens, which pose a serious threat to plants and aquaculture farming worldwide, causing tremendous economic damage every year [18].
Methods
Genomes analyzed
To analyze the representative species across all kingdoms, we first downloaded several publicly available completely sequenced genomes from each of the major kingdoms (Opisthokonta, Plantae, and Chromalveolates) from 2 sources (JGI and NCBI). We also downloaded all of the available bacterial and archaeal genomes from NCBI. When LRR-RK sequences were found in one genome, we downloaded all of the available complete genomes in that phylum. Thus, the complete proteomes of 77 eukaryotic, 50 archaeal and 884 bacterial species have been downloaded from their respective databases. See Additional file 1 for details regarding the genomes analyzed.
Sequence retrieval and domain predictions
We retrieved genes containing leucine-rich repeats (LRRs) and a kinase domain (KD) by running the hmmsearch program (HMMER 2.3.2) to search for the kinase Hidden Markov Model (HMM) profile (PF00069.16) within the proteomic sequences of completely sequenced genomes. Within this set of kinase proteins, we then searched for LRR-domain HMM profiles (PF00560.24) (E value cut-off < 1) [19, 20]. Signal peptides (SPs) and transmembrane domains (TMs) were predicted using the SignalP http://www.cbs.dtu.dk/services/SignalP/ and TMHMM http://www.cbs.dtu.dk/services/TMHMM/ websites, respectively, hosted at the Center for Biological Sequence Analysis, Technical University of Denmark [21]. Proteins containing LRRs, a TM and a KD were then considered to be putative LRR-RKs. We used the SMART web site http://smart.embl-heidelberg.de/ to check whether domains other than LRRs were predicted in the extracellular domain (ECD) of each protein [22]. If other domains were detected, the protein was rejected. Proteins containing only LRRs in their extracellular domain (ECD), a TM domain and a KD were classified as LRR-RKs. For the phylogenetic analysis presented in Figure 2, we first retrieved all of the peptide sequences of eukaryotic protein kinases used in [5] to establish the phylogenetic relationship between plant and animal protein kinases. We next retrieved one LRR-RLK protein per subgroup from the Arabidopsis genome. Finally, we included all of the newly identified LRR-RK proteins from the oomycete, Monosiga, Ectocarpus and Chlorella genomes. In oomycetes, Ectocarpus, Monosiga and Chlorella, we also retrieved proteins sequences of closely related kinases found using a Blastp search. This search was performed using the KD of the LRR-RKs, and we selected the non-LRR-RK best hit. Accessions numbers of all of these sequences are listed in the 'Accession number' section below.
Alignment and phylogenetic analysis
Peptide KD sequences of all of the kinases to be analyzed (plus four bacterial kinase genes [YP_003956736.1, ZP_04777056.1, ZP_06621294.1 and P0A5S4.1] used as the outgroup [23]) were aligned using the MAFFT program (v6.525 b, einsi parameters, 1000 iterations maximum) and manually curated [24]. Phylogenetic trees were generated under the maximum likelihood criterion using PhyML 3.0 (LG model, NNI topological moves, optimizing branch lengths and branch supports). For the approximate likelihood ratio test (aLRT), we used the minimum value between the parametric approximate likelihood ratio test (aLRT, Chi2-based) and the non-parametric aLRT (based on a Shimodaira-Hasegawa-like procedure) [25, 26]. All of the branches with support values less than 90 were collapsed. All of the manipulations of phylogenetic trees were performed using the TreeDyn [27] and MEGA4 [28] programs.
Expression analysis
We used the NCBI tBLASTn web interface to search for expressed ESTs that were similar (identity > 95%) to our set of oomycete LRR-RK proteins [29]. Only the Phytophthora infestans, Phytophthora sojae, Pythium ultimum and Saprolegnia parasitica EST databases have been searched, as the EST database of the Phytophthora ramorum genome was not available. Each EST sequence retrieved was validated by a BLASTn search using the library of nucleotide sequences from that species and from the Phytophthora infestans nucleotide sequences in GenBank. To search for ESTs from Phytophthora parasitica, the 24 LRR-RK peptide sequences of Phytophthora infestans have been used as query for a tBLASTn search on the VBI microbial database http://vmd.vbi.vt.edu/toolkit/index.php. We used the Phytophthora infestans LRR-RK proteins as queries because it is the most complete dataset thus far. This search revealed that at least 25 Phytophthora parasitica genes are homologous to the 24 Phytophthora infestans LRR-RK genes. We searched for ESTs for each of these 25 Phytophthora parasitica LRR-RKs on the NCBI Phytophthora parasitica EST database. The qRT-PCR expression analysis of 5 of the 6 Phytophthora parasitica ESTs retrieved was performed as described in Kebdani et al. (2010) using UBC, WS21 and Mago nashi protein encoding sequences used as reference genes [30–32]. Note that one of the 6 ESTs (DR440392.1) has not been analyzed by qRT-PCR because we did not succeed in designing oligonucleotides sets for this gene.
Accession numbers
The accession numbers for Arabidopsis thaliana are as follows: AtCKI1 [GenBank, CAA55395]; AtCDC2a [GenBank, AAB23643]; AtCPK7 [GenBank, AAB03247]; AtCKA1 [GenBank, BAA01090]; AtCTR1_Raf [GenBank, AAA32779]; AtAME2 [GenBank, BAA08215]; AtMKK3 [GenBank, BAA28829]; AtMEKK1 [GenBank, BAA09057]; AtNAK [GenBank, AAA18853]; AtNPH1 [GenBank, AAC01753]; AtPVPKlikePK5 [GenBank, BAA01715]; AtGSK3b [GenBank, CAA64408]; AtGSK3i [GenBank, CAA68027]; AtSnRK2 [GenBank, AAA32845]; AtMPK1 [TAIR, AT1G10210]; AtS6KlikePK1 [GenBank, AAA21142] and AtTousled [GenBank, AAA32874]. The accession numbers for Homo sapiens are as follows: hAXL [GenBank, NP_001690]; hRYK [GenBank, P34925]; hTRKalpha [GenBank, BAA34355]; hMuSK [GenBank, AAB63044]; hKLGlikePTK7 [GenBank, AAC50484]; hIR [GenBank, NP_000199]; hLTK [GenBank, P29376.3]; hRET [GenBank, AAH04257]; hTIE1 [GenBank, P35590]; hPDGFRbeta [GenBank, NM_002600.1]; hVGFR1 [GenBank, P17948.2]; hTousledLK1 [GenBank, NP_036422]; hMAPKKK1 [GenBank, Q13233]; hCLK1 [GenBank, P49759]; hMAPK1 [GenBank, NP_002736.3]; hCDK3 [GenBank, NP_001249]; hCKIalpha2 [GenBank, NP_001883]; hCaMK1 [GenBank, BAG70221]; hCK2a [GenBank, CAB65624]; hGRK6 [GenBank, P43250]; hEGFR [GenBank, P00533]; hFGFR2 [GenBank, P21802]; hHGFR [GenBank, P08581]; hEPH [GenBank, P21709]; hDDR [GenBank, Q08345]; hRaf1 [GenBank, AAA60247]; TGF beta receptors, hTGFbRI [GenBank, P36897] and hTGFbRII [GenBank, P37173]; hIRAK1 [GenBank, AAH54000] and hMAPKK1 [GenBank, Q02750]. Additional accession numbers are as follows: mCKIalpha [GenBank, NP_666199] for Mus musculus; xtPELLE [GenBank, NP_001006713] for Xenopus tropicalis; drIRAK1 [GenBank, CAP19555] for Danio rerio and dmPELLE [GenBank, NP_476971] for Drosophila melanogaster. The accessions of representative Arabidopsis thaliana LRR-RLK genes in each subfamily are as follows: [TAIR: AT4G29180] for LRRI, AtNIK1 for LRRII [TAIR: AT5G16000], AtIMK3 for LRRIII [TAIR: AT3G56100], [TAIR: AT2G45340] for LRRIV, AtSCM_SUB for LRRV [TAIR: AT1G11130], [TAIR: AT1G14390] for LRRVI-1, [TAIR: AT5G41180] for LRRVI-2, [TAIR: AT2G24230] for LRRVII, [TAIR: AT1G06840] for LRRVIII-1, [TAIR: AT3G14840] for LRRVIII-2, AtTMK1 for LRRIX [TAIR: AT1G66150], [TAIR: AT3G28450] for LRRXa, AtBRI1 for LRRXb [TAIR: AT4G39400), AtCLV1 for LRRXI [TAIR: AT1G75820], AtFLS2 for LRRXII [TAIR: AT5G46330], AtFEI1 for LRRXIIIa [TAIR: AT1G31420], AtER for LRRXIIIb [TAIR: AT2G26330], [TAIR: AT3G14840] for LRRXIV and AtRPK1 for LRRXV [TAIR: AT1G69270]. We followed the subfamily nomenclature of a previous report [33].
Abbreviations
- LRR:
-
leucine-rich repeat
- RLK:
-
receptor-like kinases
- RK:
-
receptor kinase
- LRR-RLK:
-
leucine-rich repeat receptor-like kinase
- LRR-RK:
-
leucine-rich repeat receptor kinase
- TM:
-
transmembrane
- KD:
-
kinase domain
- ECD:
-
extracellular domain
- RKG:
-
receptor kinase group
- RTK:
-
receptor tyrosine kinase
- RSK:
-
receptor serine/threonine kinase
- EST:
-
expressed sequence tag
- TM:
-
transmembrane domain
- SP:
-
signal peptide
- hpi:
-
hours post inoculation.
References
Afzal AJ, Wood AJ, Lightfoot DA: Plant receptor-like serine threonine kinases: roles in signaling and plant defense. Mol Plant Microbe Interact. 2008, 21 (5): 507-517. 10.1094/MPMI-21-5-0507.
Gish LA, Clark SE: The RLK/Pelle family of kinases. Plant J. 2011, 66 (1): 117-127. 10.1111/j.1365-313X.2011.04518.x.
Johnson KL, Ingram GC: Sending the right signals: regulating receptor kinase activity. Curr Opin Plant Biol. 2005, 8 (6): 648-656. 10.1016/j.pbi.2005.09.007.
Boller T, Felix G: A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu Rev Plant Biol. 2009, 60: 379-406. 10.1146/annurev.arplant.57.032905.105346.
Shiu SH, Bleecker AB: Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc Natl Acad Sci USA. 2001, 98 (19): 10763-10768. 10.1073/pnas.181141598.
Shiu SH, Bleecker AB: Plant receptor-like kinase gene family: diversity, function, and signaling. Sci STKE. 2001, 2001 (113): RE22-
Chinchilla D, Shan L, He P, de Vries S, Kemmerling B: One for all: the receptor-associated kinase BAK1. Trends Plant Sci. 2009, 14 (10): 535-541. 10.1016/j.tplants.2009.08.002.
Dievart A, Clark SE: LRR-containing receptors regulating plant development and defense. Development. 2004, 131 (2): 251-261.
Trinchieri G, Sher A: Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007, 7 (3): 179-190. 10.1038/nri2038.
Protein evolution. Edited by: Patthy L. 1999, Blackwell Science Ltd
Cock JM, Sterck L, Rouze P, Scornet D, Allen AE, Amoutzias G, Anthouard V, Artiguenave F, Aury JM, Badger JH, et al: The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature. 2010, 465 (7298): 617-621. 10.1038/nature09016.
Soanes DM, Talbot NJ: Comparative genome analysis reveals an absence of leucine-rich repeat pattern-recognition receptor proteins in the kingdom fungi. PLoS One. 2010, 5 (9): e12725-10.1371/journal.pone.0012725.
Tor M, Lotze MT, Holton N: Receptor-mediated signalling in plants: molecular patterns and programmes. J Exp Bot. 2009, 60 (13): 3645-3654. 10.1093/jxb/erp233.
Chae L, Sudat S, Dudoit S, Zhu T, Luan S: Diverse transcriptional programs associated with environmental stress and hormones in the Arabidopsis receptor-like kinase gene family. Mol Plant. 2009, 2 (1): 84-107. 10.1093/mp/ssn083.
Attard A, Gourgues M, Callemeyn-Torre N, Keller H: The immediate activation of defense responses in Arabidopsis roots is not sufficient to prevent Phytophthora parasitica infection. New Phytol. 2010, 187 (2): 449-460. 10.1111/j.1469-8137.2010.03272.x.
Lehti-Shiu MD, Zou C, Hanada K, Shiu SH: Evolutionary history and stress regulation of plant receptor-like kinase/pelle genes. Plant Physiol. 2009, 150 (1): 12-26. 10.1104/pp.108.134353.
De Smet I, Voss U, Jurgens G, Beeckman T: Receptor-like kinases shape the plant. Nat Cell Biol. 2009, 11 (10): 1166-1173. 10.1038/ncb1009-1166.
Stokstad E: Genetics. Genomes highlight plant pathogens' powerful arsenal. Science. 2006, 313 (5791): 1217-
Sonnhammer EL, Eddy SR, Birney E, Bateman A, Durbin R: Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 1998, 26 (1): 320-322. 10.1093/nar/26.1.320.
Eddy SR: A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009, 23 (1): 205-211.
Emanuelsson O, Brunak S, von Heijne G, Nielsen H: Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protocols. 2007, 2 (4): 953-971. 10.1038/nprot.2007.131.
Letunic I, Doerks T, Bork P: SMART 6: recent updates and new developments. Nucleic Acids Res. 2009, D229-232. 37 Database
Hanks SK, Hunter T: Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995, 9 (8): 576-596.
Katoh K, Misawa K, Kuma K, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30 (14): 3059-3066. 10.1093/nar/gkf436.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52 (5): 696-704. 10.1080/10635150390235520.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O: New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010, 59 (3): 307-321. 10.1093/sysbio/syq010.
Chevenet F, Brun C, Banuls AL, Jacq B, Christen R: TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics. 2006, 7: 439-10.1186/1471-2105-7-439.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24 (8): 1596-1599. 10.1093/molbev/msm092.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215 (3): 403-410.
Kebdani N, Pieuchot L, Deleury E, Panabieres F, Le Berre JY, Gourgues M: Cellular and molecular characterization of Phytophthora parasitica appressorium-mediated penetration. New Phytol. 2010, 185 (1): 248-257. 10.1111/j.1469-8137.2009.03048.x.
Yan HZ, Liou RF: Selection of internal control genes for real-time quantitative RT-PCR assays in the oomycete plant pathogen Phytophthora parasitica. Fungal Genet Biol. 2006, 43 (6): 430-438. 10.1016/j.fgb.2006.01.010.
Judelson HS, Ah-Fong AM, Aux G, Avrova AO, Bruce C, Cakir C, da Cunha L, Grenville-Briggs L, Latijnhouwers M, Ligterink W, et al: Gene expression profiling during asexual development of the late blight pathogen Phytophthora infestans reveals a highly dynamic transcriptome. Mol Plant Microbe Interact. 2008, 21 (4): 433-447. 10.1094/MPMI-21-4-0433.
Shiu SH, Bleecker AB: Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol. 2003, 132 (2): 530-543. 10.1104/pp.103.021964.
Baurain D, Brinkmann H, Petersen J, Rodriguez-Ezpeleta N, Stechmann A, Demoulin V, Roger AJ, Burger G, Lang BF, Philippe H: Phylogenomic evidence for separate acquisition of plastids in cryptophytes, haptophytes, and stramenopiles. Mol Biol Evol. 2010, 27 (7): 1698-1709. 10.1093/molbev/msq059.
Finet C, Timme RE, Delwiche CF, Marletaz F: Multigene phylogeny of the green lineage reveals the origin and diversification of land plants. Curr Biol. 2010, 20 (24): 2217-2222. 10.1016/j.cub.2010.11.035.
Acknowledgements
This research was funded by the French Agence Nationale de la Recherche (RICE RLK.O.me, 2008).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AD, NG and CP designed the experiments and discussed the results. AD and GD performed the bioinformatics analyses. AA and MG performed the qRT-PCR experiments. AD, NG, EG and CP drafted the manuscript. All of the authors have read and approved the final manuscript.
Electronic supplementary material
12862_2011_1960_MOESM2_ESM.DOC
Additional file 2: Step-by-step procedure to determine the number of LRR-RKs per genome. The last column of the table shows the number of proteins that we considered to be LRR-RKs in our analysis. The accession number of each gene is listed in Additional file 3. KD, kinase domain; LRRs, leucine-rich repeats; TM, transmembrane domain; ECD, extracellular domain; LRR-RKs, Leucine-rich repeat receptor kinase. (DOC 38 KB)
12862_2011_1960_MOESM3_ESM.DOC
Additional file 3: Structural features of the oomycete, Monosiga , Chlorella and Ectocarpus LRR-RKs and closely related receptors lacking LRRs and/or TM domains. All sequences except one sequence of Phytophthora ramorum (Pr81779) and one sequence of Phytophthora sojae (Ps136026) have been used for the phylogenetic analysis presented in Figure 2 and Additional file 5. These sequences were excluded because their KDs did not align with those of the other kinases. In addition, one sequence of Monosiga brevicollis (Mb34608) and one sequence of Chlorella variabilis NC64A (Ch136834) produced inconsistent results in the phylogenetic analysis and do not appear in the tree. These sequences may represent misannotated proteins or pseudogenes. Abbreviations: S, Saprolegnia parasitica; Pi, Phytophthora infestans; Pr, Phytophthora ramorum; Ps, Phytophthora sojae; Pu, Pythium ultimum; Mb, Monosiga brevicollis; Ch, Chlorella variabilis NC64A; Esi, Ectocarpus siliculosus; SP, signal peptide; KD, kinase domain; LRRs, leucine-rich repeats; +, presence; -, absence. See Materials and Methods for details about predicted SPs, LRR numbers, TM domains and KDs. (DOC 284 KB)
12862_2011_1960_MOESM4_ESM.DOC
Additional file 4: Alignment of LRR-RKs and reference eukaryotic KDs. The KDs of LRR-RKs from Ectocarpus siliculosus (Esi), Phytophthora ramorum (Pr), Phytophthora sojae (Ps), Phytophthora infestans (Pi), Saprolegnia parasitica (S), Pythium ultimum (Pu), Chlorella variabilis NC64A (Ch) and Monosiga brevicollis (Mb) were aligned with reference eukaryotic kinases from human (h), Arabidopsis thaliana (At), Drosophila melanogaster (dm), Danio rerio (dr), mouse (m) and Xenopus tropicalis (Xt). The positions of the 12 kinase subdomains are shown in roman numerals. (DOC 886 KB)
12862_2011_1960_MOESM5_ESM.DOC
Additional file 5: Details of the maximum likelihood tree representing the phylogenetic relationships among the LRR-RK KDs and other eukaryotic kinases. A. General view of the tree presented in Figure 2. The phylogenetic tree was generated from an alignment of the KDs (Additional file 4) of representative Arabidopsis and animal protein kinases, LRR-containing RKs (☼) and closely related receptors lacking TM domains and/or LRRs. These sequences, devoid of LRRs and/or TM domains, have been included to highlight the versatility of these domains with respect to their associations with KDs. The tree branches are colored as follows: Monosiga brevicollis (Monosiga, brown); Chlorella variabilis NC64A (Chlorella, greenish blue); Ectocarpus siliculosus (Ectocarpus, gray) and oomycetes (oomycetes, blue). Oomycete LRR-RKs, Monosiga LRR-RKs, Chlorella LRR-RKs and Ectocarpus LRR-RKs are included in the monophyletic receptor kinase group (RKG, red box) consisting of the plant receptor-like kinase (RLK) and animal cytoplasmic Pelle (forming the RLK/Pelle subfamily, green), receptor serine/threonine kinase (RSK, light blue), receptor tyrosine kinase (RTK, khaki) and Raf (Raf, pink) proteins. The RKG members are distinct from other the eukaryotic kinases. Note that the tree is not fully resolved. The addition of more RKs lacking LRRs to this phylogenetic analysis did not improve the resolution of the tree (data not shown); similarly, neither did the addition of non-RKs from the oomycete species (data not shown). Branch support values are shown at the nodes. B. A detailed view of the oomycete clade. In the oomycete LRR-RK subfamily, some subdivisions are Saprolegnia or Phytophthora specific (black boxes), suggesting the lineage-specific amplification by duplications in the Saprolegnia and Phytophthora genomes. Three subgroups contain both of these lineages with or without Pythium (orange boxes), suggesting that at least three genes were present in the last common ancestor of these species. Oomycete genes with evidence of expression (expressed sequence tags) are indicated with a plus sign (details in Additional file 7). C. A detailed view of other clades. (DOC 296 KB)
12862_2011_1960_MOESM6_ESM.DOC
Additional file 6: Number of gains and losses of oomycete LRR-RK genes in orthologous groups. We counted the number of genes gained and lost for each orthologous group defined in the oomycete clades of the phylogenetic tree (Additional file 5B). A. Orthologous groups containing Saprolegnia, Pythium and Phytophthora species, B. orthologous groups containing only Pythium and Phytophthora species and C. orthologous groups containing only Phytophthora species. The number on the first branch represents the number of genes present in the last common ancestor. Numbers preceded by a plus sign are the number of genes gained; numbers preceded by a minus sign are the number of genes lost. (DOC 24 KB)
12862_2011_1960_MOESM7_ESM.DOC
Additional file 7: Oomycete LRR-RK subgroups with evidence of expression. Abbreviations: S, Saprolegnia parasitica; Pi, Phytophthora infestans; Ps, Phytophthora sojae; Pp, Phytophthora parasitica; Pp, Phytophthora parasitica; EST, expressed sequence tags. (DOC 27 KB)
12862_2011_1960_MOESM8_ESM.DOC
Additional file 8: Phytophthora parasitica LRR-RKs with evidence of expression. Abbreviations: Pi, Phytophthora infestans; EST, expressed sequence tags. (DOC 24 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Diévart, A., Gilbert, N., Droc, G. et al. Leucine-Rich repeat receptor kinases are sporadically distributed in eukaryotic genomes. BMC Evol Biol 11, 367 (2011). https://doi.org/10.1186/1471-2148-11-367
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-11-367