
Abstract
Background
Oysters are morphologically plastic and hence difficult subjects for taxonomic and evolutionary studies. It is long been suspected, based on the extraordinary species diversity observed, that Asia Pacific is the epicenter of oyster speciation. To understand the species diversity and its evolutionary history, we collected five Crassostrea species from Asia and sequenced their complete mitochondrial (mt) genomes in addition to two newly released Asian oysters (C. iredalei and Saccostrea mordax) for a comprehensive analysis.
Results
The six Asian Crassostrea mt genomes ranged from 18,226 to 22,446 bp in size, and all coded for 39 genes (12 proteins, 2 rRNAs and 25 tRNAs) on the same strand. Their genomes contained a split of the rrnL gene and duplication of trnM, trnK and trnQ genes. They shared the same gene order that differed from an Atlantic sister species by as many as nine tRNA changes (6 transpositions and 3 duplications) and even differed significantly from S. mordax in protein-coding genes. Phylogenetic analysis indicates that the six Asian Crassostrea species emerged between 3 and 43 Myr ago, while the Atlantic species evolved 83 Myr ago.
Conclusions
The complete conservation of gene order in the six Asian Crassostrea species over 43 Myr is highly unusual given the remarkable rate of rearrangements in their sister species and other bivalves. It provides strong evidence for the recent speciation of the six Crassostrea species in Asia. It further indicates that changes in mt gene order may not be strictly a function of time but subject to other constraints that are presently not well understood.
Similar content being viewed by others


Background
Mitochondrial (mt) DNA is widely used for phylogenetic analysis because of its unique architecture, inheritance and small size. Metazoan mtDNA is nearly always a circular molecule except for some cnidarians [1]. It contains the same 37 genes, specifying 13 proteins of the respiratory chain [cytochrome c oxidase subunits I-III (cox1-cox3), apocytochrome b (cob), ATP synthase subunits 6 and 8 (atp6 and atp8), and NADH dehydrogenase subunits 1-6 and 4L (nad1-6, nad4L)], 2 ribosomal RNAs and 22 transfer RNAs. Although there are exceptions, most mtDNAs range in size from 14 to 17 kb. Typically, there are few intergenic nucleotides except for a single large non-coding region generally thought to contain elements that control the initiation of replication and transcription [2]. Size variation in mtDNA is usually due to the different length of the non-coding regions. Gene order is generally conserved in most metazoan taxa although some groups show considerable variation.
Variations in both mtDNA sequence and gene order have been used for phylogenetic analysis. Because mtDNA is fast evolving and nucleotide mutations may return to an early state, mtDNA sequences may not allow deep phylogenetic reconstruction. Gene order, on the other hand, has very small probability of back-mutation and may be particularly useful for high level phylogenetic analysis. Although the mechanism of mtDNA rearrangement is poorly understood, mt gene order has been increasingly used for phylogenetic studies [3–6].
As the second species-rich phylum of the animal kingdom after Arthropoda, Mollusca exhibits tremendous variation in their mt genomes. Seven bivalve lineages (Mytilidae, Unionidae, Margaritiferidae, Hyriidae, Donacidae, Solenidae, and Veneridae) have been found to have an unusual mode of inheritance for mtDNA, termed doubly uniparental inheritance (DUI) [7]. Some pulmonate gastropods have unusual tRNAs lacking the T-stem or the D-stem, similar to nematode mt tRNAs [8]. The atp6 and atp8 genes are separated in the scaphopods and two groups of gastropods (Patellogastropoda and Heterobranchia). In addition, several mt genes are duplicated in cephalopods Watasenia scintillans and Todarodes pacificus [9]. Unlike the general conservation in gene order in most other metazoan groups, most molluscan mt genomes reported so far contain considerable rearrangements especially in Bivalvia and Scaphopoda [10]. At the same time, the phylogeny of molluscs is poorly studied. Phylogenetic relationships among major molluscan groups are not well understood. The species identity and classification of some most common molluscs remain questionable.
Oysters are bivalve molluscs widely distributed in world oceans. They are benthic, sessile filter-feeders with important roles in estuary ecology. Some species support major fishery and aquaculture industries worldwide. Despite the abundance, ecological and economic significance of oysters, we know little about their species diversity and evolutionary history. Classification of oysters remains a challenge partly due to the lack of well-defined morphological characters. Shell morphology, the main character used in oyster classification, is known to be plastic and subject to environmental variation. Much of the oyster classification to date is based on shell characteristics and has resulted in considerable errors and confusion. The difficulty of oyster classification is particularly pronounced in China and other parts of Asia where a large number of species are sympatric [11, 12]. About thirty species have been recorded along the coast of China [13]. The presence of a large number of oyster species has led many to believe that Asia Pacific is the epicenter of oyster speciation, but the inability to reliably identifying them has hindered our understanding of oyster evolution. Among the extent oysters, five Crassostrea oysters, namely C. gigas, C. angulata, C. sikamea, C. hongkongensis and C. ariakensis are commonly found in China and other parts of Asia and yet, they have been difficult to identify by shell morphology alone. Recently, some confusion in the oyster identification have been resolved using DNA sequence data [12, 14–20], and the five Crassostrea oysters can be reliably identified [21]. Still, we know little about the evolutionary history of the Crassostrea species and how they relate to each other and the other oysters. For example, it is not clear whether C. angulata should be considered as an independent species and which species is the closest relative of the newly described C. hongkongensis. Most of the phylogenetic analyses so far are based on short DNA fragments, yielding variable results [12, 16, 22].
The ability to sequence and compare whole mt genomes provides a new impetus for phylogenetic analysis of oysters and other molluscs. Complete mt genome sequences have been obtained for two Crassostrea oysters from Asia (C. gigas and C. hongkongensis) and one species from the Atlantic (C. virginica) [23–25]. Comparative analysis shows that the two Asian species share the same gene order that is very different from the Atlantic species [24, 25]. To determine if gene order is conserved in other Asian species and to understand the evolution of Crassostrea oysters, we sequenced the mt genomes of four Crassostrea oysters from Asia plus one that was previously published [25], obtained newly released mt genomes of two Asian oysters (C. iredalei and Saccostrea mordax), and compared them with other molluscan genomes. Here we report the first estimates of divergence time among Crassostrea species based on complete mt sequences and the complete conservation in gene order among the six Asian Crassostrea species. The complete conservation of gene order is highly unusual considering the tremendous rearrangement of mtDNA in most marine bivalves. It provides strong evidence for recent emergence of the six Crassostrea species in Asia. Our analysis also suggests that rearrangement of the mt genome may not be strictly a function of time but subject to some other constraints that are presently not well understood.
Results and Discussion
Genome composition
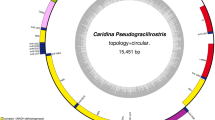
Genome composition and organization of the six Asian Crassostrea oysters is summarized in Figure 1 and additional file 1: Table S1. Organization of the American oyster C. virginica, a sister species from the Atlantic Ocean, is listed for comparative analysis (additional file 1: Table S1). The complete mt genomes of C. gigas, C. angulata, C. sikamea, C. hongkongensis and C. ariakensis are 18,225 bp, 18,225 bp, 18,243 bp, 18,622 bp and 18,414 bp in length, respectively [24, 25]. These sequences have been deposited in GenBank under the accession number EU672831, NC_012648-NC_012650, NC_011518. The American oyster C. virginica mtDNA is 17,244 bp in length. The mt genome of S. mordax is 16,532 bp in length. However, the mt genome of C. iredalei, 22,446 bp in length, is obviously longer than all other oysters [54]. The size of molluscan mt genomes varies dramatically, ranging from 13,670 bp in the snail Biomphalaria glabrata to 40,725 bp in the sea scallop Placopecten magellanicus [41, 42]. The six Asian Crassostrea oyster genomes encode 39 genes including 12 protein-coding genes, 2 rRNAs and 25 tRNAs on the same strand (Figure 1). In contrast to the typical animal mt genomes, they lack one protein-coding gene atp8 and have duplications of three tRNAs: trnM, trnK and trnQ. Another unique character is that rrnL gene is split into two segments (also in C. virginica and S. mordax) and rrnS has a nearly identical duplication, which has never been reported in other animal mt genomes [23]. Among the seven Crassostrea oysters, the lowest A+T content is 61.1% in C. virginica, while the highest A+T content is 65.3% in C. hongkongensis. In the other five species, the A+T content varies from 62.9% in C. ariakensis to 64.5% in C. iredalei [54] (Table 1). The highest A+T content of C. hongkongensis is mainly caused by the increased total length and high A+T content of the non-coding region. The length of coding regions in the Asian-Pacific oyster genomes is similar and larger than that in C. virginica, because of duplications of two tRNAs (trnK and trnQ) and one rrnS.

Mitochondrial genome map of Asian Crassostrea oysters. All 39 genes are coded on the same DNA strand. Genes for proteins and rRNAs are shown with standard abbreviations. Genes for tRNAs are designated by a single letter for the corresponding amino acid with two leucine tRNAs and two serine tRNAs differentiated by numeral. The two tRNA duplications of methionine, lysine and glutamine are named trnM1, trnM2, trnK1, trnK2, trnQ1 and trnQ2, respectively. The length of five mt genomes is indicated at the lower right-hand corner.
Gene arrangement
Molluscs, especially bivalves, display an extraordinary amount of variation in gene arrangement. Gene order of selected molluscs was presented and compared with that of oysters in Figure 2. The black chiton Katharina tunicata is the only sequenced representative of the Polyplacophora, an early diverged class of the Mollusca [43]. Its gene order may represent the pleisomorphic gene arrangement in Mollusca. At high taxonomic levels, the gene order tends to be conserved across polyplacophoran, cephalopods and gastropods. The gene order of K. tunicata differs from that of the common octopus Octopus vulgaris by merely the inversion of trnP and translocation of trnD. Gene order of other cephalopods resembles that of O. vulgaris with some translocations of tRNA or the switch of large gene blocks [9]. Additionally, the gene arrangement of K. tunicata differs from that of the blacklip abalone Haliotis rubra merely by the inversion of trnP plus transposition of four tRNAs.

Mitochondrial genome rearrangements in bivalves and representatives from other molluscan classes. All genes are transcribed from left-to-right except those indicated by underlining, which are transcribed from right to left. The bars show identical gene blocks. The arrows indicate gene translocation and the circling arrows indicate gene inversion. The grey blank boxes represent the uncompleted sequences. The non-coding regions are not presented and gene segments are not drawn to scale.
In comparison, however, mt genomes of Bivalvia show tremendous gene rearrangements. To date, all bivalves whose mt genomes are available belong to three subclasses: Palaeoheterodonta, Heterodonta and Pteriomorphia. Gene order in freshwater mussel Lampsilis ornata (Palaeoheterodonta: Unionoida) is nearly identical to that of it sister species Inversidens japanensis except for the translocation of several tRNAs, and protein-coding genes nad2 and nad3 (Figure 2) [10]. In addition, L. ornata genome contains the atp8 gene which is absent in I. japanensis (Data not shown). The remaining bivalves whose mt genomes have been sequenced are marine species, where the gene order is dramatically rearranged. Comparing the gene arrangement between Meretrix petechialis and Venerupis philippinarum (Heterodonta: Veneridae), they share four identical gene blocks: two large blocks cox1-L1-nad1-nad2-nad4L-I and cox2-P-cob-rrnL-nad4-H-E-S2-atp6-nad3-nad5, and two small blocks only containing tRNA genes. They share only one gene block cob-rrnL-nad4-atp6 with Acanchocardia tuberculata of the same order. Meanwhile, A. tuberculata and Hiatella arctica share only two small gene blocks trnM-W and nad2-cox1. Species sequenced in Pteriomorphia belong to three orders: Mytiloida, Ostreoida and Pectinoida. Few gene blocks are shared between any two pairs. Among the four scallops of the family Pectinidae (Pectinoida), gene arrangement of Mizuhopecten yessoensis closely resembles that of Chlamys farreri. They share three large gene blocks nad4L-nad6-L 2 -cob, cox3-K-F-Q-E-atp6-cox2-nad2-T-P-I-L 1 -M-nad3-nad4 and N-nad1-R-rrnL-M. But their gene order is very different from that of the other two scallops, despite being members of the same family. The two genomes of C. farreri and Argopecten irradians only show three small shared gene blocks nad6-L2-cob, nad3-nad4 and nad1-R-rrnL, while A. irradians and P. magellanicus only share one gene block L 2 -cob-cox2.
On the other hand, the mt gene order in the six Asian Crassostrea oysters is completely identical to each other (Figure 2). This is highly unusual considering the tremendous rearrangement observed in other groups of Bivalvia. Even within the same genus, comparison between the Asian Crassostrea oysters of the Pacific and the American oyster C. virginica of the Atlantic Ocean show that gene order has been drastically rearranged. Gene order of the protein-coding genes (PCGs) is the same, while several tRNAs has translocated after the divergence between the Asian and American species. The six Asian Crassostrea oysters have duplicated gene trnK, trnQ and rrnS compared with the American oyster. They differ from the Atlantic sister species by as many as nine tRNA changes (6 transpositions and 3 duplications), and furthermore, they differ significantly from S. mordax in PCGs. If the tRNA is not considered, they share three PCGs blocks cox1-cox3-cob-cox2, nad3-nad1-nad4L, nad5-nad6 and atp6-nad2. It is amazing that Crassostrea sister species from the two oceans have so much difference in gene order and close relatives even have more difference in gene order, while gene order is completely conserved in the six Crassostrea species from Asia. This raises the questions what causes rearrangements of mt genomes and how do they evolve over time. Additional studies of mt genomes of other oyster species would be interesting to see if this pattern should preserve, which may further our understanding of mt genome evolution.
Protein-coding genes
Of the 13 typical PCGs (cox1-cox3, nad1-nad6, nad4L, cob, atp6 and atp8), twelve genes were determined and the atp8 gene was absent in all oyster genomes. All PCGs are encoded on and transcribed from the same strand. These features have been observed in all other marine bivalve genomes published so far except for H. arctica where the atp8 gene has been reported, and for two species of Palaeoheterodonta (L. ornate and I. japanensis) where genes are coded on both strands (Figure 2). Thus, coding genes on the same strand and missing atp8 gene are the most distinct features of marine bivalve mt genomes, though from a recent publication from Breton et al [57] this might only hold true for Ostreoida and a few other bivalves. The two Palaeoheterodonta species that use both strands for coding are the only known members of Bivalvia showing significant conservation in gene order with other molluscan classes that also use both strands for coding. We suspect that coding on both strands may be inhibitory to mt genome rearrangement, and marine bivalves show a tremendous amount of mt genome rearrangement because they only use one strand for coding. Rearranging a genome with dual-strand coding may be more complicated and cause more harm than rearranging a genome that codes on one strand.
Mt genomes often use a variety of non-standard initiation codons [44]. In some cases, identification of the very clear initiation codon is difficult when several alternatives are inferred representing the start of coding sequence in a region. Most of PCGs initiate with the standard start codon ATG. Standard start codon ATA is used for cox3 in C. virginica, nad4 in C. hongkongensis and nad4L in C. sikamea and in C. ariakensis. Non-standard initiation codon GTG is used for nad5 in C. ariakensis, and ATT is used for nad6 in C. hongkongensis (Table 2). There are no obvious patterns in termination codon usage; the usage frequency of stop codon TAG is similar to that of TAA. Incomplete termination codons T and TA were also used. Termination codon TAA is used in 9 PCGs of C. virginica; if the incomplete termination codon is considered, the number of TAA is up to eleven. The PCGs with identical termination codon in the six genomes are nad1 and nad4L, which ended with TAA and incomplete stop codon T, respectively (Table 2).
The number of amino acids coded by each of the mt genomes is approximately equal. Excluding the stop codons, the C. virginica mtDNA encodes the least amino acids (3,696), while the C. gigas mtDNA encodes the most amino acids (3,718). Oysters with the lowest (60.1%) and highest (64.5%) A+T compositions of protein-coding region are C. virginica and C. hongkongensis, respectively. Similarly, oysters showing the lowest and highest A+T content of the first and the third positions are also C. virginica and C. hongkongensis: 55.0% and 58.3% for the first position, and 64.7% and 75.3% for the third position, respectively. However, the A+T content of the second position is approximately the same in the six genomes ranging from 60.0% in C. hongkongensis to 60.5% in C. virginica. It is obvious that the A+T content of the third codon position is higher than that of the first and the second positions. The genomic features of 19 bivalve sequences are presented in Table 1. The statistics of A+T content, start and stop codon, and amino acid number of PCGs in C. iredalei is already described in Wu et al's paper [54].
Nonsynonymous and synonymous substitutions
The estimation of nonsynonymous (Ka) and synonymous (Ks) substitution rates is of great significance in understanding evolutionary dynamics of protein-coding sequences across closely related species [55, 56]. To detect the influence of selection pressure in Crassostrea species, the numbers of Ka and Ks were calculated and their ratios were plotted for all pairwise comparisons among the seven oysters (Figure 3 and additional file 2: Table S2). The ratio of Ka/Ks in all 12 protein-coding genes varied from 0.001 for cox2 in C. angulata_C. sikamea and C. gigas_C. sikamea to 0.2657 for nad6 in C. angulata_C. ariakensis, which supports the existence of different mutation constraints among genes. Most of the amino acid substitutions are localized in the NADH complex genes. It suggests a relaxation of purifying selection in the nad6, nad2, nad3, nad5 genes compared with the more conservative genes such as cox1, cox2 and atp6.

The ratio of nonsynonymous and synonymous substitutions (Ka/Ks) estimated in all 12 protein genes of seven Crassostrea oysters.
Transfer and ribosomal RNA genes
The mt genomes of the six Asian Crassostrea oysters encode 25 tRNA genes. In addition to the 22 typical tRNAs, there are three duplications of trnM, trnK and trnQ. In comparison, however, there is no duplication of trnK and trnQ in C. virginica. The tRNA structure of C. gigas is referred to as the standard when the nucleotide variation in other four Asian oysters is examined (C. iredalei is not examined as it was not sequenced in this study). Nucleotide variations including transition/transversion and insertion/deletion mostly occur at DHU and TΨC loops (Additional file 3: Figure S1).
Duplication of tRNA is common in molluscs. A second trnM is also presented in the mussel Mytilus edulis, M. galloprovincialis and M. trossulus [45–47], and the clam V. philippinarum, H. arctica and A. tuberculata [48]. In P. magellanicus, there are up to ten copies of trnM [42]. In M. trossulus mtDNA, an additional copy of trnQ has also been reported [47]. The anticodon usage of oyster genomes is congruent to the corresponding tRNA of other molluscs with one exception. The anticodon of two trnMs in the oysters is CAU while that of two trnMs is CAU and UAU in Mytilus. The difference in anticodon corresponds to the third wobble position.
The boundaries of both the small and the large ribosomal genes were determined by BLAST with the revised annotation of C. gigas genome [23]. The rrnL gene is split into two segments, one segment of 5' end is distributed between trnQ1 and trnN, and the other segment of 3' end is located between cox1 and cox3. The split rrnL is first reported in C. virginica, but so far not reported in other metazoan mt genomes. The mt genome of S. mordax also has the split rrnL, however, it does not have the duplicated rrnS. The duplication of rrnS is only found in the six Asian Crassostrea oysters and the similarity between intraspecies and interspecies is high (95-100%) in the conserved 940 nucleotides at the center of rrnS sequences. The size of rrnL in the Asian Crassostrea oysters is nearly equal, and smaller than that in C. virginica (Table 1). The length of rrnS in C. virginica is 989 bp, while total length of rrnS in the Asian Crassostrea oysters varies from 2,232 bp (C. sikamea) to 2,264 bp (C. hongkongensis).
Non-coding regions
As in most bivalves, the oyster mtDNAs contain a large number of unassigned nucleotides. There are more than 30 non-coding regions throughout the seven Crassostrea genomes. The unassigned nucleotides vary from 1,788 bp in C. gigas to 5,873 bp in C. iredalei. The proportion of the unassigned nucleotides in the whole genome varies from 9.81% in C. gigas to 26.17% in C. iredalei. The largest non-coding region in the Asian oysters is located between gene trnG and trnV, while it is between gene trnP and trnG in C. virginica. The sequence of each corresponding non-coding region from the six Asian Crassostrea oysters was listed separately and regions large than 30 nucleotides were aligned using ClustalX 1.83. There are 14 non-coding regions aligned (See the Supplement to the non-coding regions). The alignment shows that there are sequence conservation in the non-coding region, especially, higher sequence conservation in the regions between trns2 and trnP, trnV and nad2. The sequence similarity among species in the non-coding region is positively correlated with their relatedness.
Phylogenetic analysis and divergence time estimation
Phylogenetic analysis using all mt coding sequences provides clear evolutionary relationships among the seven Crassostrea and one Saccostrea oyster species (Figure 4). C. gigas is first clustered with C. angulata and then united with C. sikamea, meanwhile C. hongkongensis and C. ariakensis formed a clade. Finally, C. iredalei with other five Crassostrea oysters form a large Asian clade. Together with Asian Crassostrea oysters, the C. virginica formed the Crassostrea clade. S. mordax diverged early from Crassostrea oysters and was positioned at the base of the large oyster clade. All the phylogenetic relationships are supported with high values. The close relationships between C. gigas and C. angulata, and C. hongkongensis and C. ariakensis are clearly demonstrated on the phylogenetic trees (Figure 4), which has been the subject of debate for some time [14, 12, 22]. The complete mt genomes of C. gigas and C. angulata differed by 3%, providing strong support for their status as two independent species.

Phylogenetic trees based on the concatenated amino acid (a) and nucleotide sequences (b) of 12 protein-coding genes (The branch length determined with NJ analysis). The chiton Katharina tunicata was used as outgroup. NJ (left number) and ML (right number) bootstrap values are given for each branch.
The first appearance of Gastropoda and Bivalvia in fossil record is approximately 542 Myr ago [40], providing the calibration constraint for divergence estimation. Divergence time estimates between species and/or clades are given in Table 3 and Figure 5. Our estimation shows that C. virginica and the Asian oysters diverged about 82.7 Myr ago. The six Asian Crassostrea oysters started their divergence about 42.8 Myr ago with the separation of C. iredalei from other species first. Other two large clades C. hongkongensis and C. ariakensis, and C. sikamea, C. gigas and C. angulata diverged 28.8 Myr ago. C. hongkongensis and C. ariakensis diverged 22.3 Myr ago. In the other clade, C. sikamea diverged from C. gigas and C. angulata 16.5 Myr ago. The two most closely related species, C. angulata and C. gigas, diverged about 2.7 Myr ago.

A phylogenetic tree with divergence time (in million years) of 19 molluscan species rooted with the chiton Katharina tunicata as determined by Bayesian phylogenetic analysis.
These estimates indicate that, relative to the divergence with C. virginica about 83 Myr ago, the speciation of the six Asian Crassostrea species is rather recent or 3-43 Myr ago. Still, it is difficult to reconcile molecular divergence with genome rearrangement data observed here. Assuming the divergence time estimates are correct, it is intriguing that the mt genomes of C. virginica and the Asian species diverged with nine structure rearrangements (6 transpositions and 3 duplications) in about 83 Myr, while there was not a single rearrangement event among the six Asian Crassostrea species in the past 43 Myr. During the same time frame, numerous rearrangement events occurred among scallop species (Figure 2). Thus, the complete conservation of gene order among the Asian Crassostrea oysters in the past 43 Myr is highly unusual, given the amazing rate of mt genome rearrangements in marine bivalves in general and within the genus Crassostrea itself. While this finding provides strong evidence for recent speciation of the six Asian Crassostrea species, it also indicates that mt genome rearrangement may not be strictly a function of time, but constrained by other factors. Detailed mechanisms of mt genome rearrangement are unknown. We suspect that coding on both strands may be one of the factors inhibiting mt genome rearrangement. This hypothesis is supported by the fact that the only two bivalve molluscs (L. ornata and I. japanensis) with dual-strand coding have relatively fewer rearrangements of gene order compared with other molluscan classes (Figure 2). All marine bivalves use one strand for coding and show tremendous rearrangements. The extensive rearrangement of bivalve mt genomes may be a consequence of coding on one strand. Furthermore, the fact that massive rearrangements in marine bivalves have not disrupted single-strand coding or changed the direction of transcription suggests that the rearrangements are not caused by inversion or reverse transposition. Thus, transposition or "tandem repeat-random loss"[49] may be important for mt genome rearrangement [29].
Dual-strand coding may not be the only factor limiting mt genome rearrangement, as it cannot explain the absence of rearrangements in the six Asian oysters over 43 Myr. We speculate that some other unknown features in mt genomes of the six Asia species may also limit genome rearrangements. The most noticeable structure change is the duplication of rrnS in the six Asian species, though it is difficult to determine if it has any effects on genome rearrangement at current stage. Further studies on the mt genomes of other oysters and marine bivalves may shed light on the origin and evolution of genome rearrangements. Gene order data have been shown to be valuable in phylogenetic analysis [3–6]. While our analysis demonstrates the power of rearrangement data, it also argues for a better understanding of mt genome rearrangement before using them to infer divergence time.
Conclusions
Asia Pacific has long been suspected to be the center of oyster speciation. Our analysis of complete mt genomes provides strong evidence that Crassostrea oysters have diversified in Asia and the divergence was rather recent or within the last 3-43 Myr. The complete conservation of gene order in the six Asian Crassostrea species over a period of 43 Myr is highly unusual given the remarkable rate of rearrangements in their sister species and other bivalves during the same time frame. It provides strong evidence, in addition to sequence data, for the recent speciation of Crassostrea oysters in Asia. It also indicates that changes in mt gene order may not be strictly a function of time but subject to other constraints that are presently not well understood.
Methods
Sample collection and DNA extraction
Specimens for the five Asian Crassostrea species were mostly collected from coastal waters of China, except that C. gigas was collected from a cultured population in Oregon, USA. C. gigas is a native species of Asia and was introduced to the West Coast of USA for aquaculture production. The oyster ID, geographic origin and collection date of each specimen are provided in Table 4. All oysters were identified according to their morphological characteristics first and confirmed with species-specific molecular markers [21]. Total genomic DNA was extracted from ethanol-fixed tissue with the CTAB method and dissolved in TE (10 mM Tris-HCl 1 mM EDTA pH 8.0) buffer before being stored at -20°C.
PCR amplification and DNA sequencing
Four pairs of primers were designed to amplify the complete mt genomes of C. gigas and C. angulata according to the sequence of C. gigas in GenBank (Additional file 3: Table S3). Sequencing primers were designed at intervals of about 500 bp. All PCR products were directly sequenced by primer-walking. Partial sequences of C. sikamea and C. ariakensis were obtained with the combined primers of the amplifying and sequencing primers designed for C. gigas. The remaining gaps were amplified with the species-specific primers designed according to the obtained sequences. As for C. hongkongensis, two short fragments of cox1 and cox2 were first amplified with the universal primer sets of LCO1490+ HCO2198 [53] and cox2F+cox2R [26, 27]. In addition, partial sequence of nad5 was amplified with the primers of C. gigas. Then, the whole mt genome was amplified based on three pairs of primers (Additional file 4: Table S3).
PCR reactions were performed with a Mastercycler gradient machine (Eppendorf). The cycling was set up with an initial denature step at 94°C for 2 min, followed by 35 cycles comprising denaturing at 94°C for 20 sec, annealing at 52-58°C for 1 min and elongation at 68°C or 72°C for 6 or 10 min depending on the expected length of the PCR products. The process was completed with a final elongation at 72°C for 10 min. The reaction volume amounted 25 μl containing 18.8 μl sterile deionized water, 2.5 μl 10×LA PCR buffer (Mg2+ plus, Takara), 1 μl dNTP mix (10 mM each), 1 μl each primer (5 μM), 0.2 μl LA Taq DNA polymerase (5 U/μl, Takara) and 0.5 μl DNA template (50 ng/μl). A negative control (no template) was included during each PCR run. PCR products were directly purified with MultiScreen-PCR96 Filter Plate (Millipore) and sequenced with ABI 3730x1 DNA Analyzer (Applied Biosystems).
Sequence analysis and gene annotation
Raw sequencing reads were first processed using Phred with the quality score 20 and assembled in Phrap with default parameters [28, 29]. Then, all assemblies and sequence quality were verified manually in Consed to remove misassemblies [30]. The accurate boundary of each gene was determined according to the annotated C. gigas mt genome [23] with minor revisions. The tRNA genes were identified by tRNAscan-SE 1.21 [31] employing the cove only search mode and the invertebrate mt genetic code. The ratio of nonsynonymous and synonymous substitutions rates (Ka/Ks) was estimated with Ka_Ks calculator in all 12 protein genes of seven Crassostrea oysters.
Phylogeny analysis and divergence time estimation
Nineteen molluscan mt genomes including four obtained in this study were used for phylogenetic analysis (Table 5). The black chiton Katharina tunicata (Polyplacophora) was rooted as the outgroup. The amino acid sequence from each of 12 protein-coding genes (excluding atp8) was aligned separately using ClustalX 1.83 [33], and then trimmed to the same length and concatenated for further analysis. The nucleotide sequence was substituted from the concatenated amino acid alignment. The final nucleotide sequence consisted of 9,537 sites. Two phylogenetic reconstruction approaches were performed including Maximum Likelihood (ML) with PHYML 3.0 [34] and Neighbor-Joining (NJ) with MEGA 4.0 [35]. The assessments of node reliability in both the ML and NJ analyses were done by using 1,000 bootstrap replicates.
Molecular estimates of divergence time for multiple gene data were performed using the relaxed Bayesian molecular clock approach as implemented in MULTIDISTRIBUTE package [36]. Key features of this program are that posterior distribution of molecular time estimates and rates of molecular evolution are approximated while simultaneously taking account of uncertainty in branch length estimates from each gene [37]. The F84+gamma evolution model, incorporating different rates of transition/transversion, variable nucleotide frequencies, and nucleotide variation across sites, was used to estimate the maximum likelihood parameter in PAML version 3.15 [38]. The Multidivtime program used the output from Estbranches analyses to estimate node divergence times for the ingroup, given time constraints, various parameters, and estimated priors. The median of all the tip-to-root branch lengths was calculated using ape and LAGOPUS package in R project [39]. Gamma priors were chosen as the following procedure outlined in the Multidivtime manual: expected time between the tip and the ingroup root (rttime) = 542 Myr (million years) ago, with standard deviation (SD) = 30 Myr ago; rate of the root node (rtrate) and its SD = 0.46 substitution per site per 100 Myr determined as the median of all the tip-to-root branch lengths divided by rttime; and rate of change between ancestral and descendant nodes (brownmean) = 0.18. Katharina tunicata was considered to be outgroup to Bivalvia and Gastropoda as required by the program. All divergence time were calculated assuming the topology of the consensus tree, which was derived from previous ML/Bayesian analyses based on protein sequences. The first appearance of skeletons in the fossil record, indicating the maximum for the origin of Gastropoda + Bivalvia is approximately 542 Myr ago, provides the calibration constraint for divergence estimation [40]. The parameters for the Markov Chain Monte Carlo (MCMC) simulation were set as follows: number of samples = 10,000, sample frequency = 200 and burn-in period = 2,000. Finally, Multidivtime analyses considering variance-covariance matrices from each gene partition were run twice ensure convergence, each one starting with a different random initial seed number.
References
Bridge D, Cunningham CW, Schierwater B, Desalle R, Buss LW: Class-level relationships in the phylum Cnidaria: evidence from mitochondrial genome structure. Proc Natl Acad Sci USA. 1992, 89: 8750-8753. 10.1073/pnas.89.18.8750.
Shadel GS, Clayton DA: Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997, 66: 409-435. 10.1146/annurev.biochem.66.1.409.
Watterson WA, Ewens WJ, Hall TE, Morgan A: The chromosome inversion problem. J Theor Biol. 1982, 99: 1-7. 10.1016/0022-5193(82)90384-8.
Sankoff D, Leduc G, Antoine N, Paquin B, Lang BF: Gene order comparisons for phylogenetic inference: evolution of the mitochondrial genome. Proc Natl Acad Sci USA. 1992, 89: 6575-6579. 10.1073/pnas.89.14.6575.
Fritzsch G, Schlegel M, Stadler P: Alignments of mitochondrial genome arrangements: applications to metazoan phylogeny. J Theor Biol. 2006, 240: 511-520. 10.1016/j.jtbi.2005.10.010.
Perseke M, Fritzsch G, Ramsch K, Bernt M, Merkle D, Middendorf M, Bernhard D, Stadler PF, Schlegel M: Evolution of mitochondrial gene orders in echinoderms. Mol Phylogenet Evol. 2008, 47: 855-864. 10.1016/j.ympev.2007.11.034.
Theologidis I, Fodelianakis S, Gaspar MB, Zouros E: Doubly uniparental inheritance (DUI) of mitochondrial DNA in Donax trunculus (Bivalvia: Donacidae) and the problem of its sporadic detection in Bivalvia. Evolution. 2008, 62: 959-970. 10.1111/j.1558-5646.2008.00329.x.
Yamazaki N, Ueshima R, Terrett JA, Yokobori SI, Kaifu M, Segawa R, Kobayashi T, Numachi KI, Ueda T, Nishikawa K, Watanabe K, Thomas RH: Evolution of pulmonate gastropod mitochondrial genomes: Comparisons of gene organizations of Euhadra, Cepaea and Albinaria and implications of unusual tRNA secondary structures. Genetics. 1997, 145: 749-758.
Akasaki T, Nikaido M, Tsuchiya K, Segawa S, Hasegawa M, Okada N: Extensive mitochondrial gene arrangements in coleoid Cephalopoda and their phylogenetic implications. Mol Phylogenet Evol. 2006, 38: 648-658. 10.1016/j.ympev.2005.10.018.
Serb JM, Lydeard C: Complete mtDNA sequence of the North American freshwater mussel, Lampsilis ornata (Unionidae): An examination of the evolution and phylogenetic utility of mitochondrial genome organization in Bivalvia (Mollusca). Mol Biol Evol. 2003, 20: 1854-1866. 10.1093/molbev/msg218.
Guo XM, Ford SE, Zhang FS: Molluscan aquaculture in China. J Shellfish Res. 1999, 18: 19-31.
Wang HY, Guo XM, Zhang GF, Zhang FS: Classification of jinjiang oysters Crassostrea rivularis (Gould, 1861) from China based on morphology and phylogenetic analysis. Aquaculture. 2004, 242: 137-155. 10.1016/j.aquaculture.2004.09.014.
Bernard FR, Cai YY, Morton B: Catalogue of living marine bivalve molluscs of China. 1993, Hong Kong: Hong Kong University Press
Lam K, Morton B: Mitochondrial DNA and morphological identification of a new species of Crassostrea (Bivalvia: Ostreidae) cultured for centuries in the Pearl River Delta, Hong Kong, China. Aquaculture. 2003, 228: 1-13. 10.1016/S0044-8486(03)00215-1.
Banks MA, McGoldrick DJ, Borgeson W, Hedgecock D: Discrimination between closely related Pacific oysters species (Crassostrea) via mitochondrial DNA sequences coding for large subunit rRNA. Mol Mar Biol Biotechnol. 1993, 2: 129-136.
Littlewood DT: Molecular phylogenetics of cupped oysters based on partial 28 S rRNA gene sequences. Mol Phylogenet Evol. 1994, 3: 221-229. 10.1006/mpev.1994.1024.
Ó Foighil D, Gaffney PM, Hilbish TJ: Differences in mitochondrial 16 S ribosomal gene sequences allow discrimination among American (Crassostrea virginica) and Asian (C. gigas, C. ariakensis) oyster species. J Exp Mar Biol Ecol. 1995, 192: 211-220. 10.1016/0022-0981(95)00065-Y.
Ó Foighil D, Gaffney PM, Wilbur WA, Hilbish TJ: Mitochondrial cytochrome oxidase I gene sequence support an Asian origin for the Portuguese oyster Crassostrea angulata. Mar Biol. 1998, 131: 497-503. 10.1007/s002270050341.
Ó Foighil D, Taylor DJ: Evolution of parental care and ovulation behavior in oysters. Mol Phylogenet Evol. 2000, 15: 301-313. 10.1006/mpev.1999.0755.
Lapègue S, Boutet I, Leitão A, Heurtebise S, Garcia P, Thiriot-Quiévreux C, Boudry P: Trans-Atlantic distribution of a mangrove oyster species revealed by 16 S mtDNA and karyological analyses. Biol Bull. 2002, 202: 232-242. 10.2307/1543473.
Wang HY, Guo XM: Identification of Crassostrea ariakensis and related oysters by multiplex species-specific PCR. J Shellfish Res. 2008, 27: 481-487. 10.2983/0730-8000(2008)27[481:IOCAAR]2.0.CO;2.
Reece KS, Cordes JF, Stubbs JB, Francis E, Hudson K: Molecular phylogenies help resolve taxonomic confusion with Asian Crassostrea oyster species. Mar Biol. 2008, 153: 709-721. 10.1007/s00227-007-0846-2.
Milbury CA, Gaffney PM: Complete mitochondrial DNA sequence of the eastern oyster Crassostrea virginica. Mar Biotechnol. 2005, 7: 697-712. 10.1007/s10126-005-0004-0.
Yu ZN, Wei ZP, Kong XY, Shi W: Complete mitochondrial DNA sequence of 3 rtraud Lydeardoyster Crassostrea hongkongensis - a case of "Tandem duplication-random loss" for genome rearrangement in Crassostrea?. BMC Genomics. 2008, 9: 477-10.1186/1471-2164-9-477.
Ren JF, Liu X, Zhang GF, Liu B, Guo XM: "Tandem duplication-random loss" is not a real feature of oyster mitochondrial genomes. BMC Genomics. 2009, 10: 84-10.1186/1471-2164-10-84.
Burger G, Lavr DV, Forget L, Lang BF: Sequencing complete mitochondrial and plastid genomes. Nature Protocols. 2007, 2: 603-614. 10.1038/nprot.2007.59.
Boore JL, Brown WM: Mitochondrial genomes of Galathealinum, Helobdella, and Platynereis: Sequence and gene arrangement comparisons indicate that Pogonophora is nota phylum and Annelida and Arthropoda are not sister taxa. Mol Biol Evol. 2000, 17: 87-106.
Ewing B, Green P: Basecalling of automated sequencer traces using phred. II Error probabilities Genome Res. 1998, 8: 186-194.
Ewing B, Hillier L, Wendl M, Green P: Basecalling of automated sequencer traces using phred. I Accuracy assessment Genome Res. 1998, 8: 175-185.
Gordon D, Abajian C, Green P: Consed: a graphical tool for sequence finishing. Genome Res. 1998, 8: 195-202.
Lowe TM, Eddy SR: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25: 955-964. 10.1093/nar/25.5.955.
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Thorne JL, Kishino H: Divergence time and evolutionary rate estimation with multilocus data. Syst Biol. 2002, 51: 689-702. 10.1080/10635150290102456.
Pereira SL, Baker AJ: A mitogenomic timescale for birds detects variable phylogenetic rates of molecular evolution and refutes the standard molecular clock. Mol Biol Evol. 2006, 23: 1731-1740. 10.1093/molbev/msl038.
Yang Z: PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997, 13: 555-556.
Paradis E, Claude J, Strimmer K: APE: Analyses of phylogenetics and evolution in R language. Bioinformatics. 2004, 20: 289-290. 10.1093/bioinformatics/btg412.
Peterson KJ, Cotton JA, Gehling JG, Pisani D: The Ediacaran emergence of bilaterians: congruence between the genetic and the geological fossil records. Philos Trans R Soc Lond B Biol Sci. 2008, 363: 1435-1443. 10.1098/rstb.2007.2233.
DeJong RJ, Emery AM, Adema CM: The mitochondrial genome of Biomphalaria glabrata (Gastropoda: Basommatophora), intermediate host of Schistosoma mansoni. J Parasitol. 2004, 90: 991-997. 10.1645/GE-284R.
Smith DR, Snyder M: Complete mitochondrial DNA sequence of the scallop Placopecten magellanicus: evidence of transposition leading to an uncharacteristically large mitochondrial genome. J Mol Evol. 2007, 65: 380-391. 10.1007/s00239-007-9016-x.
Boore JL, Brown WM: The complete DNA sequence of the mitochondrial genome of the black chiton Katharina tunicata. Genetics. 1994, 138: 423-443.
Wolstenholme DR: Animal mitochondrial DNA: structure and evolution. Intl Rev Cytology. 1992, 141: 73-216.
Hoffmann RJ, Boore JL, Brown WM: A novel mitochondrial genome organization for the blue mussel, Mytilus edulis. Genetics. 1992, 131: 397-412.
Mizi A, Zouros E, Moschonas N, Rodakis GC: The complete maternal and paternal mitochondrial genomes of the Mediterranean mussel Mytilus galloprovincialis: implications for the doubly uniparental inheritances mode of mtDNA. Mol Biol Evol. 2005, 22: 952-967. 10.1093/molbev/msi079.
Breton S, Burger G, Stewart DT, Blier PU: Comparative analysis of gender-associated complete mitochondrial genomes in marine mussels (Mytilus spp). Genetics. 2006, 172: 1107-1119. 10.1534/genetics.105.047159.
Dreyer H, Steiner G: The complete sequences and gene organisation of the mitochondrial genomes of the heterodont bivalves Acanthocardia tuberculata and Hiatella arctica and the first record for a putative Atpase subunit 8 gene in marine bivalves. Front Zool. 2006, 1: 3-13.
Chaudhuri K, Chen K, Mihaescu R, Rao S: On the tandem duplication-random loss model of genome rearrangement. Proceedings of the 17th Annual ACM-SIAM Symposium on Discrete Algorithms. 2006, 564-570. full_text.
Ren JF, Shen X, Sun MA, Jiang F, Yu Y, Chi ZF, Liu B: The complete mitochondrial genome of the clam Meretrix petechialis (Mollusca: Bivalvia: Veneridae) Mitochondrial DNA. 2009, 20: 78-87.
Boore JL, Medina M, Rosenberg LA: Complete sequences of the highly rearranged molluscan mitochondrial genomes of the scaphopod Graptacme eborea and the bivalve Mytilus edulis. Mol Biol Evol. 2004, 21: 1492-1503. 10.1093/molbev/msh090.
Ren JF, Shen X, Jiang F, Liu B: The mitochondrial genomes of two scallops, Argopecten irradians and Chlamys farreri (Mollusca: Bivalvia): the most highly rearranged gene order in the family Pectinidae. J Mol Evol. 2010, 70: 57-68. 10.1007/s00239-009-9308-4.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R: DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech. 1994, 3: 294-299.
Wu XY, Xu XD, Yu ZN, Wei ZP, Xia JJ: Comparison of seven Crassostrea mitogenomes and phylogenetic analyses. Mol Phylogenet Evol. 2010
Ohta T: Synonymous and nonsynonymous substitutions in mammalian genes and the nearly neutral theory. J Mol Evol. 1995, 40: 56-63. 10.1007/BF00166595.
Fay JC, Wu CI: Sequence divergence, functional constraint, and selection in protein evolution. Annu Rev Genomics Hum Genet. 2003, 4: 213-235. 10.1146/annurev.genom.4.020303.162528.
Acknowledgements
We thank Mengjie Qiu for DNA sequencing. We thank the editors and the anonymous reviewers for their insightful comments. This work was supported by the Bairen program of Chinese Academy of Sciences (to BL) and by the MFG Fund of Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
JR did PCR, sequencing and initial analysis; FJ did some data analysis; XG and XL provided the samples; BL, XG and XL conceived the study; JR, BL and XG wrote and revised the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2010_1609_MOESM2_ESM.XLS
Additional file 2: Table S2: The ratio of nonsynonymous and synonymous substitutions (Ka/Ks) estimated with Ka_Ks calculator in all 12 protein genes of seven Crassostrea oysters. (XLS 18 KB)
12862_2010_1609_MOESM3_ESM.DOC
Additional file 3: Figure S1: Comparison of the potential secondary structures of the 25 inferred tRNAs among five Crassostreaoyster mtDNAs. (DOC 9 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ren, J., Liu, X., Jiang, F. et al. Unusual conservation of mitochondrial gene order in Crassostreaoysters: evidence for recent speciation in Asia. BMC Evol Biol 10, 394 (2010). https://doi.org/10.1186/1471-2148-10-394
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-10-394