
Abstract
Background
Divergence within cis-regulatory sequences may contribute to the adaptive evolution of gene expression, but functional alleles in these regions are difficult to identify without abundant genomic resources. Among African cichlid fishes, the differential expression of seven opsin genes has produced adaptive differences in visual sensitivity. Quantitative genetic analysis suggests that cis-regulatory alleles near the SWS2-LWS opsins may contribute to this variation. Here, we sequence BACs containing the opsin genes of two cichlids, Oreochromis niloticus and Metriaclima zebra. We use phylogenetic footprinting and shadowing to examine divergence in conserved non-coding elements, promoter sequences, and 3'-UTRs surrounding each opsin in search of candidate cis-regulatory sequences that influence cichlid opsin expression.
Results
We identified 20 conserved non-coding elements surrounding the opsins of cichlids and other teleosts, including one known enhancer and a retinal microRNA. Most conserved elements contained computationally-predicted binding sites that correspond to transcription factors that function in vertebrate opsin expression; O. niloticus and M. zebra were significantly divergent in two of these. Similarly, we found a large number of relevant transcription factor binding sites within each opsin's proximal promoter, and identified five opsins that were considerably divergent in both expression and the number of transcription factor binding sites shared between O. niloticus and M. zebra. We also found several microRNA target sites within the 3'-UTR of each opsin, including two 3'-UTRs that differ significantly between O. niloticus and M. zebra. Finally, we examined interspecific divergence among 18 phenotypically diverse cichlids from Lake Malawi for one conserved non-coding element, two 3'-UTRs, and five opsin proximal promoters. We found that all regions were highly conserved with some evidence of CRX transcription factor binding site turnover. We also found three SNPs within two opsin promoters and one non-coding element that had weak association with cichlid opsin expression.
Conclusions
This study is the first to systematically search the opsins of cichlids for putative cis-regulatory sequences. Although many putative regulatory regions are highly conserved across a large number of phenotypically diverse cichlids, we found at least nine divergent sequences that could contribute to opsin expression differences in cis and stand out as candidates for future functional analyses.
Similar content being viewed by others
Background
Adaptive phenotypic evolution may result either from protein-coding mutations that modify the structure and function of genes, or from regulatory mutations that alter the timing, location, or expression of genes [1–3]. Although examples of protein-coding mutations that contribute to phenotypic evolution are well known (e.g., [4–6]), examples of regulatory mutations that also affect phenotypic adaptation are less well known, but no less important (e.g., [7–9]). One class of regulatory mutations, cis-regulatory mutations, are found in close proximity to the genes they regulate and function by altering the binding of transcription factors necessary for gene expression. Cis-regulatory mutations exhibit several features that make them ideally suited for adaptive phenotypic evolution, including codominance [10] and modularity [8]. These features make cis-regulatory mutations efficient targets for natural selection [11] and limit the negative consequences of pleiotropy that presumably affect many trans-regulatory and protein-coding mutations. Finally, since cis-regulatory mutations may underlie many of the adaptive and disease phenotypes found in nature, identifying these alleles remains an important goal of evolutionary genetics. However, identifying cis-regulatory mutations can be challenging without abundant functional genomic resources, since the transcription factor binding sites (TFBS) they affect are small, lack strict conservation, and are found in difficult-to-annotate regions of the genome [2, 3].
The location of cis-regulatory sequences can be near-to or far-from the genes they regulate. Promoter sequences found directly upstream of genes can harbor cis-regulatory alleles [12, 13], as can enhancer or repressor elements located many kilobases away [14, 15]. Cis-regulatory sequences can even reside within the untranslated regions (UTRs) of genes, where they alter the binding of microRNAs (miRNAs) that regulate gene expression following transcription [16, 17]. But where ever their location, two methods commonly used to identify cis-regulatory sequences and alleles are phylogenetic footprinting and phylogenetic shadowing [18]. In phylogenetic footprinting, one compares DNA surrounding some gene(s) of interest among numerous divergent taxa in hopes of identifying non-coding regions that are highly conserved. By the very nature of their conservation, these conserved non-coding elements (CNEs) stand out as candidate regulatory sequences, since conservation is often used to indicate function. Once candidate regulatory sequences have been identified via phylogenetic footprinting, the method used to identify putative cis-regulatory alleles within them is differential phylogenetic footprinting, or phylogenetic shadowing [18, 19]. In phylogenetic shadowing, one compares putative regulatory sequences among closely related taxa in hopes of identifying sequence polymorphisms correlated with the divergent expression of some target gene(s). Following their application, functional genomic analyses are necessary to validate the function of any candidate sequences or alleles identified by the phylogenetic footprinting and shadowing methods; but even by themselves, both methods can provide valuable insights into the location of potential cis-regulatory sequences and the transcription factors that bind them.
The goal of this study is to identify candidate cis-regulatory sequences that control opsin gene expression in African cichlid fishes. Opsins are a group of G protein-coupled receptors that confer sensitivity to light and mediate color vision [20]. African cichlids comprise a diverse clade of freshwater, teleost fish found throughout the lakes and rivers of Africa, including the three African Great Lakes, Lakes Tanganyika, Malawi, and Victoria [21, 22]. Cichlids from Lakes Tanganyika and Malawi exhibit dramatic variation in their sensitivity to colored light [23–25]. Species from these lakes exhibit retinal sensitivities that are maximally sensitive to short, middle, or long-wavelength spectra; in some cases, closely related species can differ in their maximal retinal sensitivity by over 100 nm [25–27]. This striking variation makes the cichlid visual system one of the most diverse vertebrate visual systems so far identified. Most variation in cichlid color sensitivity is due to changes in the regulation of their cone opsin genes [26, 27]. Cichlids have seven cone opsin genes used for color vision; these opsins are SWS1 (ultraviolet-sensitive), SWS2B (violet-sensitive), SWS2A (blue-sensitive), RH2B (blue-green-sensitive), RH2A and RH2A (green-sensitive), and LWS (red-sensitive) [28]. Additionally, these opsins are located in three regions of the cichlid genome: SWS1 is found on cichlid linkage group (LG) 17; RH2B, RH2A and RH2A are found together in a tandem array on LG 5; and SWS2A, SWS2B, and LWS form a second tandem array on LG 5 (Lee et al. 2005) (Figure 1). Among different cichlid species, these opsins are alternatively co-expressed in three predominant groups, or palettes, to produce the three common visual pigment sets: SWS1-RH2B-RH2A (short wavelength-sensitive), SWS2B-RH2B-RH2A (middle wavelength-sensitive), and SWS2A-RH2A-LWS (long wavelength-sensitive) [26]. Cichlids exhibit several correlations between the expression of their opsins and important ecological variables, including foraging preference and ambient light intensity [26, 27]. These correlations suggest that opsin gene expression varies adaptively in cichlids, especially since some expression-ecology correlations have evolved independently among cichlids in different lakes [27]. A recent quantitative genetic analysis of opsin expression in two Lake Malawi cichlids found a quantitative trait locus (QTL) located near the opsin genes [29]. The proximity of this QTL to the opsins suggests that mutations within one or more cis-regulatory sequences may contribute to variation in cichlid opsin expression. But like many non-model systems, few genomic resources are currently available for cichlids, making it difficult to identify potential cis-regulatory alleles and test their association with opsin gene expression.

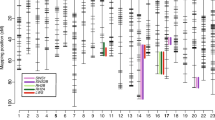
Conservation between O. niloticus opsin-containing BAC regions and four fish genomes. A) SWS1 opsin-containing region. B) SWS2-LWS opsin-containing region. C) RH2 opsin-containing region. Top line represents O. niloticus BAC sequence. Conserved non-coding elements (CNEs) are numbered and highlighted in red; repetitive sequences are highlighted in green; promoter sequences later examined for interspecific polymorphism are highlighted in blue.
Here, we sequence and analyze bacterial artificial chromosome (BAC) clones containing the opsin genes of two African cichlid species, Oreochromis niloticus [30] and Metriaclima zebra [31]. Oreochromis niloticus (the Nile tilapia) is a riverine cichlid that expresses the long wavelength-sensitive opsin palette as adults but also expresses the other palettes as fry and juveniles [32]. O. niloticus is an outgroup to the diverse haplochromine cichlids endemic to Lakes Tanganyika, Malawi, and Victoria. Metriaclima zebra (the 'classic' Zebra cichlid) is one such haplochromine cichlid found in Lake Malawi. M. zebra expresses the short wavelength-sensitive opsin palette as an adult and during all developmental stages [32]. Both species last shared a common ancestor ~ 18 MYA, whereas M. zebra diverged from other phenotypically diverse Lake Malawi cichlids less than 2 MYA [33]. After sequencing the opsin-containing BAC clones from these species, we used the resulting sequences for several analyses, including:
-
(1)
Annotation and comparison of the opsin-containing regions from the genome assemblies of several model teleosts. We perform phylogenetic footprinting by comparing the opsin-containing regions of O. niloticus and several model fish genomes. We use this comparison to locate conserved non-coding elements (CNEs) that serve as candidate cis-regulatory sequences for the opsins.
-
(2)
Computational prediction of binding sites for 12 transcription factors important for vertebrate opsin expression [34–41] (Table 1). We perform this search in each CNE as well as within the proximal promoter of each opsin. We also perform an analogous search for miRNA target sites within the 3'-UTR of each opsin.
-
(3)
Phylogenetic shadowing between O. niloticus and M. zebra using the TFBS and miRNA target site profiles found in each CNE, promoter, and 3'-UTR sequence. In each region we compare the proportion of divergent TFBS/miRNA target sites with the amount expected given the over-all sequence divergence of the opsin BACs and introns (a measure of neutral evolutionary divergence [42, 43]). These comparisons are used to identify putative cis-regulatory sequences that have undergone significant evolutionary divergence among African cichlids.
-
(4)
Following phylogenetic shadowing, we re-sequence the most divergent regions in a panel of 18 phenotypically diverse cichlids from Lake Malawi. We search these sequences for polymorphisms that may indicate the presence of cis-regulatory alleles. This final analysis allows us to determine whether the divergent regions we identify between O. niloticus and M. zebra also contain polymorphisms correlated with opsin expression in the more closely related cichlids of Lake Malawi.
We use the final results of this study to examine which regulatory regions are most likely to contain functional regulatory alleles that determine opsin expression in African cichlids. We find that many non-coding regions are highly conserved between O. niloticus and M. zebra, as well as among the closely related cichlids of Lake Malawi. However, we find at least two CNEs, five proximal promoters, and two 3'-UTRs that exhibit significant divergence in the number and type of TFBS and miRNA targets found between O. niloticus and M. zebra. We also identify at least three alleles that are weakly associated with SWS2A, RH2B, and LWS expression - three opsins that show strong differential expression among cichlid species. These results suggest that cis-regulatory sequences may contribute to opsin expression differences among African cichlids, and provide numerous candidates for future functional studies.
Results and Discussion
BAC Sequencing and Analysis
BAC identification, sequencing, assembly, and comparison
Within the cichlid genome, the opsins are found in three separate tandem arrays. SWS1 is found alone on cichlid linkage group (LG) 17; SWS2A, SWS2B, and LWS are found together in a tandem array on LG 5 [44]; and RH2B, RH2Aα, and RH2Aβ are found in a second tandem array on LG 5 approximately 30 cM from the SWS2-LWS array (KL Carleton, unpublished data) [44]. We identified opsin-containing BAC clones for O. niloticus by PCR screening [30] and for M. zebra by filter hybridization [31]. We then shotgun sequenced each clone using ABI Sanger or 454 Life Sciences technology. Clone IDs, estimated sizes, sequencing methods, assembly statistics, final contig length, and GenBank accession numbers for resulting contigs are listed in Table 2. The average read length for ABI-generated sequences was ~700 bp, while the average read length for 454-generated sequences was ~110 bp. For the O. niloticus SWS1-containing clone, we used a combination of ABI and 454 sequences since the assemblies based on ABI-generated reads alone were poor. For all other clones, we used additional Sanger reads to fill in the gaps and join all contigs into their final BAC assemblies (Table 2). Overall, the final assemblies of each clone based on ABI and 454 technology joined an average of 85% of reads into a single contig that was within 10 - 40 kb of the estimated clone size (Table 2). All assemblies successfully covered the opsin-containing regions in O. niloticus and M. zebra.
We aligned each BAC assembly from O. niloticus and M. zebra and found them to be highly similar. The only significant difference was a 6.1 kb insertion in the M. zebra RH2-containing BAC, located between the RH2Aα, and RH2Aβ opsins (Additional file 1). This insertion is likely a transposon. The average pairwise Jukes-Cantor-corrected sequence divergence (Dxy) across each BAC assembly was 8.4% (± 3.1% s.e.). This rate of sequence divergence is consistent with comparisons of other genes between these species, and it is one of the first large-scale estimates of sequence divergence between O. niloticus and M. zebra. We then subdivided each BAC assembly into opsin protein-coding (CDS) and intronic (INT) sequences. For O. niloticus and M. zebra, the mean Dxy across all opsin CDS was 3.8% (± 0.3%), while the divergence across all INT was 9.5% (± 1.9%). (We excluded both the first intron as well as the first and last six bases of each intron since these regions may contain regulatory sequences and splice sites that are more highly conserved than other intronic regions [43]). Comparison of the average Dxy across all regions reveals that the mean divergence of the functionally important opsin CDS is significantly lower than Dxy across either the BACs or INT sequences (t-tests: CDS vs. BAC, t8, 0.05 = 2.60, p = 0.032; CDS vs. INT, t27, 0.05 = 2.17, p = 0.039), but that Dxy between BAC and INT sequences do not differ (t23, 0.05 = 0.08, p = 0.935). In addition to evaluating which regions of each opsin-containing BAC retain the highest conservation and are most likely to be functional, these divergence estimates also provide an important null hypothesis for our subsequent analyses using phylogenetic shadowing: in general, we expect O. niloticus and M. zebra to share (e.g, exhibit orthology in) ~92% of their TFBS and miRNA target sites, and exhibit divergence in ~8%. Divergence in greater than 8% of the TFBS and miRNA target sites identified may indicate significant cis-regulatory sequence evolution in the regions examined.
BAC annotation and the opsin repertoire of teleost fishes
In order to perform phylogenetic footprinting across the opsin arrays of cichlids, we first investigated the synteny of each opsin array of O. niloticus relative to several model fish species using PipMaker [45] and MultiPipMaker [46]. We found considerable synteny in the opsin-containing regions among O. niloticus (tilapia), Gasterosteus aculeatus (stickleback), Oryzias latipes (medaka), Tetraodon nigroviridis (tetraodon), and Danio rerio (zebrafish) (Figure 1; Additional file 2A). The clearest example of this synteny was the SWS2-LWS opsin array. This array is flanked by the genes HCFC1 and GNL3L and is essentially co-linear in all five fish genomes (Figure 1; see Additional file 3 for the position and orientation of flanking genes). We found evidence for a localized duplication of the SWS2 opsins in O. latipes and O. niloticus, since both these species have two adjacent SWS2 opsin genes (Additional file 4). Closely related Poeciliid fishes also possess adjacent SWS2 paralogs [47], suggesting that this duplication event probably occurred at least 153 - 113 MYA at the base of the Acanthopterygii [48, 49].
In contrast to the SWS2-LWS array, we observed considerable variation in opsin gene content for the RH2 opsins. O. niloticus and M. zebra possess three RH2 genes while D. rerio has four [50, 51], G. aculeatus has two, and T. nigrovirdis has one functional RH2 opsin and one RH2 pseudogene [52]. We therefore used phylogenetic analyses to investigate the orthology of the RH2 and SWS2 genes among these fishes and found that most RH2 duplications are species-specific [53] (Additional File 4). Thus, synteny in the region containing the RH2 opsin array was lower than in the SWS2-LWS array, but was still largely co-linear between O. niloticus, G. aculeatus, and T. nigroviridis (Additional file 2B). The genes SLC6A13-like and SYNPR flank the RH2 opsins in these fishes (Figure 1; Additional file 3).
Synteny in the region surrounding the SWS1 opsin was difficult to assess due to species-specific deletions and poor genome assembly. The T. nigrovirdis genome assembly lacks the SWS1 opsin altogether, and this region is found within an unordered chromosome or ultracontig in both the G. aculeatus and O. latipes genomes. For G. aculeatus, we found a small 92 kb region containing the SWS1 opsin that was collinear with the O. niloticus BAC sequence, but which contained one large inversion. For O. latipes, we found an even smaller 60 kb region that was syntenic for only 11 kb surrounding the SWS1 opsin. Synteny with D. rerio was also generally low (Additional file 2C). Therefore, despite the lack of SWS1 duplicates compared to the SWS2 or RH2 opsins, the SWS1 region is still poorly assembled in the existing annotations of several teleost genomes, potentially complicating direct comparisons of synteny in this region. In these species, the SWS1 opsin appears to be flanked by the genes TNPO3 and CALUA (Figure 1; Additional file 3).
Analysis of Conserved Non-Coding Elements (CNEs)
Phylogenetic footprinting to identify CNEs
We used MultiPipMaker [46] to highlight non-coding elements surrounding each opsin gene array from O. niloticus to D. rerio, representing nearly 300 MY of fish evolution [49]. The resulting plots illustrate at least 20 conserved non-coding elements (CNEs) surrounding the opsin gene arrays of O. niloticus and the other fish species examined (red bars in Figure 1). We also found six regions of putatively high conservation that are largely composed of repetitive sequence (green bars in Figure 1), which we did not analyze further. The conservation of these CNEs over several million years of fish evolution suggests that they contain functionally important regulatory modules necessary for gene expression.
At least one CNE we identified through phylogenetic footprinting is orthologous to other vertebrate cis-regulatory sequences. CNE 7 (highlighted in Figure 1 and located between the SWS2B and LWS opsins) consists of two non-contiguous regions of high conservation in pufferfish, stickleback, medaka, swordtails, and cichlids [47] (Figure 1). The first region, CNE 7a, was also identified following a comparative analysis of opsin-containing BACs from swordtails (Xiphophorus helleri) [47]. Through BLAST and mirbase [54], we found that CNE 7a is most similar to zebrafish miRNA dre-miR-726 (score 173.3, e-value = 0.006), and the same genomic region from zebrafish is identical to this miRNA (Figure 2). Dre-miR-726 is expressed in the retina of larval and adult zebrafish [55]. Since many miRNAs are transcribed along with the genes they regulate, the proximity of miR-726 to the SWS2 and LWS opsins suggests that it could play a role in opsin regulation. The ~90 bp CNE encoding mir-726 is conserved in numerous other taxa as well, including additional fishes, frogs, and lizards [47, 56].

Alignment of two putative LWS opsin regulatory elements (CNE 7a and b) in fishes. A) Alignment of CNE 7a from five fish genomes to dre-miR-726. This region is highly similar among all fish species examined. Black box indicates the mature microRNA sequence. B) Alignment of CNE 7b from five fish genomes to the human LWS-LCR. These sequences show regions of high similarity between humans and fishes. Asterisks (*) indicate positions that are identical among all taxa; colons (:) indicate positions that are identical among four out of five taxa. Boxes highlight conserved transcription factor binding sites.
The second highly conserved region, CNE 7b, is positionally and structurally orthologous to the mammalian LWS locus control region (LWS-LCR; Figure 2B) [47, 56, 57]. This enhancer is located ~3.8 kb upstream of the LWS opsin in O. niloticus and other vertebrates, including humans. The LWS-LCR is hypothesized to enhance LWS expression in eutherian mammals by looping and binding to the LWS proximal promoter [57–59]. Wang et al. [59] demonstrated that the human ortholog of this sequence can function as an enhancer of both LWS and MWS opsin expression in mice. Additionally, a recent study of LWS regulation in zebrafish also identified a similar sequence at this position that modulates LWS expression in that species, which they named the LWS activating region (LAR) [60]. Comparison of the mammalian LWS-LCR, the zebrafish LAR, and CNE 7b from cichlids and other teleosts reveals a high degree of sequence similarity among these regions (Figure 2B). In Figure 2B, we also highlight several conserved transcription factor binding sites common to each sequence, including sites for CRX, THR, and AP-1 (Figure 2B; see also Table 1). Thus, our results demonstrate the effectiveness of the phylogenetic footprinting method for identifying functional cis-regulatory sequences necessary for vertebrate opsin expression. It is therefore possible that the remainder of the CNEs we identify also encode cis-regulatory sequences necessary for the correct spatial and developmental expression of the opsins in cichlids.
We note that our present study focuses on small regions of high conservation within a ~30 kb window of non-coding sequence surrounding the opsin arrays, but that cis-regulatory sequences may often reside tens or hundreds of kilobases from the genes they regulate. However, two recent analyses of general transcription factor binding sites found that functional binding sites generally cluster in regions 1 kb around the proximal promoter of each gene [61, 62]. This observation suggests that a focused study of conserved elements within or near the opsins is a reasonable strategy for this initial study. A FASTA file of all CNE sequences from O. niloticus and M. zebra is provided in Additional file 5.
TFBS search and phylogenetic shadowing of CNEs
We compared the 20 CNEs identified between O. niloticus and M. zebra and found many to be highly conserved; however, we found no identifiable orthologs between O. niloticus and M. zebra for CNEs 6 or 19. For the remaining CNEs, the average pairwise sequence divergence between O. niloticus and M. zebra was 4.2% (± 0.5%), which is significantly less than the mean Dxy of introns (9.5%, t-test: t38, 0.05 = 2.99, p = 0.005). This result suggests that the conserved non-coding regions identified among O. niloticus and other fishes have remained conserved among African cichlids as well.
We used the Transcription Element Search System [63] to computationally search all orthologous CNEs for binding sites corresponding to twelve transcription factors that have been associated with opsin expression in fishes and other vertebrates including thyroid hormone and retinoic acid receptors [34–37, 39, 41, 64, 65]. A complete list of these transcription factors and their associated opsins is presented in Table 1. We found computationally-predicted binding sites for these functionally important transcription factors in all but one of the CNEs surveyed (Table 3; see Additional file 6 for detailed counts of all TFBS). Only CNE 10 lacked binding sites for any of the twelve transcription factors in either species examined. Within the remaining sequences we found binding sites for all twelve transcription factors except PNR and RXRγ. After relaxing our matching criteria, we still failed to find binding sites for these two transcription factors (data not shown). In both O. niloticus and M. zebra, binding sites for AP-1 and CRX were extremely abundant, although binding sites for each of three retinoic acid receptors (RARs) and THRβ were also common (Additional file 6). We found several CNEs with a high density of transcription factor binding sites given the total sequence length surveyed - generally 9 TFBS or more (see Additional file 6). For O. niloticus these high-density CNEs are CNEs 2, 3, 13, 15, 19, and 20, and for M. zebra these are CNEs 2, 8, 11, 13, 15, and 20. Due to their potential enrichment for functional TFBSs relative to other CNEs, we believe these eight CNEs represent the most likely candidates for functional cis-regulators of opsin expression in fishes.
Consistent with the high similarity of their sequences, the results of our TFBS search differed very little between O. niloticus and M. zebra. We used exact binomial tests to compare the proportion of shared and divergent TFBSs observed between O. niloticus and M. zebra to the null ratio of 92:8 (see above). Treating each TFBS independently, we counted each non-orthologous or divergent TFBS as a success, each orthologous or shared TFBS as a failure, then tested the hypothesis that the true probability of success (proportion of divergent TFBS, Pdiv) was > 8%. Of 17 testable CNEs, we found that O. niloticus and M. zebra differed significantly from this null expectation at four CNEs: CNEs 3, 4, 15, and 18 (Table 3). After Bonferroni correction for multiple comparisons, however, only the results for CNE 3 remained significant (exact binomial test: divergent TFBS = 7, total TFBS = 8, Pdiv = 87.5%, p < 0.001). Overall, these results did not change when we used the mean divergence of introns from each CNE's nearest down-stream opsin as a null hypothesis, except that O. niloticus and M. zebra also exhibited significant divergence at CNE 4 (divergent TFBS = 2, total TFBS = 2, Pdiv = 100.0%, p = 0.001). Both CNE 3 and 4 are located upstream of the SWS2A opsin. For CNE 3, O. niloticus has 8 TFBS while M. zebra has only one; for CNE 4, M. zebra has two while O. niloticus has none. These results are consistent with what one might expect based on the expression of these opsins in adults, since SWS2A is highly expressed among O. niloticus adults, but is not expressed in M. zebra [32]. Thus, we show that O. niloticus and M. zebra have diverged significantly in the identity of their TFBS profiles for two putative cis-regulatory elements (CNEs 3 and 4), and differ in the presence/absence of two more (CNEs 6 and 19). Three of these CNEs (3, 4, and 6) are found upstream of the SWS2A opsin (Figure 1). These results offer the compelling possibility that at least some of the differences in opsin expression observed between O. niloticus and M. zebra could be due to divergence in the TFBS profiles of CNEs surrounding their opsins.
We acknowledge that our use of the overall proportion of divergent TFBS (Pdiv) to detect CNEs that have undergone significant cis-regulatory divergence ignores many nuances of TFBS evolution, such as the overall number and kind of TFBS present in each CNE and species. But because of the small number of TFBSs found within each CNE (the average number of TFBSs found in each CNE was 5.9), it is difficult to perform robust tests of divergence in the number of binding sites for individual transcription factors. Therefore, we have summed all TFBSs into orthologous (shared) and non-orthologous (divergent) groups in order to perform phylogenetic shadowing between O. niloticus and M. zebra. However, even within these broad categories, we have only enough power that CNEs with Pdiv > 25% stand out as statistical outliers, and only those with Pdiv > 80% remain significant after correction for multiple comparisons. In the future we aim to perform more nuanced, sequence-based tests of cis-regulatory divergence in cichlids. We present these tests for cis-regulatory divergence as a first step in this process.
Analysis of Proximal Promoter regions
Phylogenetic footprinting of opsin proximal promoters
The MultiPip plots shown in Figure 1 reveal 20 CNEs upstream of the opsins, but also show several regions of high conservation within the 5' proximal promoter of multiple opsins as well. In particular, SWS2A, SWS2B, and LWS all exhibit regions of high conservation in the first 1 kb of sequence upstream of their translation start site (TSS). For the LWS opsin, this region of conservation spans nearly 0.7 kb of the proximal promoter in multiple fish species, including G. aculeatus, O. latipes, and T. nigroviridis (Figure 1B). RH2A and RH2A also exhibit some small regions of high conservation just upstream of their TSSs, which probably reflect the 5'-UTR region. Additionally, the promoter upstream of RH2B also contains some conserved regions of repetitive sequence (Figure 1C). It is compelling that many of the opsins exhibit strong conservation of sequences within 1 kb of their TSSs, which we use to define the proximal promoter, because the true promoter regions for these genes are unknown in cichlids. However, important cis-regulatory sequences have been identified in close proximity to the opsin genes in other fish species. In particular, several CRX transcription factor binding sites found within 500 bp of the SWS2 opsin regulate the expression of this gene in D. rerio [41]. Therefore, the conservation we observe upstream of the SWS2A, SWS2B, and LWS opsins may indicate the presence of additional cis-regulatory sequences within the proximal promoters of these genes as well. A FASTA file of all opsin and non-opsin promoter sequences (see below) from O. niloticus and M. zebra is presented in Additional file 5.
TFBS search and phylogenetic shadowing of opsin proximal promoters
The distribution and number of TFBSs found within the proximal promoter region of each opsin was similar to those found in the CNEs. Within each opsin's proximal promoter, we found that AP-1 and CRX binding sites were nearly ubiquitous (Figure 3). Binding sites for NFκB, RARα, RARβ, RXRβ and THRβ were also common, and we once again found no binding sites for PNR and RXRγ. The absence of binding sites for PNR and RXRγ in both the CNEs and promoters may rule-out these factors as candidate trans-regulators of cichlid opsin expression differences; however the lack of these factors could also be due to biases in the way TESS identifies binding sites. Interestingly, we found several CRX binding sites directly upstream of the SWS2A and SWS2B opsins (Figure 3). These binding sites could potentially function as regulators of SWS2 opsin expression in cichlids as they do in zebrafish [41].

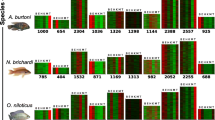
Transcription factor binding site diversity within opsin proximal promoters. A - G) Distribution of ten transcription factor binding sites (TFBS) in the proximal promoters of each opsin in O. niloticus and M. zebra. TFBS labelled in blue are present in O. niloticus only, those in red are present in M. zebra only, and those in black are found in both species. Sites labelled simply RAR correspond to all three retinoic acid paralogs. The orientation of factors above or below the central reference line has no special meaning, although O. niloticus-only sites are generally above the line, and M. zebra-only sites are below it. H) Comparison of the average number of binding sites for each transcription factor in the proximal promoters of the opsins and seven randomly-selected, non-opsin genes in O. niloticus. On average, the opsins contain significantly greater numbers of binding sites for these transcription factors compared to the non-opsin genes.
Pairwise sequence divergence in the proximal promoter regions was greater than for the other regions examined. The average Dxy of the proximal promoters was 10.2% (± 3.2%), which differed significantly from the mean of CNEs (4.2%, t-test: t23, 0.05 = 2.48, p = 0.021), but not the introns (9.5%, t-test: t27, 0.05 = 0.14, p = 0.89). This result suggests that the opsin promoter regions of cichlids may exhibit greater divergence in putative cis-regulatory sequences than the CNEs. Indeed, we found that O. niloticus and M. zebra exhibited significant divergence in their TFBS profiles for six of the seven proximal promoters examined (Figure 3); however, following correction for multiple hypothesis testing, only five of these remained significant: SWS1, SWS2A, RH2B, RH2A and RH2A (Figure 3; see also Table 3). O. niloticus and M. zebra differ dramatically in the expression of each of these genes [32], suggesting that their divergent transcription factor profiles could explain these differences. A comparison of which TFBS differ between O. niloticus and M. zebra reveals a slight over-representation of CRX sites in O. niloticus (17 vs. 7), and of THRα sites in M. zebra (4 vs. 0) (Figure 3).
Using phylogenetic shadowing, we identified five cichlid opsins with promoter sequences that exhibit significant divergence in their binding site profiles for 12 transcription factors. We note, however, that by focusing on only these TFBSs, we potentially miss many interesting patterns of divergence in transcription factors that have not already been associated with vertebrate opsin expression. A comprehensive search of all TFBSs identified by TESS could potentially pick up these missed patterns, but such a search would be extremely cumbersome and subject to many false positives [66]. Because of their small size, TFBS motifs are likely to appear throughout the genome frequently by chance, and it is difficult to determine which are likely to be functional based on sequence matches alone. Therefore, we opted to focus on genes that are obvious candidates for analysis.
We performed an additional analysis to determine the relevance of these twelve candidate factors by comparing the number of TFBSs found for each factor within the proximal promoters of the opsins and seven randomly chosen non-opsin genes. We hypothesized that if these candidates are relevant to the control of opsin expression in cichlids, then we should find a significantly greater number of TFBSs for each factor upstream of the opsin genes compared to the non-opsin genes. Indeed, we found that the opsins contain a greater number of binding sites for eight out of ten factors compared to the randomly-chosen non-opsin genes (Wilcoxon paired signed-rank test: V = 50, p = 0.0124; Figure 3H). The non-opsin promoters contained higher mean numbers of TFBSs for AP-1 and THRα only. This result suggests that the proximal promoters of the opsins are significantly enriched for the binding sites of transcription factors that influence vertebrate opsin expression. This enrichment also suggests that polymorphisms in these regions could conceivably lead to functional differences in transcription factor binding and opsin expression. However, we note that we found no significant correlation between distance matrices of the opsins based on these TFBS profiles and their expression in developing O. niloticus (data not shown). This additional result suggests that, although binding sites for many of these candidate transcription factors may be over-represented in the promoters of opsins, they do not predict which opsins are co-expressed in African cichlids.
The search parameters we have chosen aim to identify TFBSs with high confidence while still accounting for the observation that many transcription factors exhibit degenerate binding of DNA motifs [67, 68], and can bind these motifs in an orientation-independent manner [69, 70]. We are currently performing a quantitative genetic analysis of many markers located across the genome in order to identify other loci and transcription factors that may be associated with cichlid opsin expression. This quantitative genetic analysis should provide an unbiased search for additional transcription factors that may influence cichlid opsin expression.
Analysis of opsin 3'-UTRs
Phylogenetic footprinting of opsin 3'-UTRs
In addition to mutations within conserved non-coding elements and 5' promoter regions, polymorphisms within 3'-UTRs can also act as cis-regulatory alleles [16, 17]. These polymorphisms affect gene expression by altering the binding of miRNAs in a manner analogous to how mutations within TFBS can alter gene expression, except that miRNAs inhibit gene expression post-transcriptionally. Our phylogenetic footprinting analysis reveals that every opsin exhibits some conservation of the 50 - 100 bp region found directly downstream of the opsin coding sequences (Figure 1). Generally, this conservation is strongest between O. niloticus, O. latipes, and G. aculeatus, reflecting the close phylogenetic relationship among these species. For RH2A, the 3' conserved region extends nearly 700 bp past the end of the coding region. Initially, these results suggest that the opsin 3'-UTRs of cichlids will be highly conserved, reflecting the strong evolutionary constraint on UTR sequence and function seen in both flies and humans [16, 71]. However, a recent survey of polymorphisms affecting miRNA target sites in cichlids found that the 3'-UTR of some genes may in fact be under divergent selection in African cichlids [72]. Therefore, we searched the 3'-UTRs of the opsins for target sites corresponding to known fish miRNAs.
Of the 30 known miRNA targets we searched for in cichlids (see below), we found at least one target site in each opsin 3'-UTR that was conserved among cichlids and other teleosts (Table 4). Many of these conserved sites are expressed within the retina of vertebrates and play a role in retinal development [73–76]. For example, dre-miR-217, dre-miR-181a, and dre-miR-23b are all integral to the development and maintenance of the zebrafish retina [77–79], while dre-miR-96 and dre-miR-182a are sensory organ-specific [76]. Only one conserved site that was found in cichlids and other teleosts differed between O. niloticus and M. zebra. A target for dre-miR-722, found downstream of the LWS opsin in O. niloticus and the pufferfish (Takifugu rubripes), is missing in the orthologous 3'-UTR from M. zebra due to a single nucleotide polymorphism (SNP). However, the two conserved target sites for dre-miR-722 and dre-miR-728 are both found within the 3'-UTRs of several Lake Victorian cichlids (data not shown). Like O. niloticus, Lake Victoria's cichlids express the long wavelength opsin palette as adults [80], possibly indicating that these factors play a role in LWS expression. If we interpret evolutionary conservation as an indication of function, we believe the conserved sites listed in Table 4 represent those miRNA target sites that are most likely to regulate opsin expression in African cichlids. The sequences of all O. niloticus and M. zebra opsin 3'-UTRs are available in Additional file 5.
microRNA target search and phylogenetic shadowing of opsin 3'-UTRs
We searched the 3'-UTRs of each opsin in O. niloticus and M. zebra for target sites corresponding to known fish miRNAs [54]. In all, we identified 84 predicted target sites matching 30 known miRNAs from cichlids and D. rerio (Additional file 7). Like the CNEs and promoter regions analyzed earlier, all 3'-UTR sequences generally exhibited high similarity between O. niloticus and M. zebra. The average pairwise divergence (Dxy) for O. niloticus and M. zebra 3'-UTRs was 5.2% (± 1% s.e.). This small level of divergence is very similar to the level observed for opsin coding sequences, though it did not differ from the average Dxy of introns (9.5%, t-test: t27, 0.05 = 1.33, p = 0.196). Consequently, the results of our miRNA target search were once again very similar for O. niloticus and M. zebra, especially for those sites conserved in other fishes as well (Additional file 7; Table 4, see above). However, we still found that O. niloticus and M. zebra differed significantly in the proportion of divergent and shared miRNA target sites for the 3'-UTRs of the RH2B and SWS2B opsins (exact binomial tests: RH2B, divergent miRNA sites = 4, total miRNA sites = 8, Pdiv = 50.0%, p = 0.002; SWS2B, divergent miRNA sites = 4, total miRNA sites = 5, Pdiv = 80.0%, p < 0.001; see Table 3). These results did not change when we altered the null hypothesis to reflect the divergence of each opsin's intronic sequence (data not shown). For RH2B, we found that M. zebra exhibited four unique target sites for miRs-101, 144, 196, and 2184. For SWS2B, M. zebra had unique targets for miRNAs-194 and 23, while O. niloticus had targets for miRNAs -92 and 137. RH2B is strongly differentially expressed in these two species, while SWS2B is only expressed in some adults of O. niloticus [81]. Thus, we not only identified at least 8 conserved--and perhaps core--miRNA target sites in the 3'-UTR of each cichlid opsin (Table 4), we also found that O. niloticus and M. zebra are significantly divergent in at least two of these regions (SWS2B and RH2B).
It is important to note that most miRNA target sites we identified in the 3'-UTRs of the cichlid opsins correspond to miRNAs that are expressed in the vertebrate retina (Additional file 7). Of sites corresponding to 30 different miRNAs, 22 (73%) correspond to miRNAs expressed within the retinas of fish, mammals, or amphibians (Additional file 7). Notably, however, we did not find any miRNA target sites that correspond to miR-726, the miRNA found upstream of the LWS opsin and encoded by CNE 7a (see Figure 2). Further, many of the conserved and non-conserved miRNA target sites we identify also correspond to miRNAs associated with retinal development (for example, dre-miRs 23, 92, 722, and 194) [76, 82, 83], and miR-129 is also associated with retinoblastoma in humans [84]. Given that O. niloticus and M. zebra differ dramatically in their developmental patterns of opsin gene expression, it is interesting to speculate that these miRNAs could contribute to the developmental differences in opsin expression observed between these and other African cichlid species [32, 85]
In the present study we have focused on miRNA target sites found within the 3'-UTR of the cichlid opsins, but miRNA cleavage of messenger RNAs by binding to sites within core messenger RNA sequences has also been demonstrated in humans and plants [86, 87]. It is still not clear whether miRNAs regulate gene expression more often by binding to the 3'-UTR or messenger RNA sequence, although a review by Bartel [88] suggested that translational repression by binding to UTR sequences is more prominent. Finally, we note also that the cellular machinery cannot distinguish between functional and non-functional miRNA target sites based on their evolutionary conservation in other species, as we do here [88] (see Table 4). However, given that scans for miRNA target sites can have a high rate of false positives, evolutionary conservation is currently the best way to avoid high error rates and to infer function. The fact that we identified a high percentage of target sites that correspond to miRNAs found within the vertebrate eye suggests that many of these sites are not false-positives; therefore, it is plausible that they may actually function to regulate opsin expression in cichlids. In the future we will determine whether these and other miRNAs are actually expressed in the retinas of African cichlids. If so, then heterologous reporter assays could be used to verify what role divergence in miRNA target sites may play in the evolution of cichlid opsin expression [89, 90].
Phylogenetic shadowing among the cichlids of Lake Malawi
Resequencing and analysis of LWS-LCR, opsin promoters, and 3'-UTRs
Two broad goals of this study have been to (1) identify potential cis-regulatory sequences surrounding the opsin gene arrays of African cichlids (phylogenetic footprinting), and (2) identify those sequences whose divergence may explain patterns of differential opsin gene expression among African cichlids (phylogenetic shadowing). For both goals we have relied on sequenced BAC clones of Oreochromis niloticus and Metriaclima zebra--two species that have BAC libraries available, but that also differ dramatically in their evolutionary age (~18 MY [33]) and adult and developmental patterns of opsin expression [32]. Therefore, as a final goal, we wanted to determine whether the candidate cis-regulatory sites we identified via phylogenetic shadowing also vary among a more closely related (~2 MY [33]) panel of 18 cichlid species from Lake Malawi. Although much more closely related to M. zebra than O. niloticus, adults of these species exhibit the same opsin expression patterns as adult and juvenile O. niloticus [23, 24, 26]. Our panel included one individual from six species for each of the three adult opsin expression palettes observed among Lake Malawi's cichlids (short-, middle-, and long-wavelength sensitive) (see Additional file 8 for a list of the species used). The regions we re-sequenced included the proximal promoters upstream of the SWS1, SWS2A, SWS2B, RH2B, and LWS opsins (highlighted in blue in Figure 1), the LWS-LCR (CNE 7), and the 3'-UTRs of the SWS2B and LWS opsins. After sequencing, we examined these regions for levels of interspecific polymorphism and performed a test of association for cis-regulatory alleles.
Although the 18 Lake Malawi cichlid species we use have been previously characterized with regard to opsin gene expression, we confirmed these gene expression results by measuring the expression of each opsin in all species via RT-qPCR (see Additional File 8 for opsin expression results). These expression results were highly concordant with previous measurements [26]. Following qPCR, we re-sequenced the entire 1 kb region upstream of both the SWS1 and SWS2A opsins, 956 bp upstream of the LWS opsin, 951 bp upstream of the RH2B opsin, and 694 bp upstream of the SWS2B opsin. We also re-sequenced 900 bp surrounding the LWS-LCR (CNE 7) and 450 bp downstream of the SWS2B and LWS opsins. As expected given the young age of Lake Malawi cichlids, we found that all regions were highly conserved among the species sampled. Overall, we identified fewer than 15 single nucleotide polymorphisms (SNPs) and insertion/deletions (indels) per region examined (Table 5). In each case, most SNPs were found in only one individual. Other diversity statistics--including the total number of segregating sites (S), total number of singletons (s), number of haplotypes (H), nucleotide diversity (π), sequence conservation (C), and Tajima's D (TD)--also indicate low levels of polymorphism, despite our use of alternate species and genera as sampling units (see Additional file 8 for a list of all polymorphisms found among the 18 species sampled). Nevertheless, following a sliding window analysis of nucleotide diversity (π) and minor allele frequency (MAF), we were able to identify several peaks of relatively highπ and MAF within each region (Figure 4). These peaks correspond to SNPs and indels segregating at high frequency within the species and genera sampled, and therefore represent potential cis-regulatory alleles.

Interspecific polymorphism in eight putative cis -regulatory regions from 18 Lake Malawi cichlid species. A - H) Minor allele frequency (MAF; in red) and nucleotide diversity (π; in black) calculated in a sliding window across the proximal promoter regions of five opsins (A - E), CNE 7 (LWS-LCR) (F), and two opsin 3'-UTRs (G - H) using 18 Lake Malawi cichlid species. Numbers above peaks of MAF and π denote the position of SNPs analyzed for allelic-association with opsin expression (see Table 6); asterisks (*) denote polymorphisms that interrupt CRX binding sites.
Several peaks of relatively high nucleotide diversity and MAF correspond to polymorphisms within predicted CRX binding sites, but none correspond to any other TFBS or miRNA target sites (Table 5). Specifically, two peaks of π and MAF located -217 and -224 bp upstream of the SWS2A translation start site (TSS) correspond to a single SNP and 8 bp indel that both disrupt putative CRX binding sites. The 8 bp indel located at SWS2A-217 completely eliminates the CRX binding site in several species (Additional file 8). We identified at least three other polymorphisms upstream of the SWS1 and RH2B opsins that also disrupt CRX binding sites--each present in only a single species--but no polymorphisms that interrupt the binding sites of any other candidate transcription factors. One peak of nucleotide diversity at position 183 of CNE 7 (see Additional file 5 for this sequence) corresponds to a SNP within the miRNA miR-726; however, this mutation does not occur within the mature miRNA sequence. Finally, we found only three polymorphisms total within the 3'-UTR of both the SWS2B and LWS opsins (Figure 4; Additional file 8), none of which interrupted predicted miRNA target sites. The polymorphism that segregates between O. niloticus and M. zebra within the LWS 3'-UTR was fixed in all Lake Malawi cichlid species (see Table 4). Thus, few of the polymorphisms we identify in the putative cis-regulatory sequences of the opsins are predicted to alter opsin expression among 18 Lake Malawi cichlid species. However, since mutations within transcription factor binding sites have been shown to alter gene expression [91], our results suggest that polymorphisms within CRX TFBSs could contribute to the differential patterns of SWS2A expression observed among Lake Malawi cichlids.
Association between polymorphisms and cichlid opsin expression
To test this hypothesis, we performed allelic association tests between these and other SNPs underlying peaks of nucleotide diversity and high MAF (see Figure 4) with the expression of their nearest downstream opsin (Table 6). Three polymorphisms (SWS2A-217, RH2B-161, CNE 7-570) exhibited significant or marginally non-significant associations with the expression of their downstream opsins (Table 6); however only RH2B-161 is significant following Bonferroni correction for multiple comparisons (t-tests: RH2B-161: t17, 0.05 = 3.447, p = 0.0036). Despite this limitation, we believe these preliminary results are compelling since all three polymorphisms occur on the same linkage group (LG 5) believed to contain a cis-regulatory element that modulates cichlid opsin expression, and all three are associated with opsins whose expression is significantly associated with this QTL in African cichlids [29]. SWS2A-217 obliterates a CRX binding site in numerous cichlids, and polymorphisms affecting CRX binding sites have been shown to modulate SWS2 opsin expression in zebrafish [41]. CNE 7-570 is found very near the LWS-LCR and could potentially affect LCR binding. It is therefore possible that all three alleles acts as, or are linked to, cis-regulatory elements that modulate opsin expression in cichlids.
We acknowledge that the sample sizes we use for phylogenetic shadowing among Lake Malawi's cichlids are small and at best provide a weak test for cis-regulatory alleles associated with opsin expression. Additionally, we use cross species and genera comparisons for an analysis that is generally based on individual variation within populations. However, Lake Malawi cichlids are extremely similar at the genetic level and share many ancestral polymorphisms [92]. For this reason, genetic analyses across cichlid species are analogous to within-species polymorphism studies in other vertebrates, such as chimps and humans [72, 92]. Additionally, recent work in cichlids has successfully used cross-species comparisons to fine-map cis-regulatory alleles underlying pigmentation differences, so long as these differences have a common origin among the different species sampled [93]. It is hard to predict which traits will have a common origin among different African cichlid species, as previous work [94] suggested that the pigmentation trait mapped in Roberts et al. [93] had evolved several times. Our recent work reconstructing the evolution of opsin regulatory changes in cichlids revealed that the three opsin expression palettes have evolved repeatedly among cichlids in Lakes Tanganyika and Malawi [27], but it is still unclear whether or not the three palettes have a common origin among Lake Malawi's cichlids. But despite our small sample size, we have found some evidence of binding site turnover in CRX binding sites within the 5' promoters of Lake Malawi cichlids, but no evidence of turnover in other candidates TFBS or miRNA target sites. Additionally, we also identified three putative cis-regulatory polymorphisms associated with SWS2A, RH2B, and LWS opsin expression. Although very preliminary, these results offer compelling candidates for additional functional and association analyses between more closely related cichlid populations and species.
The search for cis-regulatory sequences
Cis-regulatory sequences may reside many kilobases away from the genes they regulate, as in the case of enhancer or repressor elements; or they may be found very near their genetic targets, as in the case of promoter elements and UTRs. Given this diversity, is it possible to predict which non-coding regions are most likely to contain functional cis-regulatory alleles? If we accept estimates of pairwise sequence divergence (Dxy) as indicative of those regions most likely to contain functional opsin regulatory alleles, then our estimates of Dxy between O. niloticus and M. zebra suggest that the proximal promoter regions are most likely to contain cis-regulatory alleles that alter opsin expression (Figure 5A; see also Additional file 9 for a list of Dxy values for every region examined). The opsin promoters exhibit the highest levels of pairwise sequence divergence of all coding and non-coding regions examined, and also contain more sequences with divergent TFBS profiles (Figure 3; Table 3), and putative regulatory alleles (Table 6). However, this conclusion is undoubtedly influenced by what could be a naive choice of promoter sequences (the true functional opsin promoter regions have not yet been identified in cichlids and may be more highly conserved), increased length of the promoter sequence relative to other regions analyzed (we analyzed 1 kb for each promoter versus ~ 400 bp for each CNE and UTR), and the increased power to detect significant divergence from null expectations afforded by the large number of TFBS found within the proximal promoters (we found ~ 22 TFBS within each promoter versus ~ 6 TFBS/miRNA target sites within each CNE and UTR).

Divergence among coding and non-coding regions in O. niloticus and M. zebra BAC sequences. A) Pairwise sequence divergence (Dxy) between O.niloticus and M. zebra for different coding and non-coding regions of the opsin-containing BACs. Average Jukes-Cantor-corrected Dxy is higher among 5' proximal promoter regions for each opsin. B) Venn diagram of proportion of shared and divergent TFBS and microRNA target sites among non-coding regions examined in this study. Opsin promoter regions exhibit slightly elevated proportions of divergent sites compared to either CNEs or 3'-UTRs. C) Comparison of proportion divergent TFBS/miRNA target sites (Pdiv) and pairwise sequence divergence (Dxy). Non-coding sequences with elevated Pdiv do not necessarily exhibit increased Dxy, even among proximal promoters regions. Filled points are those sequences with Pdiv values that differ significantly from 8% (see also Table 3).
If the overall proportion of divergent TFBS/miRNA target sites (Pdiv) is used to identify those non-coding regions most likely to contain functional cis-regulatory alleles, the proximal promoter regions still exhibit the highest proportion of divergent regulatory regions, although the advantage is only slight. Only about 55% of TFBS are shared between O. niloticus and M. zebra promoters, while 45% are divergent (Figure 5B). In contrast, the CNEs and 3'-UTRs exhibit lower (and very similar) proportions of shared versus divergent TFBS/miRNA target sites (~ 67% shared and ~ 33% divergent; Figure 5B). In this case, it is difficult to confidently conclude that 5' promoter regions are more likely to contain functional alleles that regulate opsin expression, although the data are suggestive. When both pairwise divergence and the proportion divergent TFBS/miRNA target sites are taken into account, we find that regions that exhibit statistically significant divergence are not necessarily those regions that exhibit greater pairwise sequence divergence (Figure 5C). In fact, the regions with the highest Pdiv also exhibit some of the lowest Dxy values. This result suggests that the increased number of statistically divergent promoter regions we observe is not a function of sequence divergence, but rather increased statistical power afforded by the greater length of the sequences surveyed and the greater number of TFBS found.
Additionally, our results show that the majority of the non-coding regions examined exhibit Pdiv values near 37%, with a median of 30% (Figure 5C). This observation suggests that the 8% divergence criterion we used as null model for evolutionary divergence is likely too low and also suggests that our power for many regions was inadequate due to the small number of TFBS or miRNA target sites identified (see above). But even when a more liberal null divergence value of 30% is used, our results largely remain consistent: O. niloticus and M. zebra still exhibit significant divergence in their TFBS and miRNA target profiles for CNEs 3 and 4 (located near the SWS2A opsin), the proximal promoters for RH2B and SWS1, and the 3'-UTR for SWS2B (p < 0.05; see Table 3 for Pdiv values).
Finally, we note that many putative regulatory regions identified in our opsin-containing BACS are highly conserved among many phenotypically diverse cichlid species from Lake Malawi, as well as between the ~18 MY divergent Oreochromis niloticus and Metriaclima zebra. This conservation suggests that trans-acting factors may also play an important role in generating evolutionary changes in cichlid opsin expression. For example, in both yeast and humans, interspecific differences in gene expression are primarily the result of trans-regulatory factors [95, 96]. And although cis-regulatory alleles contribute more to interspecific differences in gene expression among several Drosophila species, trans-acting alleles generally contribute to these differences as well [97]. Coding mutations within trans-acting transcription factors can act in a modular fashion, thereby mitigating negative pleiotropic effects [98], and these mutations may still affect gene expression even when the sites they bind remain conserved [99], as many of the TFBSs we examine are. Also, in addition to the putative cis-regulatory factors associated with SWS2B, SWS2A, and RH2B opsin expression in cichlids, Carleton et al. [29] also identified one trans-acting locus in the same cross, as well as another trans-acting locus in a separate cross. These two loci, located on cichlid LGs 13 and 4, do not occur in linkage with the cichlid opsins and explain a higher portion of the variance in opsin expression than the single cis-associated factor on LG 5 [29]. Whether these sites represent transcription factors, miRNAs, or other trans-acting binding sites is unknown, but several good candidate genes are located in these regions. Future work will aim to map and characterize these putative trans-regulatory regions in a variety of cichlid taxa.
Conclusions
Mutations within cis-regulatory regions are compelling candidates for the adaptive evolution of gene expression [3]. Here we generated and surveyed non-coding sequences surrounding the opsin gene arrays of two African cichlids, Oreochromis niloticus and Metriaclima zebra. This study is the first to systematically survey the cichlid opsins for putative cis-regulatory sequences, and our results suggest that these regions could potentially contribute to variation in cichlid opsin expression. The results of our study reveal:
-
(1)
The presence of numerous conserved non-coding elements located up- and downstream of the opsins that may function as regulators of cichlid opsin expression, including a retinal miRNA and one known opsin enhancer (LWS-LCR). African cichlids were divergent in two of these (CNEs 3 and 4, both located upstream of the SWS2A opsin).
-
(2)
Significant divergence and enrichment of transcription factor binding sites within the proximal promoter of five of the seven opsins (SWS1, SWS2A, RH2B, RH2A, and RH2A).
-
(3)
Numerous target sites for retinal and sensory organ-specific miRNAs within the 3'-UTR of each opsin. African cichlids were divergent in two of their opsin 3'-UTRs (SWS2B and RH2B).
-
(4)
The presence of several candidate cis-regulatory alleles located within the promoters of the RH2B and SWS2A opsins, as well as one near the LWS-LCR (CNE 7).
Future work will aim to further characterize these candidate cis-regulatory sequences, as well as to identify candidate trans-acting alleles. Given that spectral sensitivity and opsin expression in vertebrates can be influenced by coding mutations [26, 80, 100], trans-regulatory mutations [29], cis-regulatory mutations [41], and possibly miRNAs as well, cichlids may represent an ideal system in which to examine how these various molecular mechanisms interact to influence the evolution of visual system diversity in vertebrates.
Methods
Sequencing and assembly of BAC clones
We isolated clones containing the opsin genes from BAC libraries of two African cichlids, Oreochromis niloticus [30] and Metriaclima zebra [31]. For O. niloticus, we used PCR to screen pooled clones from the T3 and T4 libraries [30]. Primers used for these screens were: SWS1 (F: TACCTGCAGGCTGCCTTTAT; R: CTCGCATGGAGGCTAAGAAC), RH2A (F: GCAGACCCGATCTTCTTCAA; R: AGCAGACGTGATTGTGATGG), and LWS (F: TCCTGTGCTACCTTGCTGTG; R: ACAACGACCATCCTGGAGAC). We first chose 10 super-pools, each covering 10% of the entire 35,000 pooled clones, and screened them for opsin-positive plates. We then screened row and column pools from the plates with positive results to identify the exact clones containing the opsins. Fingerprinted contigs (FPCs) corresponding to the positive clones were identified and all clones in the contig were PCR tested for the opsins (see Additional file 10). Contig geometries were confirmed by end sequencing the BACs, designing primers, and PCR testing (Additional file 10). Based on the resulting alignments, one clone for each opsin array was selected for sequencing.
DNA from the selected clones was prepared using the Qiagen® MaxiPrep Plasmid Purification kit following the manufacturer's protocols. The O. niloticus clones were sent to the Joint Genome Institute (JGI) for ABI-Sanger sequencing. Shotgun libraries were prepared and 4 × 384-well plates were sequenced using ABI technology in both forward and reverse directions. The resulting reads were base-called and assembled with phred [101] and phrap [102]. Additional reads for the SWS1-containing clone were generated using 454 Life Sciences technology [103]. We performed two different sequencing runs for this clone, assembled them into contigs, and combined them with the JGI ABI reads in Sequencher v4.9 (Gene Codes Corporation, Inc.). This resulted in several large but non-overlapping contigs. To finish joining these contigs we used BLAST [104] and Pipmaker [45] to identify and align the largest contigs to orthologous genomic regions from the genomes of other teleost fish (for an example see Additional File 2). Based on these alignments we designed PCR primers to sequence across the gaps to join the contigs.
For M. zebra we screened high-density BAC array filters using filter hybridization [31]. This search utilized PCR probes generated from M. zebra retinal cDNAs that were labeled using the ECL Nucleic Acid Labelling and Detection Kit (Amersham Biosciences). We obtained three clones from these arrays and confirmed that they contained the opsins via PCR as detailed above. DNA for these clones was prepared using the Qiagen® MaxiPrep kit following the manufacture's protocols. BAC clones were sized by pulsed field gel electrophoresis following digestion with NotI. We then sent the purified, sized samples to 454 Life Sciences (Branford, CT) for sequencing on the GS20. We performed two sequencing runs on the SWS1 and LWS-containing clones, but only one for the clone containing RH2A. All runs utilized a quarter plate. Due to the length of the 454 reads, the resulting sequences formed more, but smaller contigs relative to O. niloticus. To finish joining these contigs we aligned the largest (> 5 kb) contigs to the finished O. niloticus BAC sequences in Sequencher v4.9 and once again designed PCR primers to sequence across the gaps. We annotated the BAC sequences for both O. niloticus and M. zebra using BLAST [104].
Finally, we performed a global alignment of each BAC from O. niloticus and M. zebra in the program wgVISTA [105]. We measured sequence similarity and divergence across each BAC using the phylip program dnadist, implemented in the Mobyle online bioinformatics server [106]. When measuring pairwise sequence divergence (Dxy), we used the Jukes-Cantor nucleotide model to correct for multiple hits. We repeated these measurements for each of the CNEs, promoter regions, and 3'-UTRs. We compared Dxy among each of these regions and the entire BAC sequences using t-tests implemented in the statistical software package R v2.10.0 [107]. Prior to performing all tests, we transformed the Dxy scores by log10 in order to meet the assumption of normality of errors.
Phylogenetic analyses
We generated phylogenies of the teleost RH2 and SWS2 opsins in order to identify orthologous opsins among the focal fish genomes examined. We accessed all relevant opsin sequences from the genome assemblies listed above via BLAT. We aligned both opsin data sets using the E-INS-i strategy of the multiple alignment program MAFFT v6.0 [108] and then chose an appropriate model of nucleotide substitution via the program jModelTest v0.1.1 [109]. This model was TIM3ef+G for both the RH2 and SWS2 alignments. We then used this model and the corresponding parameters estimated by jModelTest to generate Neighbor-Joining trees for the opsins with Maximum Likelihood-corrected distances. For the RH2/SWS2 datasets, these parameters included the nucleotide substitution rate matrix (A-C: 0.601/0.617; A-G: 1.470/1.734; A-T: 1.00/1.00; C-G: 0.601/0.617; C-T: 2.729/2.877; G-T: 0.599/0.155) and the shape of the gamma distribution (0.507/0.577). We measured the nodal support of these trees with 1000 bootstrap replicates. We rooted both trees using the LWS-1 opsin of zebrafish.
Identification of conserved non-coding elements
We used phylogenetic footprinting [18] to identify putative cis-regulatory elements by searching for conserved non-coding elements (CNEs) surrounding the opsin gene arrays. To do this, we identified 100-300 kb regions of orthology between the O. niloticus BAC sequences and the genome assemblies of four teleost fishes using BLAT and the UCSC genome browser. The additional genomes were stickleback (Gasterosteus aculeatus, Broad Institute v1.0, February 2006), medaka (Oryzias latipes, National Institute of Genetics and the University of Tokyo v1.0, October 2005), pufferfish (Tetraodon nigroviridis, Geoscope and Broad Institute v7, February 2004), and zebrafish (Danio rerio, Trust Sanger Institute zv8, December 2008). We then determined the location of known opsin genes and examined synteny across these regions via DOT plots generated in the program PipMaker [45] (for an example see Additional File 2). Regions of high synteny surrounding the opsins were then identified using MultiPipMaker [46]. We defined a CNE as any region ≥ 50 bp long that was conserved (> 60% sequence identity) between Oreochromis niloticus and at least one other teleost species (Oryzias latipes, Gasterosteus aculeatus, and Tetraodon nigroviridis). In each case, we attempted to analyze as many CNEs as possible, but acknowledge that some small regions may have been missed.
Profiling of transcription factor binding sites and Phylogenetic shadowing
We identified binding sites within each CNE as well as the proximal promoters located approximately 1 kb upstream of each opsin's translation start site using the Transcription Element Search System, TESS v6.0 [63]. We altered the default search parameters of TESS by changing the minimum log-likelihood ratio score from 12 to 9. We then limited our search results to high quality matches by accepting only those hits that met three criteria: (1) a log-likelihood (La) score ≥ 9.0, (2) a ratio of the actual log-likelihood score to the maximum possible log-likelihood (Lq) score ≥ 80%, and (3) a probability value for the log-likelihood score (Lpv) < 0.05. Although TESS can potentially identify binding sites for many different transcription factors, we were primarily interested in those factors that have been shown to influence opsin expression in fish and other vertebrates (Table 1). Following the automated search in TESS, we manually searched the lists for duplicate sites at each position, and removed them prior to further analysis.
For phylogenetic shadowing, we analyzed the number of shared and divergent transcription factor binding sites found in each CNE and opsin proximal promoter from O. niloticus and M. zebra. We counted the total number of binding sites orthologous in both species, as well as those that were found in only one species or the other. We calculated the proportion of divergent TFBSs (Pdiv) as (D/(D+S))*100, where D is the number of divergent TFBS and S is the number of shared sites. We compared the observed proportion of divergent sites to the null proportion suggested by the global sequence similarity of the O. niloticus and M. zebra BACs (92% versus 8%). We tested the independence between these observed and expected proportions using exact binomial tests [110] implemented in the R statistical software package. To control the Type I error rate for each region examined, we calculated Bonferroni-corrected p-values for all tests in R. For phylogenetic shadowing between O. niloticus and M. zebra, the corrected significance threshold was α = 0.05/31 = 0.0016.
Finally, we also compared the average number of binding sites for each transcription factor between the proximal promoters of the O. niloticus opsins and seven randomly chosen, non-opsin genes from a draft assembly of the O. niloticus genome (available at http://www.BouillaBase.org; accessed October 2010). These genes were ACTG1, AMPD3, DHCR7, ENSGAC000000020282, IGFALS, KCNJ9, and REEP1. Proximal promoters from these randomly chosen sequences were identified based on comparison of the O. niloticus genes with orthologous regions from the stickleback genome. Comparison of the average number of binding sites across all opsins and transcription factors was performed using a Wilcoxon paired signed-rank test computed in R.
Comparison of opsin expression and TF binding site profiles
We evaluated the correlation between the transcription factor binding sites in the proximal promoter of each opsin and the expression of each opsin among developing O. niloticus fry using Mantel's test of two distance matrices. We generated Euclidean distance matrices of the total number of binding sites for 12 transcription factors within the proximal promoter region of each opsin as well as the percent of total opsin expression from developing O. niloticus fry, reported in Carleton et al. [32]. We calculated Mantel's test using the 'mantel.randtest' function from the R package ade4 [111]. Approximate p-values were calculated following 500 randomizations of each matrix. All transcription factor numbers and expression values were standardized prior to clustering. We also expanded this analysis to the entire proximal promoter region after calculating a sequence similarity matrix for the entire proximal promoter using the phylip program dnadist.
Profiling of microRNAs target sites
We searched the 3'-UTRs of each opsin for binding sites matching the target seed of known miRNAs (miRNA) via the SeedMatch algorithm previously used to identify miRNA targets in cichlid UTRs [72]. This algorithm is similar to the TargetScanS algorithm used in other studies to identify miRNA targets [112]. Briefly, non-redundant fish miRNA targets were obtained from miRBase (http://www.mirbase.org[54]; accessed June 2010) and supplemented with several miRNA target sequences identified in cichlids [72]. We searched each opsin 3'-UTR--defined as the ~500 bp region between the transcription end site and the polyadenylation site (AATAAA)--for sequences matching the seeds of miRNAs from this non-redundant library. In order to account for the high rate of false-positives generated by simply searching for matching seed sites, we aligned the 3'-UTR of each cichlid opsin with those from G. aculeatus, O. latipes, T. nigrovirdis, the Japanese pufferfish (Tetrapdon rubripes), and D. rerio in order to identify sites that were conserved across multiple fish species. For this purpose we defined the first 1 kb of sequence downstream of these latter species' opsins as the 3'-UTR and aligned these to the cichlid sequences with MLagan [113]. To account for errors in the alignment of orthologous 3'-UTRs, we counted as conserved the same miRNA target site found within 50 bp of each other across species. For cichlid opsins that lacked orthologs in the other species, we used the nearest paralog (see Additional file 4).
Resequencing of putative regulatory sequences in Lake Malawi cichlids
We generated a panel of 18 Lake Malawi cichlids that vary in opsin gene expression. In one individual per species, we sequenced approximately 1 kb of DNA upstream of the translation start site for five opsins and CNE 7, as well as 0.5 kb downstream of the SWS2B and LWS opsins. We generated primers for these regions based on the O. niloticus and M. zebra BAC assemblies. The taxa sampled are listed in Additional file 11 along with their GenBank accession numbers; the primers used to generate these sequences are listed in Additional File 12. We measured opsin expression for each individual following the protocols described in Spady et al. [28] and Hofmann et al. [26]. Described briefly, we dissected whole retinas from individual fish and extracted whole RNA from them using Qiagen Qiashredder and RNeasy RNA extraction kits (Valencia, CA). We quantified each RNA sample via spectral absorption, and then reverse transcribed 0.5 μg using Superscript III (Invitrogen). We used previously developed Taqman primers and probes to individually quantify the expression of each opsin in these samples; however, as in our previous studies [26, 28], we quantified the expression of the two RH2A paralogs jointly. Reaction efficiencies for each opsin were standardized relative to an internal construct developed especially for this purpose and described in Spady et al. [28].
Following re-sequencing of the candidate cis-regulatory regions, we estimated polymorphism statistics for the resulting sequences, and also performed a sliding-window analysis of nucleotide diversity (π), in the program DnaSP v5 [114]. For the sliding-window analysis, we ignored all gaps and specified a window length of 50 bp and a step size of 10 bp. Finally, we calculated the statistical association between polymorphisms found in CRX binding sites and other peaks of nucleotide diversity among the sampled taxa using linear regression in the program gPLINK v1.07 [115]. For each test, we estimated the association of each locus with the expression of its downstream opsin under an additive genetic model, using membership in one of two major phylogenetic clades (mbuna and utaka; see Additional file 8) as a covariate.
Abbreviations
- BAC:
-
bacterial artificial chromosome
- CDS:
-
protein-coding sequence
- CNE:
-
conserved non-coding element
- Dxy:
-
pairwise sequence divergence
- HWE:
-
Hardy-Weinberg equilibrium
- INT:
-
intronic sequence
- LG:
-
linkage group
- MAF:
-
minor allele frequency
- miRNA:
-
microRNA
- Pdiv:
-
proportion divergence TFBS/miRNA target sites
- PRO:
-
proximal promoter region
- QTL:
-
quantitative trait locus
- SNP:
-
single nucleotide polymorphism
- TFBS:
-
transcription factor binding site
- TSS:
-
translation start site
- UTR:
-
untranslated region
References
Carroll SB: Evolution at two levels:on genes and form. PLoS Biol. 2005, 3: e245-10.1371/journal.pbio.0030245.
Hoekstra HE, Coyne JA: The locus of evolution: evo devo and the genetics of adaptation. Evolution. 2007, 61: 995-1016. 10.1111/j.1558-5646.2007.00105.x.
Wray GA: The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007, 8: 206-216.
Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP: A Single Amino Acid Mutation Contributes to Adaptive Beach Mouse Color Pattern. Science. 2006, 313: 101-104. 10.1126/science.1126121.
Jessen TH, Weber RE, Fermi G, Tame J, Braunitzer G: Adaptation of bird hemoglobins to high altitudes: demonstration of molecular mechanism by protein engineering. Proceedings of the National Academy of Sciences of the United States of America. 1991, 88: 6519-6522. 10.1073/pnas.88.15.6519.
Yokoyama S, Zhang H, Radlwimmer FB, Blow NS: Adaptive evolution of color vision of the Comoran coelacanth (Latimeria chalumnae). Proceedings of the National Academy of Sciences of the United States of America. 1999, 96: 6279-6284. 10.1073/pnas.96.11.6279.
Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, Brady SD, Southwick AM, Absher DM, Grimwood J, Schmutz J, et al: Adaptive Evolution of Pelvic Reduction in Sticklebacks by Recurrent Deletion of a Pitx1 Enhancer. Science. 2010, 327: 302-305. 10.1126/science.1182213.
Jeong S, Rebeiz M, Andolfatto P, Werner T, True J, Carroll SB: The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell. 2008, 132: 783-793. 10.1016/j.cell.2008.01.014.
Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, Silverman JS, Powell K, Mortensen HM, Hirbo JB, Osman M, et al: Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet. 2007, 39: 31-40. 10.1038/ng1946.
Lemos B, Araripe LO, Fontanillas P, Hartl DL: Dominance and the evolutionary accumulation of cis- and trans-effects on gene expression. Proc Natl Acad Sci USA. 2008, 105: 14471-14476. 10.1073/pnas.0805160105.
Hartl DL, Clark AG: Principles of Population Genetics. 2006, Sunderland MA: Sinaur Associates, Inc., 4
Berman BP, Nibu Y, Pfeiffer BD, Tomancak P, Celniker SE, Levine M, Rubin GM, Eisen MB: Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proceedings of the National Academy of Sciences of the United States of America. 2002, 99: 757-762. 10.1073/pnas.231608898.
Yuh CH, Bolouri H, Davidson EH: Genomic Cis-Regulatory Logic: Experimental and Computational Analysis of a Sea Urchin Gene. Science. 1998, 279: 1896-1902. 10.1126/science.279.5358.1896.
Ebert BL, Firth JD, Ratcliffe PJ: Hypoxia and Mitochondrial Inhibitors Regulate Expression of Glucose Transporter-1 via Distinct Cis-acting Sequences. Journal of Biological Chemistry. 1995, 270: 29083-29089. 10.1074/jbc.270.49.29083.
Tuan DY, Solomon WB, London IM, Lee DP: An erythroid-specific, developmental-stage-independent enhancer far upstream of the human "beta-like globin" genes. Proceedings of the National Academy of Sciences of the United States of America. 1989, 86: 2554-2558. 10.1073/pnas.86.8.2554.
Chen K, Rajewsky N: Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006, 38: 1452-1456. 10.1038/ng1910.
Kloc M, Bilinski S, Pui-Yee Chan A, Etkin LD: The Targeting of Xcat2 mRNA to the Germinal Granules Depends on a cis-Acting Germinal Granule Localization Element within the 3'UTR. Developmental Biology. 2000, 217: 221-229. 10.1006/dbio.1999.9554.
Gumucio DL, Shelton DA, Zhu W, Millinoff D, Gray T, Bock JH, Slightom JL, Goodman M: Evolutionary strategies for the elucidation of cis and trans factors that regulate the developmental switching programs of the beta-like globin genes. Mol Phylogenet Evol. 1996, 5: 18-32. 10.1006/mpev.1996.0004.
Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, Rubin EM: Phylogenetic Shadowing of Primate Sequences to Find Functional Regions of the Human Genome. Science. 2003, 299: 1391-1394. 10.1126/science.1081331.
Wald G: The molecular basis of visual excitation. Nature. 1968, 219: 800-807. 10.1038/219800a0.
Kocher TD: Adaptive evolution and explosive speciation: the cichlid fish model. Nat Rev Genet. 2004, 5: 288-298. 10.1038/nrg1316.
Seehausen O: African cichlid fish: a model system in adaptive radiation research. Proceedings of the Royal Society B: Biological Sciences. 2006, 273: 1987-1998. 10.1098/rspb.2006.3539.
Carleton KL: Cichlid fish visual systems: mechanisms of spectral tuning. Integrative Zoology. 2009, 4: 75-86. 10.1111/j.1749-4877.2008.00137.x.
Carleton KL, Spady TC, Kocher TD: Visual communication in East African cichlid fishes: diversity in a phylogenetic context. Communication in Fishes. Edited by: Ladich F, Collin SP, P.M, G. KB. Enfield. 2006, Science Publishers, 487-515.
Jordan R, Kellogg K, Howe D, Juanes F, Stauffer J, Loew E: Photopigment spectral absorbance of Lake Malaŵi cichlids. J Fish Biol. 2006, 68: 1291-1299. 10.1111/j.0022-1112.2006.00992.x.
Hofmann CM, O'Quin KE, Marshall NJ, Cronin TW, Seehausen O, Carleton KL: The eyes have it: regulatory and structural changes both underlie cichlid visual pigment diversity. PLoS Biol. 2009, 7: e1000266-10.1371/journal.pbio.1000266.
O'Quin KE, Hofmann CM, Hofmann HA, Carleton KL: Parallel evolution of opsin gene expression in African cichlid fishes. Molecular Biology and Evolution. 2010
Spady TC, Parry JW, Robinson PR, Hunt DM, Bowmaker JK, Carleton KL: Evolution of the cichlid visual palette through ontogenetic subfunctionalization of the opsin gene arrays. Mol Biol Evol. 2006, 23: 1538-1547. 10.1093/molbev/msl014.
Carleton KL, Hofmann CM, Klisz C, Patel Z, Chircus LM, Simenauer LH, Soodoo N, Albertson RC, Ser JR: Genetic basis of differential opsin gene expression in cichlid fishes. J Evol Biol. 2010, 23 (4): 840-53. 10.1111/j.1420-9101.2010.01954.x.
Katagiri T, Asakawa S, Minagawa S, Shimizu N, Hirono I, Aoki T: Construction and characterization of BAC libraries for three fish species; rainbow trout, carp and tilapia. Anim Genet. 2001, 32: 200-204. 10.1046/j.1365-2052.2001.00764.x.
Di Palma F, Kidd C, Borowsky R, Kocher TD: Construction of bacterial artificial chromosome libraries for the Lake Malawi cichlid (Metriaclima zebra), and the blind cavefish (Astyanax mexicanus). Zebrafish. 2007, 4: 41-47. 10.1089/zeb.2006.9996.
Carleton KL, Spady TC, Streelman JT, Kidd MR, McFarland WN, Loew ER: Visual sensitivities tuned by heterochronic shifts in opsin gene expression. BMC Biol. 2008, 6: 22-10.1186/1741-7007-6-22.
Genner MJ, Seehausen O, Lunt DH, Joyce DA, Shaw PW, Carvalho GR, Turner GF: Age of Cichlids: New Dates for Ancient Lake Fish Radiations. Molecular Biology and Evolution. 2007, 24: 1269-1282. 10.1093/molbev/msm050.
Applebury ML, Farhangfar F, Glosmann M, Hashimoto K, Kage K, Robbins JT, Shibusawa N, Wondisford FE, Zhang H: Transient expression of thyroid hormone nuclear receptor TRbeta2 sets S opsin patterning during cone photoreceptor genesis. Dev Dyn. 2007, 236: 1203-1212. 10.1002/dvdy.21155.
Browman H, Hawryshyn C: Retinoic Acid Modulates Retinal Development in the Juveniles of a Teleost Fish. J Exp Biol. 1994, 193: 191-207.
Browman HI, Hawryshyn CW: The developmental trajectory of ultraviolet photosensitivity in rainbow trout is altered by thyroxine. Vision Res. 1994, 34: 1397-1406. 10.1016/0042-6989(94)90139-2.
Dann SG, Allison WT, Veldhoen K, Johnson T, Hawryshyn CW: Chromatin immunoprecipitation assay on the rainbow trout opsin proximal promoters illustrates binding of NF-kappaB and c-jun to the SWS1 promoter in the retina. Exp Eye Res. 2004, 78: 1015-1024. 10.1016/j.exer.2003.12.012.
Ng L, Hurley JB, Dierks B, Srinivas M, Salto C, Vennstrom B, Reh TA, Forrest D: A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet. 2001, 27: 94-98.
Peng GH, Ahmad O, Ahmad F, Liu J, Chen S: The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum Mol Genet. 2005, 14: 747-764. 10.1093/hmg/ddi070.
Roberts MR, Hendrickson A, McGuire CR, Reh TA: Retinoid X Receptor γ Is Necessary to Establish the S-opsin Gradient in Cone Photoreceptors of the Developing Mouse Retina. Investigative Ophthalmology & Visual Science. 2005, 46: 2897-2904. 10.1167/iovs.05-0093.
Takechi M, Seno S, Kawamura S: Identification of cis-acting elements repressing blue opsin expression in zebrafish UV cones and pineal cells. J Biol Chem. 2008, 283: 31625-31632. 10.1074/jbc.M806226200.
Halligan DL, Eyre-Walker A, Andolfatto P, Keightley PD: Patterns of Evolutionary Constraints in Intronic and Intergenic DNA of Drosophila. Genome Research. 2004, 14: 273-279. 10.1101/gr.1329204.
Keightley PD, Gaffney DJ: Functional constraints and frequency of deleterious mutations in noncoding DNA of rodents. Proceedings of the National Academy of Sciences of the United States of America. 2003, 100: 13402-13406. 10.1073/pnas.2233252100.
Lee BY, Lee WJ, Streelman JT, Carleton KL, Howe AE, Hulata G, Slettan A, Stern JE, Terai Y, Kocher TD: A second-generation genetic linkage map of tilapia (Oreochromis spp.). Genetics. 2005, 170: 237-244. 10.1534/genetics.104.035022.
Schwartz S, Zhang ZD, Smit A, Reimer C, Bouck C, Gibbs RA, Hardison RC, Miller W: PipMaker--A web server for aligning two genomic DNA sequences. Genome Research. 2000, 10: 577-586. 10.1101/gr.10.4.577.
Schwartz S, Elnitski L, Li M, Weirauch M, Riemer C, Smit A, Green ED, Hardison RC, Miller W: MultiPipMaker and supporting tools: Alignments and analysis of multiple genomic DNA sequences. Nucleic Acids Res. 2003, 31: 3518-3524. 10.1093/nar/gkg579.
Watson CT, Lubieniecki KP, Loew E, Davidson WS, Breden F: Genomic organization of duplicated short wave-sensitive and long wave-sensitive opsin genes in the green swordtail, Xiphophorus helleri. BMC Evol Biol. 2010, 10: 87-10.1186/1471-2148-10-87.
Carleton KL, Kocher TD: Cone opsin genes of African cichlid fishes: tuning spectral sensitivity by differential gene expression. Mol Biol Evol. 2001, 18: 1540-1550.
Steinke D, Salzburger W, Meyer A: Novel relationships among ten fish model species revealed based on a phylogenomic analysis using ESTs. J Mol Evol. 2006, 62: 772-784. 10.1007/s00239-005-0170-8.
Chinen A, Hamaoka T, Yamada Y, Kawamura S: Gene duplication and spectral diversification of cone visual pigments of zebrafish. Genetics. 2003, 163: 663-675.
Hofmann CM, Carleton KL: Gene duplication and differential gene expression play an important role in the diversification of visual pigments in fish. J Int Comp Biol. 2009, 49: 630-643. 10.1093/icb/icp079.
Neafsey DE, Hartl DL: Convergent loss of an anciently duplicated, functionally divergent RH2 opsin gene in the fugu and Tetraodon pufferfish lineages. Gene. 2005, 350: 161-171. 10.1016/j.gene.2005.02.011.
Yokoyama S, Tada T: Evolutionary dynamics of rhodopsin type 2 opsins in vertebrates. Mol Biol Evol. 2010, 27: 133-141. 10.1093/molbev/msp217.
Griffiths-Jones S, Saini HK, Dongen Sv, Enright AJ: miRBase: tools for microRNA genomics. Nucleic Acids Research. 2007, gkm952-
Kloosterman WP, Steiner FA, Berezikov E, de Bruijn E, van de Belt J, Verheul M, Cuppen E, Plasterk RH: Cloning and expression of new microRNAs from zebrafish. Nucleic Acids Res. 2006, 34: 2558-2569. 10.1093/nar/gkl278.
Opsin evolution: LWS PhyloSNPs [http://genomewiki.ucsc.edu/index.php/Opsin_evolution:_LWS_PhyloSNPs].
Wakefield MJ, Anderson M, Chang E, Wei KJ, Kaul R, Graves JA, Grutzner F, Deeb SS: Cone visual pigments of monotremes: filling the phylogenetic gap. Vis Neurosci. 2008, 25: 257-264.
Smallwood PM, Wang Y, Nathans J: Role of a locus control region in the mutually exclusive expression of human red and green cone pigment genes. Proc Natl Acad Sci USA. 2002, 99: 1008-1011. 10.1073/pnas.022629799.
Wang Y, Macke JP, Merbs SL, Zack DJ, Klaunberg B, Bennett J, Gearhart J, Nathans J: A locus control region adjacent to the human red and green visual pigment genes. Neuron. 1992, 9: 429-440. 10.1016/0896-6273(92)90181-C.
Tsujimura T, Hosoya T, Kawamura S: A single enhancer regulating the differential expression of duplicated red-sensitive opsin genes in zebrafish. PLoS Genet. 2010, 6: e1001245-10.1371/journal.pgen.1001245.
Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al: Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007, 447: 799-816. 10.1038/nature05874.
Rozowsky J, Euskirchen G, Auerbach RK, Zhang ZD, Gibson T, Bjornson R, Carriero N, Snyder M, Gerstein MB: PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol. 2009, 27: 66-75. 10.1038/nbt.1518.
Schug J: Using TESS to predict transcription factor binding sites in DNA sequence. Curr Protoc Bioinformatics. 2008, Chapter 2: Unit 2 6
Salbert G, Fanjul A, Piedrafita FJ, Lu XP, Kim SJ, Tran P, Pfahl M: Retinoic acid receptors and retinoid X receptor-alpha down-regulate the transforming growth factor-beta 1 promoter by antagonizing AP-1 activity. Mol Endocrinol. 1993, 7: 1347-1356. 10.1210/me.7.10.1347.
Schule R, Rangarajan P, Yang N, Kliewer S, Ransone LJ, Bolado J, Verma IM, Evans RM: Retinoic acid is a negative regulator of AP-1-responsive genes. Proc Natl Acad Sci USA. 1991, 88: 6092-6096. 10.1073/pnas.88.14.6092.
Wasserman WW, Sandelin A: Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004, 5: 276-287. 10.1038/nrg1315.
Letovsky J, Dynan WS: Measurement of the binding of transcription factor Sp1 to a single GC box recognition sequence. Nucleic Acids Res. 1989, 17: 2639-2653. 10.1093/nar/17.7.2639.
Stormo GD: DNA binding sites: representation and discovery. Bioinformatics. 2000, 16: 16-23. 10.1093/bioinformatics/16.1.16.
Baker DL, Dave V, Reed T, Periasamy M: Multiple Sp1 binding sites in the cardiac/slow twitch muscle sarcoplasmic reticulum Ca2+-ATPase gene promoter are required for expression in Sol8 muscle cells. J Biol Chem. 1996, 271: 5921-5928. 10.1074/jbc.271.10.5921.
Latchman DS: Eukaryotic transcription factors. 2004, Amsterdam; Boston: Elsevier/Academic Press, 4
Andolfatto P: Adaptive evolution of non-coding DNA in Drosophila. Nature. 2005, 437: 1149-1152. 10.1038/nature04107.
Loh YH, Yi SV, Streelman JT: Evolution of microRNAs and the diversification of species. Genome Biol Evol. 2010
Arora A, McKay GJ, Simpson DA: Prediction and verification of miRNA expression in human and rat retinas. Invest Ophthalmol Vis Sci. 2007, 48: 3962-3967. 10.1167/iovs.06-1221.
Ryan DG, Oliveira-Fernandes M, Lavker RM: MicroRNAs of the mammalian eye display distinct and overlaping tissue specificity. Mol Vis. 2006, 12: 1175-1184.
Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RHA: MicroRNA Expression in Zebrafish Embryonic Development. Science. 2005, 309: 310-311. 10.1126/science.1114519.
Xu S, Witmer PD, Lumayag S, Kovacs B, Valle D: MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J Biol Chem. 2007, 282: 25053-25066. 10.1074/jbc.M700501200.
Guerin MB, McKernan DP, O'Brien CJ, Cotter TG: Retinal ganglion cells: dying to survive. Int J Dev Biol. 2006, 50: 665-674. 10.1387/ijdb.062159mg.
Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, et al: TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009, 11: 881-889. 10.1038/ncb1897.
Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, et al: miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007, 129: 147-161. 10.1016/j.cell.2007.03.008.
Terai Y, Seehausen O, Sasaki T, Takahashi K, Mizoiri S, Sugawara T, Sato T, Watanabe M, Konijnendijk N, Mrosso HD, et al: Divergent selection on opsins drives incipient speciation in Lake Victoria cichlids. PLoS Biol. 2006, 4: e433-10.1371/journal.pbio.0040433.
Lisney TJ, Studd E, Hawryshyn CW: Electrophysiological assessment of spectral sensitivity in adult Nile tilapia Oreochromis niloticus: evidence for violet sensitivity. J Exp Biol. 2010, 213: 1453-1463. 10.1242/jeb.036897.
Calissano M, Diss JK, Latchman DS: Post-transcriptional regulation of the Brn-3b transcription factor in differentiating neuroblastoma cells. FEBS Lett. 2007, 581: 2490-2496. 10.1016/j.febslet.2007.04.066.
Decembrini S, Bressan D, Vignali R, Pitto L, Mariotti S, Rainaldi G, Wang X, Evangelista M, Barsacchi G, Cremisi F: MicroRNAs couple cell fate and developmental timing in retina. Proc Natl Acad Sci USA. 2009, 106: 21179-21184. 10.1073/pnas.0909167106.
Zhao JJ, Yang J, Lin J, Yao N, Zhu Y, Zheng J, Xu J, Cheng JQ, Lin JY, Ma X: Identification of miRNAs associated with tumorigenesis of retinoblastoma by miRNA microarray analysis. Childs Nerv Syst. 2009, 25: 13-20. 10.1007/s00381-008-0701-x.
O'Quin KE, Smith AR, Sharma A, Carleton KL: New evidence for the role of heterochrony in the repeated evolution of cichlid opsin expression. Evolution & Development. 2011, 13 (2): 193-203.
Hutvagner G, Zamore PD: A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002, 297: 2056-2060. 10.1126/science.1073827.
Llave C, Xie Z, Kasschau KD, Carrington JC: Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002, 297: 2053-2056. 10.1126/science.1076311.
Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004, 116: 281-297. 10.1016/S0092-8674(04)00045-5.
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB: Prediction of mammalian microRNA targets. Cell. 2003, 115: 787-798. 10.1016/S0092-8674(03)01018-3.
Stark A, Brennecke J, Russell RB, Cohen SM: Identification of Drosophila MicroRNA targets. PLoS Biol. 2003, 1: E60-10.1371/journal.pbio.0000060.
Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, et al: Variation in transcription factor binding among humans. Science. 2010, 328: 232-235. 10.1126/science.1183621.
Loh YH, Katz LS, Mims MC, Kocher TD, Yi SV, Streelman JT: Comparative analysis reveals signatures of differentiation amid genomic polymorphism in Lake Malawi cichlids. Genome Biol. 2008, 9: R113-10.1186/gb-2008-9-7-r113.
Roberts RB, Ser JR, Kocher TD: Sexual Conflict Resolved by Invasion of a Novel Sex Determiner in Lake Malawi Cichlid Fishes. Science. 2009, 326: 998-1001. 10.1126/science.1174705.
Allender CJ, Seehausen O, Knight ME, Turner GF, Maclean N: Divergent selection during speciation of Lake Malawi cichlid fishes inferred from parallel radiations in nuptial coloration. Proceedings of the National Academy of Sciences of the United States of America. 2003, 100: 14074-14079. 10.1073/pnas.2332665100.
Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, Cheung VG: Genetic analysis of genome-wide variation in human gene expression. Nature. 2004, 430: 743-747. 10.1038/nature02797.
Sung HM, Wang TY, Wang D, Huang YS, Wu JP, Tsai HK, Tzeng J, Huang CJ, Lee YC, Yang P, et al: Roles of trans and cis variation in yeast intraspecies evolution of gene expression. Mol Biol Evol. 2009, 26: 2533-2538. 10.1093/molbev/msp171.
Wittkopp PJ, Haerum BK, Clark AG: Evolutionary changes in cis and trans gene regulation. Nature. 2004, 430: 85-88. 10.1038/nature02698.
Hsia CC, McGinnis W: Evolution of transcription factor function. Curr Opin Genet Dev. 2003, 13: 199-206. 10.1016/S0959-437X(03)00017-0.
Levine M, Tjian R: Transcription regulation and animal diversity. Nature. 2003, 424: 147-151. 10.1038/nature01763.
Spady TC, Seehausen O, Loew ER, Jordan RC, Kocher TD, Carleton KL: Adaptive molecular evolution in the opsin genes of rapidly speciating cichlid species. Mol Biol Evol. 2005, 22: 1412-1422. 10.1093/molbev/msi137.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-185.
Phrap: phragment assembly program [http://www.phrap.org].
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al: Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005, 437: 376-380.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Couronne O, Poliakov A, Bray N, Ishkhanov T, Ryaboy D, Rubin E, Pachter L, Dubchak I: Strategies and Tools for Whole-Genome Alignments. Genome Research. 2003, 13: 73-80. 10.1101/gr.762503.
Néron B, Ménager H, Maufrais C, Joly N, Maupetit J, Letort S, Carrere S, Tuffery P, Letondal C: Mobyle: a new full web bioinformatics framework. Bioinformatics. 2009, 25: 3005-3011. 10.1093/bioinformatics/btp493.
R: A language and environment for statistical computing. [http://www.R-project.org]
Katoh K, Toh H: Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008, 9: 286-298. 10.1093/bib/bbn013.
Posada D: jModelTest: Phylogenetic model averaging. Molecular Biology and Evolution. 2008, 25: 1253-1256. 10.1093/molbev/msn083.
Sokal RR, Rohlf FJ: Biometry. 1995, New York: W. H. Freeman and Company
Chessel D, Dufor AB, Thioulouse J: The ade4 package - I: One-table methods. R News. 2004, 4: 5-10.
Lewis BP, Burge CB, Bartel DP: Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005, 120: 15-20. 10.1016/j.cell.2004.12.035.
Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Green ED, Sidow A, Batzoglou S: LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13: 721-731. 10.1101/gr.926603.
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452. 10.1093/bioinformatics/btp187.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007, 81: 559-575. 10.1086/519795.
Hackler L, Wan J, Swaroop A, Qian J, Zack DJ: MicroRNA profile of the developing mouse retina. Invest Ophthalmol Vis Sci. 2010, 51: 1823-1831. 10.1167/iovs.09-4657.
Karali M, Peluso I, Marigo V, Banfi S: Identification and characterization of microRNAs expressed in the mouse eye. Invest Ophthalmol Vis Sci. 2007, 48: 509-515. 10.1167/iovs.06-0866.
Hoegg S, Boore J, Kuehl J, Meyer A: Comparative phylogenomic analyses of teleost fish Hox gene clusters: lessons from the cichlid fish Astatotilapia burtoni. BMC Genomics. 2007, 8: 317-10.1186/1471-2164-8-317.
Acknowledgements
We thank Takayuki Katagiri for making the Oreochromis niloticus BAC clone library and Bo Young Lee for pooling this library for PCR screening. We also thank Frederica DiPalma for generating the Meteriaclima zebra library and Celeste Kidd for screening this library for the opsin-containing BACs. This work was supported with grants to KLC from NSF (IOS-0841270), NIH (R15 EY016721-01) and the University of Maryland. KEO was supported by a Wayne T. and Mary T. Hockmeyer Doctoral Fellowship and an Ann G. Wylie Dissertation Fellowship from the University of Maryland.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
KEO participated in BAC annotation, carried out the survey of transcription factor binding sites, participated in the sequencing of opsin proximal promoters, participated in the survey of miRNA target sites, performed all statistical analysis, and wrote the manuscript. DS participated in the BAC assembly and annotation. ZN and JS both participated in the sequencing of opsin proximal promoters. SDE sequenced the LWS and SWS2B 3'-UTRs. YHL and JTS performed the search of microRNA target sites. JLB performed the BAC sequencing. KLC designed the study; aided in the BAC screening, sequencing, and assembly; participated in BAC annotation; carried out the analysis of opsin gene expression, and participated in the drafting of the manuscript. All authors read and approved the final manuscript.
Daniel Smith, Zan Naseer, Jane Schulte, Samuel D Engel contributed equally to this work.
Electronic supplementary material
12862_2011_1735_MOESM2_ESM.PDF
Additional file 2: Synteny (Pip plots) of O. niloticusopsin-containing BACs against the genome assemblies of five teleost species. (PDF 2 MB)
12862_2011_1735_MOESM3_ESM.PDF
Additional file 3: Opsin gene content of five teleost genomes. Phylogeny of the teleost taxa is recreated from [118]. (PDF 171 KB)
12862_2011_1735_MOESM4_ESM.PDF
Additional file 4: Orthology of RH2 and SWS2 opsin paralogs from five teleost fish genomes. A) RH2 phylogeny. B) SWS2 phylogeny. In both cases, broken lines indicate branches leading from the outgroup that were shortened to fit each tree into the figure; these do not represent missing or incomplete branch length information. (PDF 244 KB)
12862_2011_1735_MOESM5_ESM.FAS
Additional file 5: FASTA file of 20 conserved non-coding elements (CNEs), promoter sequences, 3-UTRs, and seven non-opsin promoters from O. niloticusand M. zebra(80 sequences total). (FAS 42 KB)
12862_2011_1735_MOESM6_ESM.XLS
Additional file 6: Complete transcription factor binding site profiles for 20 CNEs in O. niloticusand M. zebra. (XLS 32 KB)
12862_2011_1735_MOESM7_ESM.XLS
Additional file 7: Complete list of miRNA target sites identified within the 3'-UTR of each opsin in O. niloticusand M. zebra. (XLS 30 KB)
12862_2011_1735_MOESM8_ESM.XLS
Additional file 8: Names, opsin expression values, and polymorphisms found within the proximal promoters of 18 Lake Malawi cichlid species. (XLS 44 KB)
12862_2011_1735_MOESM9_ESM.XLS
Additional file 9: Length and Dxy scores between O. niloticusand M. zebrafor each coding and non-coding region examined. (XLS 30 KB)
12862_2011_1735_MOESM10_ESM.PDF
Additional file 10: Identification of opsin-containing BACs from Finger Printed Contigs. A-C) BACs fingerprinted contig containing the SWS2A-SWS2B-LWS (A) RH2 (B) and SWS1 (C) genes. Arrows indicate PCR products successfully amplified using primers designed to BAC end sequences for clones whose names are shown in the corresponding color. Colored circles are the approximate locates of each gene. (PDF 2 MB)
Additional file 11: GenBank accession numbers for all sequences generated in this study. (XLS 20 KB)
12862_2011_1735_MOESM12_ESM.XLS
Additional file 12: Primers used to amplify and sequence the proximal promoter regions and 3'-UTR of several opsins from 18 Lake Malawi cichlid species. (XLS 16 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
O'Quin, K.E., Smith, D., Naseer, Z. et al. Divergence in cis-regulatory sequences surrounding the opsin gene arrays of African cichlid fishes. BMC Evol Biol 11, 120 (2011). https://doi.org/10.1186/1471-2148-11-120
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-11-120