
Abstract
Background
The decline in amphibian populations across the world is frequently linked to the infection of the chytrid fungus Batrachochytrium dendrobatidis (Bd). This is particularly perplexing because Bd was only recently discovered in 1999 and no chytrid fungus had previously been identified as a vertebrate pathogen.
Results
In this study, we show that two large families of known virulence effector genes, crinkler (CRN) proteins and serine peptidases, were acquired by Bd from oomycete pathogens and bacteria, respectively. These two families have been duplicated after their acquisition by Bd. Additional selection analyses indicate that both families evolved under strong positive selection, suggesting that they are involved in the adaptation of Bd to its hosts.
Conclusions
We propose that the acquisition of virulence effectors, in combination with habitat disruption and climate change, may have driven the Bd epidemics and the decline in amphibian populations. This finding provides a starting point for biochemical investigations of chytridiomycosis.
Similar content being viewed by others

Background
A recent report by IUCN indicates that amphibians are the most endangered group of vertebrates worldwide, with 32 percent listed as threatened with extinction and 159 species already believed to be extinct [1]. Among the several factors often linked to the decline in amphibian populations is the chytrid fungus Batrachochytrium dendrobatidis (Bd), the causative agent of chytridiomycosis. Ever since the discovery of Bd in 1999 [2], numerous cases have been reported for the destruction associated with chytridiomycosis [3, 4]. The destructive path of chytridiomycosis fits all of the descriptors of a novel epidemic (i.e. sudden appearance, strong virulence, and rapid transmission) [5]. Investigations of museum specimens collected from as far back as 1902 in Japan [6] and 1938 in Africa [7] have detected Bd in skin cross sections. On the other hand, long-term population studies since the 1900's had no records of mass mortalities until the late 1970's when the first Bd epidemics were reported [8]. This leads to an interesting question about how pathogenesis suddenly evolved in this previously benign organism.
Many disease organisms existed in non-virulent/commensal forms until they acquired some novel traits allowing them to exploit new niches [9]. Among the several possible underlying mechanisms, horizontal gene transfer (HGT) is the most rapid way for an organism to gain novel phenotypes [10] and it has contributed to the origin and spread of pathogenesis in numerous bacteria [9, 11] and occasionally in eukaryotes [12, 13]. In this study, we show that homologs of two large families of known virulence effector genes were likely acquired by Bd. These families have been rapidly duplicated after their acquisition in Bd and evolved under strong positive selection. We hypothesize that the acquisition of these virulence effector homologs may partly explain the evolution of chytridiomycosis.
Results and Discussion
Phylogenomic analyses identified homologs of two large families of known virulence effectors in Bd, including serine peptidases and CRN proteins (Table 1; Additional file 1). Of these, members of the serine peptidase family have been implicated as virulence effectors in Bd because of their potential role in degrading host antimicrobial peptides [14]. Previous microarray data also indicated that many serine peptidase genes in Bd are highly expressed in the sporangia stage, which is associated with the infection of keratinized host tissue [14]. Our analyses show that at least 32 serine peptidase genes are present in the Bd genome (Additional file 1). Other identifiable homologs of Bd serine peptidases are only found in bacteria, where they are more broadly distributed.
The CRN protein family consists of cytoplasmic virulence effectors only known in oomycete plant pathogens [15, 16]. These effectors are referred to as "crinkler" proteins because of their association with cell death and leaf crinkling, which parallels the effects of Bd on amphibian skin [17]. In oomycete pathogens, the N-terminal region of CRN proteins contains a highly conserved LFLAK domain that is characteristic of all CRN proteins. The C-terminal region is diverse in domain and motif structure, based on which 24 subfamilies were identified in the oomycete Phytophthora infestans [16]. The C-terminal region of CRN proteins also controls virulence and, when expressed in plant cells, may induce cell death [16]. Our analyses identified 84 genes encoding CRN protein homologs in Bd (Table 1). These genes are present in genomes of both Bd isolates (JAM81 and JEL423) and include eight of the 24 subfamilies of CRN proteins known in oomycetes (Table 2).
Compared to oomycetes, the number of genes in each subfamily is often higher in Bd. In particular, the DXX-DHA subfamily of CRN proteins is highly duplicated in Bd and contains 30 and 44 gene copies from the two respective genomes, many of which show very little sequence variation. The homology of CRN proteins from Bd and oomycetes is most obvious in the C-terminal effector region, with a sequence identity of up to 46.5%. The N-terminal LFLAK domain characteristic of oomycete CRN proteins also is largely conserved in some identified CRN homologs in Bd (Additional file 2). As in oomycete pathogens [16, 18], only a fraction of CRN homologs identified in Bd are predicted to contain signal peptides (Table 1). However, almost all other identified CRN homologs in Bd have short N-terminal extensions of about 20 amino acids in positions corresponding to signal peptides (Table 1 and Additional file 2).
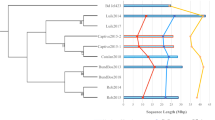
The observation that a gene is uniquely shared by distantly related organisms can be attributed to multiple possible scenarios, in particular differential loss and HGT [19, 20]. Although differential loss and HGT can always be invoked as alternative explanations to each other, each scenario becomes increasingly less likely if it requires considerably more corresponding (gene loss or HGT) events [19, 21]. In the cases of serine peptidases and CRN proteins, because chytrids and oomycetes belong to opisthokonts and heterokonts respectively, the gene loss scenario entails numerous independent loss events in related taxa. A more parsimonious and plausible scenario is the occurrence of HGT between Bd and oomycetes. To further investigate the number and the direction of potential HGT events involved, we performed phylogenetic analyses on serine peptidases and each of the eight CRN subfamilies shared between Bd and oomycetes. Our analyses indicate that all serine peptidase homologs in Bd form a weakly supported clade (Figure 1A). Given that serine peptidases are more widely distributed in bacteria, it is most likely that serine peptidases in Bd are derived from a single HGT event from bacteria, followed by gene duplication. Analyses of CRN subfamilies show that, in most cases, sequences from Bd form a distinct clade whereas those from oomycetes form another, suggesting independent HGT for each CRN subfamily. With paralogous sequence clades as outgroup, our analyses of the DN17 subfamily also show that Bd sequences were likely acquired from oomycetes, though the support is still modest (Figure 1B). Such low support is expected given the short length of sequences used in our phylogenetic analyses. Additional evidence consistent with the direction of HGT from oomycetes comes from their higher CRN gene diversity. The fewer CRN subfamilies in Bd (8 versus 24 in oomycetes) are in line with the transfer from oomycetes, as we would expect the recipient organism to receive a subset of the gene repertoire of the donor. Nevertheless, for both serine peptidases and the CRN family, we were unable to determine the timing of these HGT events because of the paucity of sequence data from other chytrids and oomycetes.

Molecular phylogenies of serine peptidases (A) and the DN17 subfamily of CRN proteins (B). Numbers above major branches show bootstrap values from maximum likelihood and distance analyses, respectively. The DN17 tree is rooted at the two distinct sequence copies (groups) in oomycetes. Genes from Bd, oomycetes and bacteria are indicated in different colors. P = Phytophthora; A = Aphanomyces.
Because both the serine peptidase and CRN families are highly duplicated in Bd, we reasoned that they may confer important functions and, therefore, investigated the possibility that strong selection has been involved in their evolution. The estimate of d N / d S for the serine peptidase family in Bd is 0.46, while this value is 0.01 in bacteria. Similarly, d N / d S for each of seven CRN subfamilies (with more than three sequences) is significantly higher in Bd than in oomycetes (Table 3). These results indicate that both the serine peptidase and CRN families evolved faster in Bd than in oomycetes and bacteria. We further used three likelihood tests (M2a vs. M1a, M8 vs. M7 and M8 vs. M8a) [22] to identify the role of positive selection on the serine peptidase family and seven CRN subfamilies in Bd and in oomycetes or bacteria, respectively. A priori we stipulated that evidence for positive selection will be sufficient only if all three tests performed show statistical significance. In total, all eight families and subfamilies in Bd show strong evidence of positive selection (Table 3; Additional file 3). In contrast, we found that only four subfamilies of CRN proteins (DX8, DXX-DXV, DXX-DHA, and DN17) evolved under positive selection in oomycetes. For the subfamilies that were influenced by positive selection in both Bd and oomycetes, the improved branch-site model [23] was used to identify positive selection specifically on the diverging Bd lineages. The genes in Bd were used as foreground branches and those in oomycetes were used as background. Except for the subfamily DXX-DHA, likelihood tests for the other three CRN subfamilies show statistically significant support for positive selection in the Bd branch (Additional file 3). Furthermore, multiple positively selected amino acid sites were identified using the Bayes Empirical Bayes (BEB) estimation procedure [24], suggesting that positive selection acted specifically on certain amino acid sites of the serine peptidase family and the CRN subfamilies after their acquisition by Bd.
Most genes of the serine peptidase family in Bd were assigned to two clades in the phylogeny with strong bootstrap support (Figure 1A). We estimated the coefficients of both type I (a site-specific shift in evolutionary rate) [25] and type II functional divergence (a cluster-specific shift of amino acid property) [26] between them. The results showed that only the type I coefficient of functional divergence is significantly greater than zero (θ I = 0.5744 ± 0.0589, LRT = 220.3270, p < 0.01). These results suggest that site-specific selective constraints have been altered for most of the members of this family, leading to group-specific functional evolution after their diversification. Furthermore, 26 amino acid residues were found to be critical for type I functional divergence with a posterior probability higher than 0.95 (Additional file 3), indicating that the evolutionary rates for these sites have shifted between these two clades. Some amino acid residues in the region of the Peptidase_S41 domain may also have contributed to the functional divergence, as there are 11 critical residues that are located in this region.
The acquisition of the serine peptidase and CRN families followed by rapid duplication and positive selection suggests that these acquired genes are likely important in the adaptation of Bd. Because both families are known virulence effectors, their presence in Bd provides a starting point for biochemical investigations of chytrid pathogenesis. If members of these two families are indeed related to Bd virulence, their acquisition may at least partly explain the emergence of chytridiomycosis. Peptidases are ubiquitous tools of bacterial parasites that enable host infection, and have also been identified as key factors in fungal pathogenesis. For example, Microsporum canis, a dermatophytic fungal parasite, infects keratinized tissue (e.g. skin and nails) in mammals. This fungus secretes dipeptidyl peptidases, which are homologous to serine peptidases of the S9 family in bacteria [27]. These and other proteases enable the fungus to obtain nutrients from the complex network of insoluble proteins that comprise keratin-rich tissues. As mentioned above, serine peptidases were identified as a class of proteins that are associated with virulence in Bd using microarray expression analyses [14]. The CRN genes were previously only known in oomycetes, which themselves also are important pathogens. Although most attention has been paid to oomycete plant pathogens, there is also a diverse group of oomycetes that infect animals [28]. Some of these pathogens are responsible for epidemics in aquatic animals such as crayfish [28]. The oomycete parasite Saproglenia may induce high mortality in salmon populations, both in aquaculture and in the wild [29], with effects similar to Bd [30]. Hence, the oomycete lineage is well adapted to parasitize vertebrate hosts. It is not surprising that oomycetes have an arsenal of genes associated with parasitism and that the acquisition of such genes may help convert a commensal chytrid to a parasitic pathogen.
Conclusions
Our data show that two large families of known virulence effector genes, CRN proteins and serine peptidases, were likely transferred to Bd from oomycete pathogens and bacteria, respectively. These acquired virulence effector homologs have been under rapid gene duplication and strong positive selection, suggesting that they may play an important role in the adaptation of Bd. Given that pathogenesis is the outcome of microbe-host interactions, the impact of any virulence effector is likely more severe when the host becomes stressed. We hypothesize that the acquisition of virulence effectors, in combination with habitat disruption and climate change, led to the Bd epidemics and the decline in amphibian populations. Further investigations are warranted to fully understand the biological functions of virulence effector homologs identified in our analyses.
Methods
Data source and genome screening for HGT candidates
Phylogenomic analyses used a customized database combining sequences from the NCBI non-redundant (nr) database, dbEST, the Taxonomically Broad EST Database (TBestDB) [31] and additional eukaryotic genomes. Annotated protein sequences of the heterokont Aureococcus anophagefferens, haptophyte Emiliania huxleyi, green algae Chlorella sp. and C. vulgaris, metazoans Daphnia pulex, Capitella sp. and Lottia gigantea were obtained from the Joint Genome Institute http://www.jgi.doe.gov. The annotated genome of the red alga Cyanidioschyzon merolae was downloaded from the Cyanidioschyzon merolae Genome Project http://merolae.biol.s.u-tokyo.ac.jp, and those of fungi Rhizopus oryzae, Allomyces macrogynus, and Spizellomyces punctatus were obtained from their sequencing projects at the Broad Institute of Harvard and MIT http://www.broadinstitute.org. Detailed sources for annotated protein sequences and ESTs of two Bd isolates and seven oomycetes used in our analyses are shown in Additional file 4. A comprehensive database was created using above data.
Genome screening for horizontally acquired genes was performed using AlienG [32]. AlienG is based on the observation that sequence similarity is correlated to sequence relatedness, where sequence similarity is measured by BLAST bit scores. A query sequence is considered to be a candidate of HGT-derived gene if it is significantly more similar to homologs from a potential donor (S d ) than to those from closely related taxa (S r ). Specifically, a bit score ratio S d /S r = 1.5 was used as threshold to identify candidates of HGT-derived genes in Bd. To eliminate potential artifacts, only candidates with expression evidence from ESTs or located in contigs containing more than 5 genes are retained for further analyses.
Identification of serine peptidase and CRN protein families in Bd and oomycetes
Phylogenomic analyses identified homologs of serine peptidases and CRN proteins among the HGT candidates. To retrieve other CRN and serine peptidase homologs in Bd, we first downloaded bacterial serine peptidases and annotated oomycete CRNs from both GenBank and oomycete genome projects [16, 18]; these downloaded sequences were then used as queries to search (BLASTP) against Bd JAM81 annotated protein sequences. All hits with E-values lower than 1 × 10-5 were retained. To classify the retrieved CRN homologs in Bd, we followed the criteria used for the oomycete P. infestans [16], which is based on the structures of C-terminal specific domains. As a result, eight subfamilies of CRN homologs were identified in Bd. To further estimate the copy numbers for homologs of serine peptidases and each CRN subfamily in Bd, we first constructed profiles for the Bd serine peptidase domain and the C-terminal specific domain regions of each CRN subfamily using HMMER [33]. The generated profiles were used to search the Bd genome, and sequences with E-values lower than 1 × 10-5 were retrieved and further aligned using conserved domain regions as references. Extremely divergent or partial sequences were removed from further analyses. The remaining genes were considered to be putative serine peptidases or CRN proteins. To exclude the possibility of contamination in Bd JAM81, the same process was also used to identify genes in the isolate Bd JEL423. Because the CRN protein family from four oomycetes (P. infestans, P. sojae, P. ramorum, and Pythium ultimum) have been well annotated [16, 18], we also used their C-terminal specific domain regions as references to identify CRN proteins in other oomycete datasets, including the P. capsici genome and ESTs for three other oomycetes (P. brassicae, P. parasitica, and Aphanomyces euteiches). Partial sequences were excluded and the remaining genes were regarded as CRN proteins. Secretion signals of serine peptidases and CRN proteins were identified using three tools (SignalP [34], TargetP [35], and iPSORT [36]) with default settings. The conserved N-terminal profiles were generated using WebLogo http://weblogo.berkeley.edu.
Phylogenetic analyses
Protein sequence alignment was carried out using MUSCLE [37] and clustalX [38], followed by visual inspection and manual refinement. Gaps and ambiguously aligned sites were removed manually. The most appropriate protein substitution matrix, rate heterogeneity and invariant sites were determined using ModelGenerator [39] for serine peptidases and each of the eight CRN subfamilies identified in Bd. Phylogenetic analyses were performed with a maximum likelihood method using PHYML [40] and a distance method using neighbor of PHYLIPNEW v.3.68 [41] in EMBOSS package [42]. Bootstrap support values were estimated using 100 pseudo-replicates. For distance analyses, maximum likelihood distances were calculated using TREE-PUZZLE v.5.2 [43] and PUZZLEBOOT v.1.03 (A. Roger and M. Holder, http://www.tree-puzzle.de. The models used in TREE-PUZZLE/BUZZLEBOOT to calculate the maximum likelihood distances were the same as those determined by ModelGenerator. All other parameters in the analyses used default settings.
Detection of positive selection
The d N /d S values for the serine peptidase family and seven CRN subfamilies (with more than three members in Bd) were estimated both in Bd and in oomycetes or bacteria, respectively. Genes with partial sequences were removed from the analyses. The program PAL2NAL [44] was used to convert protein sequence alignments into the corresponding codon-based DNA alignments. The program CODEML [22] was then used to calculate the d N /d S values.
Maximum likelihood analyses of selective constraints on the serine peptidase family and seven CRN subfamilies were performed using PAML [22] with their phylogenies and the codon alignments as input. For the serine peptidase family and each CRN subfamily, we firstly used the site-specific models [22, 45, 46] to identify positively selected codon sites in Bd and in oomycetes or bacteria, respectively. In this analysis, we compared models M1a vs. M2a, M7 vs. M8 and M8a vs. M8 to identify positive selection. To assess whether positive selection acted on the evolution of each group, the log likelihood values for each pair of nested models were compared using a likelihood ratio test (LRT). In the LRT, twice the log likelihood difference (2Δ/) between the two models was compared with the χ 2 statistics, with degrees of freedom (DF) equal to the difference of the number of parameters. The DFs were 3, 2, and 1 for comparisons M1a vs. M2a, M7 vs. M8 and M8a vs. M8, respectively. If the LRT was statistically significant and d N /d S estimate greater than 1, evidence for positive selection would be considered sufficient for the genes analyzed. Then, the Bayes Empirical Bayes method [24] was used to determine the amino acids most likely responsible for positive selection.
The improved branch-site models [23] were also used to search for positive selection for the serine peptidase family and each CRN subfamily. In these tests, we compared the null model A (model = 2, NSsites = 2, with ω fixed to 1) with the alternative model (model = 2, NSsites = 2). For each family or subfamily, the Bd branch on the phylogeny was used as foreground, while the oomycetes or bacteria branch used as background. When the LRT suggests positive selection under this model, the codon sites likely evolved under positive selection can be identified through posterior probability from Bayes Empirical Bays method [24].
Analysis of functional divergence
The program DIVERGE [47] was used to estimate the coefficients of both type I (θ I ) and type II (θ II ) functional divergence between serine peptidase clades A and B. Because subfamilies with fewer than four sequences cannot be analyzed using this method, three other serine peptidase proteins were excluded from this analysis. A likelihood ratio test was performed to test the null hypothesis θ = 0 using χ 2 distribution with 1 DF. A significant θ I indicates that site-specific altered selection constraints after the divergence of clades, while a significant θ II means site-specific shift of amino acid physiochemical property [25, 26].
References
Vié J-C, Hilton-Taylor C, Stuart SN: Wildlife in a Changing World - An Analysis of the 2008 IUCN Red List of Threatened Species. 2009, Gland, Switzerland: UCN
Longcore JE, Pessier AP, Nichols DK: Batrochochytrium dendrobatidis gen. et sp. nov., a chytrid pathogenic to amphibians. Mycologia. 1999, 219-227.
Fisher MC, Garner TW, Walker SF: Global emergence of Batrachochytrium dendrobatidis and amphibian chytridiomycosis in space, time, and host. Annu Rev Microbiol. 2009, 63: 291-310. 10.1146/annurev.micro.091208.073435.
James TY, Litvintseva AP, Vilgalys R, Morgan JA, Taylor JW, Fisher MC, Berger L, Weldon C, du Preez L, Longcore JE: Rapid global expansion of the fungal disease chytridiomycosis into declining and healthy amphibian populations. PLoS Pathog. 2009, 5: e1000458-10.1371/journal.ppat.1000458.
Skerratt LF, Berger L, Speare R, Cashings S, McDonald KR, Phillott AD, Hines HB, Kenyon N: Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. EcoHealth. 2007, 4: 125-134. 10.1007/s10393-007-0093-5.
Goka K, Yokoyama J, Une Y, Kuroki T, Suzuki K, Nakahara M, Kobayashi A, Inaba S, Mizutani T, Hyatt AD: Amphibian chytridiomycosis in Japan: distribution, haplotypes and possible route of entry into Japan. Mol Ecol. 2009, 18: 4757-4774. 10.1111/j.1365-294X.2009.04384.x.
Weldon C, du Preez LH, Hyatt AD, Muller R, Spears R: Origin of the amphibian chytrid fungus. Emerg Infect Dis. 2004, 10: 2100-2105.
Barinaga M: Where have all the froggies gone?. Science. 1990, 247: 1033-1034. 10.1126/science.247.4946.1033.
Ziebuhr W, Ohlsen K, Karch H, Korhonen T, Hacker J: Evolution of bacterial pathogenesis. Cell Mol Life Sci. 1999, 56: 719-728. 10.1007/s000180050018.
Gogarten JP, Doolittle WF, Lawrence JG: Prokaryotic evolution in light of gene transfer. Mol Biol Evol. 2002, 19: 2226-2238.
Ochman H, Lawrence JG, Groisman EA: Lateral gene transfer and the nature of bacterial innovation. Nature. 2000, 405: 299-304. 10.1038/35012500.
Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, Houterman PM, Kang S, Shim WB, Woloshuk C, Xie X, Xu JR, Antoniw J, Baker SE, Bluhm BH, Breakspear A, Brown DW, Butchko RA, Chapman S, Coulson R, Coutinho PM, Danchin EG, Diener A, Gale LR, Gardiner DM, Goff S, et al: Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 464: 367-373.
Friesen TL, Stukenbrock EH, Liu Z, Meinhardt S, Ling H, Faris JD, Rasmussen JB, Solomon PS, McDonald BA, Oliver RP: Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet. 2006, 38: 953-956. 10.1038/ng1839.
Rosenblum EB, Stajich JE, Maddox N, Eisen MB: Global gene expression profiles for life stages of the deadly amphibian pathogen Batrachochytrium dendrobatidis. Proc Natl Acad Sci USA. 2008, 105: 17034-17039. 10.1073/pnas.0804173105.
Torto TA, Li S, Styer A, Huitema E, Testa A, Gow NA, van West P, Kamoun S: EST mining and functional expression assays identify extracellular effector proteins from the plant pathogen Phytophthora. Genome Res. 2003, 13: 1675-1685. 10.1101/gr.910003.
Haas BJ, Kamoun S, Zody MC, Jiang RH, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, Bozkurt TO, Ah-Fong AM, Alvarado L, Anderson VL, Armstrong MR, Avrova A, Baxter L, Beynon J, Boevink PC, Bollmann SR, Bos JI, Bulone V, Cai G, Cakir C, Carrington JC, Chawner M, Conti L, Costanzo S, Ewan R, Fahlgren N, et al: Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009, 461: 393-398. 10.1038/nature08358.
Berger L, Speare R, Skerratt LF: Distribution of Batrachochytrium dendrobatidis and pathology in the skin of green tree frogs Litoria caerulea with severe chytridiomycosis. Dis Aquat Organ. 2005, 68: 65-70.
Levesque CA, Brouwer H, Cano L, Hamilton JP, Holt C, Huitema E, Raffaele S, Robideau GP, Thines M, Win J, Zerillo MM, Beakes GW, Boore JL, Busam D, Dumas B, Ferriera S, Fuerstenberg SI, Gachon CM, Gaulin E, Govers F, Grenville-Briggs L, Horner N, Hostetler J, Jiang RH, Johnson J, Krajaejun T, Lin H, Meijer HJ, Moore B, Morris P, et al: Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 11: R73-
Huang J, Gogarten JP: Ancient horizontal gene transfer can benefit phylogenetic reconstruction. Trends Genet. 2006, 22: 361-366. 10.1016/j.tig.2006.05.004.
Keeling PJ, Palmer JD: Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet. 2008, 9: 605-618. 10.1038/nrg2386.
Gogarten JP, Townsend JP: Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol. 2005, 3: 679-687. 10.1038/nrmicro1204.
Yang Z: PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007, 24: 1586-1591. 10.1093/molbev/msm088.
Zhang J, Nielsen R, Yang Z: Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005, 22: 2472-2479. 10.1093/molbev/msi237.
Yang Z, Wong WS, Nielsen R: Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005, 22: 1107-1118. 10.1093/molbev/msi097.
Gu X: Functional divergence in protein (family) sequence evolution. Genetica. 2003, 118: 133-141. 10.1023/A:1024197424306.
Gu X: A simple statistical method for estimating type-II (cluster-specific) functional divergence of protein sequences. Mol Biol Evol. 2006, 23: 1937-1945. 10.1093/molbev/msl056.
Vermout S, Baldo A, Tabart J, Losson B, Mignon B: Secreted dipeptidyl peptidases as potential virulence factors for Microsporum canis. FEMS Immunol Med Microbiol. 2008, 54: 299-308. 10.1111/j.1574-695X.2008.00479.x.
Phillips AJ, Anderson VL, Robertson EJ, Secombes CJ, van West P: New insights into animal pathogenic oomycetes. Trends Microbiol. 2008, 16: 13-19. 10.1016/j.tim.2007.10.013.
van West P: Saprolegnia parasitica, an oomycete pathogen with a fishy appetite: new challenges for an old problem. Mycologist. 2006, 20: 99-104. 10.1016/j.mycol.2006.06.004.
Kiesecker JM, Blaustein AR, Belden LK: Complex causes of amphibian population declines. Nature. 2001, 410: 681-684. 10.1038/35070552.
O'Brien EA, Koski LB, Zhang Y, Yang L, Wang E, Gray MW, Burger G, Lang BF: TBestDB: a taxonomically broad database of expressed sequence tags (ESTs). Nucleic Acids Res. 2007, 35: D445-451. 10.1093/nar/gkl770.
Tian J, Sun G, Ding Q, Huang J, Oruganti S, Xie B: AlienG: an effective computational tool for phylogenetic identification of horizontally transferred genes. The third International Conference on Bioinformatics and Computational Biology (BICoB): 23-25 March 2011; New Orleans, Louisiana. 2011, 13-18.
Eddy SR: Profile hidden Markov models. Bioinformatics. 1998, 14: 755-763. 10.1093/bioinformatics/14.9.755.
Bendtsen JD, Nielsen H, von Heijne G, Brunak S: Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004, 340: 783-795. 10.1016/j.jmb.2004.05.028.
Emanuelsson O, Brunak S, von Heijne G, Nielsen H: Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007, 2: 953-971. 10.1038/nprot.2007.131.
Bannai H, Tamada Y, Maruyama O, Nakai K, Miyano S: Extensive feature detection of N-terminal protein sorting signals. Bioinformatics. 2002, 18: 298-305. 10.1093/bioinformatics/18.2.298.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32: 1792-1797. 10.1093/nar/gkh340.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23: 2947-2948. 10.1093/bioinformatics/btm404.
Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McLnerney JO: Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol. 2006, 6: 29-10.1186/1471-2148-6-29.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Felsenstein J: PHYLIP (Phylogeny Inference Package) version 3.61. Distributed by the author. Book PHYLIP (Phylogeny Inference Package) version 3.61. Distributed by the author (Editor ed.^eds). City. 2005
Rice P, Longden I, Bleasby A: EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000, 16: 276-277. 10.1016/S0168-9525(00)02024-2.
Schmidt HA, Strimmer K, Vingron M, von Haeseler A: TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002, 18: 502-504. 10.1093/bioinformatics/18.3.502.
Suyama M, Torrents D, Bork P: PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34: W609-612. 10.1093/nar/gkl315.
Yang Z, Nielsen R, Goldman N, Pedersen AM: Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000, 155: 431-449.
Wong WS, Yang Z, Goldman N, Nielsen R: Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004, 168: 1041-1051. 10.1534/genetics.104.031153.
Gu X, Vander Velden K: DIVERGE: phylogeny-based analysis for functional-structural divergence of a protein family. Bioinformatics. 2002, 18: 500-501. 10.1093/bioinformatics/18.3.500.
Acknowledgements
This work is supported in part by a NSF Assembling the Tree of Life (ATOL) grant (DEB 0830024).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JH designed the study and wrote the manuscript. GS and ZY performed the analyses and participated in manuscript writing. SK and KS wrote the manuscript and contributed to data analyses. All authors read and approved the final manuscript.
Guiling Sun, Zefeng Yang contributed equally to this work.
Electronic supplementary material
12862_2011_1821_MOESM1_ESM.PDF
Additional file 1: Gene structures and subcellular localizations of serine peptidases in Bd. This file contains information about gene identifiers, numbers of exon, and predicted subcellular localizations for serine peptidases that were identified in Bd JAM 81. (PDF 75 KB)
12862_2011_1821_MOESM2_ESM.PDF
Additional file 2: LFLAK domain of the CRN protein family in oomycetes and Bd. This file shows that the LFLAX domain of the CRN protein family is largely conserved between oomycetes and Bd. (PDF 122 KB)
12862_2011_1821_MOESM3_ESM.DOC
Additional file 3: Results of selection analyses. This file contains results of positive selection analyses using site-specific and branch-site models, respectively. It also shows amino acid residues that are critical to functional divergence between serine peptidase clades A and B. (DOC 4 MB)
12862_2011_1821_MOESM4_ESM.PDF
Additional file 4: Data sources for Bd isolates and oomycetes used in the analyses. This file shows data types and download sites for two Bd isolates and seven oomycetes used in the analyses. (PDF 62 KB)
12862_2011_1821_MOESM5_ESM.PDF
Additional file 5: Two different conserved N-terminal domains of Bd CRN proteins. This file shows two different types of N-terminal domain for Bd CRN proteins. These domain types are also indicated in Table 1. (PDF 179 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sun, G., Yang, Z., Kosch, T. et al. Evidence for acquisition of virulence effectors in pathogenic chytrids. BMC Evol Biol 11, 195 (2011). https://doi.org/10.1186/1471-2148-11-195
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-11-195