
Abstract
Background
In the large angiosperm order Lamiales, a diverse array of highly specialized life strategies such as carnivory, parasitism, epiphytism, and desiccation tolerance occur, and some lineages possess drastically accelerated DNA substitutional rates or miniaturized genomes. However, understanding the evolution of these phenomena in the order, and clarifying borders of and relationships among lamialean families, has been hindered by largely unresolved trees in the past.
Results
Our analysis of the rapidly evolving trnK/matK, trnL-F and rps16 chloroplast regions enabled us to infer more precise phylogenetic hypotheses for the Lamiales. Relationships among the nine first-branching families in the Lamiales tree are now resolved with very strong support. Subsequent to Plocospermataceae, a clade consisting of Carlemanniaceae plus Oleaceae branches, followed by Tetrachondraceae and a newly inferred clade composed of Gesneriaceae plus Calceolariaceae, which is also supported by morphological characters. Plantaginaceae (incl. Gratioleae) and Scrophulariaceae are well separated in the backbone grade; Lamiaceae and Verbenaceae appear in distant clades, while the recently described Linderniaceae are confirmed to be monophyletic and in an isolated position.
Conclusions
Confidence about deep nodes of the Lamiales tree is an important step towards understanding the evolutionary diversification of a major clade of flowering plants. The degree of resolution obtained here now provides a first opportunity to discuss the evolution of morphological and biochemical traits in Lamiales. The multiple independent evolution of the carnivorous syndrome, once in Lentibulariaceae and a second time in Byblidaceae, is strongly supported by all analyses and topological tests. The evolution of selected morphological characters such as flower symmetry is discussed. The addition of further sequence data from introns and spacers holds promise to eventually obtain a fully resolved plastid tree of Lamiales.
Similar content being viewed by others


Background
With more than 23,000 species in at least 23 families [1], Lamiales (eudicots/asterids) are one of the largest orders of flowering plants, with representatives found all over the world. The highest diversity is contributed by herbaceous plants with mono-symmetric flowers. Some members are economically important, such as Lamiaceae (pot-herbs like mint, sage, oregano or basil), Oleaceae (olives), Pedaliaceae (sesame), Verbenaceae (timber, medicinal) Plantaginaceae (drugs like digitalis, ornamentals) and Scrophulariaceae (ornamentals). The order contains lineages with highly specialized life forms and traits of particular scientific interest. So far, their comparative study has been limited by the lack of a robust phylogenetic framework for Lamiales. Desiccation-tolerant members (so-called "resurrection plants", see Figure 1a) of the recently described family Linderniaceae [2] are a focus of molecular and evolutionary studies [3, 2]. Extreme metabolic and genomic shifts are exhibited by parasitic plants. With Orobanchaceae, Lamiales harbor the largest number of parasitic angiosperms (Figure 1b). The family comprises both hemi- and holoparasites [4], with some species causing serious damage in agriculture [5]. Chloroplast genomes of members of Orobanchaceae show gene order rearrangements, high evolutionary rates and gene losses, potentially as a consequence of parasitism in this family. One line of current research in the family concentrates on gradual plastid evolution under increasingly relaxed functional constraints [Wicke et al., in prep].

Example taxa from Lamiales, showing representatives of desiccation-tolerant, parasitic, and carnivorous lineages, as well as members from families frequently referred to in the text. a: the desiccation-tolerant Craterostigma pumilum from Linderniaceae; b: the holoparasitic Orobanche gracilis from Orobanchaceae, a family that contains all hemi- and holoparasites from Lamiales; c: Pinguicula leptoceras from Lentibulariaceae, the largest family of carnivorous plants in angiosperms; d: Pinguicula filifolia, with a habit resembling Byblis; e: Byblis gigantea from Byblidaceae, another carnivorous lineage previously suspected to be the closest relative of Lentibulariaceae; f: Rhynchoglossum gardneri from Gesneriaceae and g Calceolaria andina from Calceolariaceae, two families inferred here as sister groups based on molecular data, alveolated seeds and pair-flowered cymes; h Prunella grandiflora (Lamiaceae), i: Verbena bonariensis (Verbenaceae); both families were long regarded as close relatives but are inferred as only distantly related (Figure 2). Photos: a: E.F.; c, d, e: A.F.; f: Nadja Korotkova; g: D.C.A.; b, h, i: K.F.M.
Carnivory in Lamiales
Lentibulariaceae, the most species-rich family of carnivorous plants (ca. 350 spp.) belongs to Lamiales (Figure 1c, d). This family is unique for a variety of reasons: traps of Utricularia (bladderworts) are regarded as a complex modification of leaves [6, 7], and the typical angiosperm body plan is strongly relaxed in members of this genus [8–10]. Utricularia and its sister genus, Genlisea (the corkscrew plants), are the only carnivorous angiosperms known to feed on protozoa [11]. They have the smallest holoploid genome sizes among angiosperms, with some nuclear genomes as small as 63 Mbp or less [12], and exhibit the highest relative DNA substitution rates for some of the investigated chloroplast genome regions [13, 14]. Pinguicula (butterworts), the third genus of Lentibulariaceae, is far less extreme in genome size, substitution rate and morphology, and exhibits glandular leaves that function as adhesive ("flypaper") traps (Figure 1c, d).
Apart from Lentibulariaceae, the monogeneric Australian family Byblidaceae (Figure 1e) also attracts and catches insects with simple flypaper traps comparable in function to those of Pinguicula. The carnivorous syndrome of Byblis was questioned by some authors, as the plants were considered to lack their own digestive enzymes and have not been demonstrated to be able to take up released nutrients, thus being ranked as merely "protocarnivorous" [15]. However, a recent study [16] detected phosphatase activity, thereby restoring the rank of carnivory to Byblis. Morphological links - flypaper trap leaves that are densely covered with multicellular, non-vascularized epidermal glands, as well as embryology [17, 18] - and early phylogenetic studies suggested a sister relationship of Byblidaceae and Lentibulariaceae [19], thus hypothesizing a single origin of carnivory in the order, which was questioned later [14]. With the recently described genus Philcoxia [20], a further supposedly "protocarnivorous" lineage emerged and was placed in Lamiales [21]. Although a first test of enzymatic activity was negative [21], this might have been an artifact caused by the minuteness of the leaves, and further experiments to test its status as potentially fully carnivorous are underway.
Understanding the evolution of the morphological, ecological, and genomic peculiarities in the order heavily relies on having robust hypotheses on organismal relationships. For example, knowledge of the closest relatives of resurrection plants, parasites, and carnivores, respectively, would enable us to infer (pre-) adaptations and genomic changes on the evolutionary path leading to each of these specialized groups.
Phylogeny and systematics of Lamiales: current state of knowledge
While the monophyly of many of the currently accepted families has been inferred with confidence by a number of molecular phylogenetic studies [22, 23], there has been only little progress on understanding the relationships among families. Nearly all phylogenetic trees produced so far lacked resolution and support for inter-familiar relationships of Lamiales [24–26]. This has earned Lamiales the reputation of being among the most difficult angiosperm clades to resolve [27].
Circumscription of Lamiales and the inclusion of Hydrostachys
The current concept of Lamiales [28] expands the earlier order Lamiales from pre-cladistic classification systems [29, 30] to also include former Scrophulariales and Oleales. While there is overwhelming evidence for the monophyly of Lamiales circumscribed like this [28], the surprising inclusion of Hydrostachys as an early branch in Lamiales was recently proposed [31]. Hydrostachys is a rheophyte from Africa and Madagascar suggested to be related to Cornales in most previous analyses of DNA sequence data, albeit without consistent placement in this order [32–34].
Most studies converged on a set of most likely candidates for the first branches of the Lamiales tree. Oleaceae have been consistently identified as being among the first branches [2, 14, 24, 35]. Whenever the monotypic Plocospermataceae from Central America had been included in the sampling [26, 35], they were found to be sister to the remaining Lamiales. In contrast, the Carlemanniaceae-suspected to have affinities of some kind to early branching Lamiales - have never been analyzed in the context of a broad Lamiales sampling. Tetrachondraceae have been resolved as a branch following Oleaceae [36, 26].
No clear picture in more derived parts of tree
In contrast, there has not been any consistent hypothesis on the "backbone" of the remainder of the Lamiales tree [37, 31]. Conflicting hypotheses have been put forward with regard to the relationships of Gesneriaceae and Calceolariaceae (Figure 1f, g) to each other and to remaining Lamiales. A successive branching order of Oleaceae, Calceolariaceae, Gesneriaceae, and remaining Lamiales was originally suggested [38, 39], but support for the placement of Gesneriaceae and for the monophyly of the more derived remaining Lamiales was always negligible. On the other hand, a clade including Gesneriaceae and Calceolariaceae was hypothesized [2, 40, 41]. Consequently, relationships of Calceolariaceae remained indistinct, and until now there has been no study sampling all families from early branching Lamiales with a sufficient amount of sequence data to provide a clear picture.
The situation is even worse for the more derived, remaining lineages of the Lamiales tree - as far as the backbone and relationship among families is concerned, almost no resolution could be obtained by previous studies [42, 31, 43].
The new circumscription of many traditional families
Lamiales are also known for the decomposition of previously widely accepted families due to phylogenetic insights.
Scrophulariaceae and Plantaginaceae
The most prominent case for a family that turned out to be polyphyletic are the Scrophulariaceae. In their traditional circumscription they used to be the largest family (more than 5000 spp. [44]) among Lamiales. In the first report on the polyphyly of Scrophulariaceae [45], members of the "old" Scrophulariaceae sensu lato were found in two different clades, named "scroph I" (including Scrophularia) and "scroph II" (containing Plantago, Antirrhinum, Digitalis, Veronica, Hippuris and Callitriche). The first clade was later [38] referred to as Scrophulariaceae sensu stricto (s. str.), while the "scroph II" clade was called Veronicaceae. However, since Plantago is contained in that clade, Plantaginaceae as the older name should be given priority and meanwhile became accepted for this clade [46, 28]. Plantaginaceae experienced an enormous inflation since these early studies, when more and more genera from former Scrophulariaceae s. l. were included in phylogenetic studies and identified as members of this newly circumscribed family [22, 37–39]. Some genera from tribe Gratioleae, including Gratiola itself, have been found in a well supported clade. Based on the unknown relationships to the the other lamialean families, it has been suggested to separate this part of the inflated Plantaginaceae by restoring family rank to former tribe Gratioleae from Scrophulariaceae as traditionally circumscribed [2].
Orobanchaceae
Initial molecular phylogenetic studies [47, 48] showed that all hemi-parasitic members of the former Scrophulariaceae s. l. should be included in a newly circumscribed Orobanchaceae while the non-parasitic genus Lindenbergia was found sister to all hemi- and holoparasites and also included in Orobanchaceae. In this expanded circumscription [4, 49], the monophyly of Orobanchaceae is strongly supported by all studies, and the family now comprises 89 genera with about 2000 species [49] and unites phototrophic, hemi- and holoparasitic plants. As next relatives to Orobanchaceae, a clade consisting of the East Asian genera Rehmannia (six species) and Triaenophora (one or two species) was identified recently [43, 50].
Phrymaceae
Shortly after the first reports on the polyphyly of Scrophulariaceae [45], it was noticed that Mimulus (tribe Mimuleae) neither clustered with the "scroph I" nor the "scroph II" clade, but instead was found in a group together with Lamiaceae, Paulownia and Orobanchaceae [38]. Sampling the taxonomically isolated Phryma (Phrymaceae), but not Mimulus, Phryma appeared as sister to Orobanchaceae plus Paulownia [26]. In an attempt to redefine the Phrymaceae, their circumscription was expanded to include Mimulus, Hemichaena, Berendtiella, Leucocarpus, Glossostigma, Peplidium, Elacholomia, Lancea, and Mazus [51]. However, relationships to other families of Lamiales remained unclear. Sampling six genera from Phrymaceae [39], two clades emerged: one comprising Mimulus, Phryma, Hemichaena and Berendita, the other including Mazus and Lancea being sister to Rehmannia. Thus, the monophyly of Phrymaceae was put into question.
Linderniaceae
Linderniaceae were described as a new family independent from Scrophulariaceae, comprising genera formerly classified in the tribe Lindernieae of Scrophulariaceae s. l. and are characterized by stamens in which the abaxial filaments are conspicuously geniculate, zigzag shaped or spurred [2, 52, 53]. The original recognition as a distinct clade was based upon a taxon set including the genera Artanema, Craterostigma, Crepidorhopalon, Torenia and Lindernia. The existence of a Linderniaceae clade was confirmed by other studies comprising Craterostigma, Lindernia, Torenia and Micranthemum [22] or Stemodiopsis, Micranthemum, Torenia and Picria [39].
Calceolariaceae
Jovellana and Calceolaria (formerly Calceolarieae/Scrophulariaceae) were identified as another lineage separate from Scrophulariaceae, which led to recognizing them at family level (Calceolariaceae) [38]. The authors of this study initially also listed Porodittia as genus of this new family, but a subsequent study [41] showed Porodittia to be nested in Calceolaria.
Schlegeliaceae, Paulowniaceae, and Stilbaceae
The genera Paulownia and Schlegelia, which had been traditionally included either in Bignoniaceae or Scrophulariaceae, were not found to be related to any of these families based on molecular data [54] and therefore treated as families of their own [55, 56]. In addition, Halleria was transferred from Scrophulariaceae to Stilbaceae [38]. Molecular phylogenetic studies later expanded the circumscription of Stilbaceae to a total of 11 genera [37, 39].
Aims of this study
Using a dataset representing all major lineages from Lamiales, the goal of the present study was to investigate inter-familial relationships within Lamiales, in the hope to come up with a better resolved tree that provides the basis for an interpretation of the evolution of the above-mentioned morphological, ecological, and molecular peculiarities observed in the order.
Since the protein-coding genes usually applied to the inference problem in Lamiales have not provided satisfactory resolution in the past, the approach in the current study was to employ non-coding and rapidly evolving chloroplast DNA. Introns and spacers have been demonstrated to be a valuable source of phylogenetic signal even on deeper taxonomic levels than they used to be applied to [57–59]. Mutational dynamics of non-coding regions also include microstructural changes in addition to substitutions, and generally are less constrained than coding genes [60]. Non-coding markers have been shown to be significantly more informative than coding regions [57]. Even more, non-coding markers have been successfully applied to disentangle deep nodes in angiosperm evolution [58].
Methods
Taxon sampling and plant material
Sequences from the plastid markers trnK/matK, trnL-F and rps16 were newly generated or downloaded from GenBank for 98 taxa from Lamiales, two outgroup taxa from Solanaceae, and one from Rubiaceae. All 23 families currently accepted for Lamiales [28] were sampled. Since one of the specific questions in our study was the relationship between Lentibulariaceae and Byblidaceae, which might have been blurred by long branch attraction (LBA) problems in previous studies, we slightly enhanced sampling for both families in one set of analyses and included two to three species for each genus. The complete material sampled is shown in Table 1. Using fewer representatives for either family did not change results. We also used a somewhat denser taxon sampling for Gratioleae (Plantaginaceae) in order to (i) examine whether the distinctness of this tribe [2] can be confirmed after taxan sampling enhancement and (ii) doublecheck the position of the apparently "protocarnivorous" genus Philcoxia.
Amplification and sequencing
Total genomic DNA was isolated using the AVE Gene Plant Genomics DNA Mini Kit (AVE Gene, Korea), according to the manufacturer's protocol. As phylogenetic markers, the trnK intron including the coding matK, the trnL-F region, and the rps16 intron were amplified using standard PCR protocols. Primers used for amplification and sequencing are given in Table 2. Reactions were performed in 50 μl volumes containing 2 μl template DNA (10 ng/μl), 10 μl dNTP mix (1.25 mM each), 2 μl of each forward and reverse primer (20 pm/μl), and 0.25 μl Taq polymerase (5 U/μl, Peqlab). Thermal cycling was performed on an Biometra T3 thermocycler using the following PCR profiles: 1:30 min at 96°C, 1 min at 50°C, 1:30 min at 72°C, 35 cycles of 30 sec at 96°C, 1 min at 50°C, 1:30 min at 72°C, and a final extension time of 10 min at 72°C for the trnK intron; 35 cycles of 1 min at 94°C, 1 min at 52°C and 2 min at 72°C, followed by a final extension time of 15 min at 72°C for the trnL-F region; 1:30 min at 94°C, 30 cycles of 30 sec at 94°C, 30 sec at 56°C and 1 min at 72°C, and a final extension time of 15 min at 72°C for the rps16 intron. Fragments were gel-purified on a 1.2% agarose gel (Neeo-agarose, Roth), extracted with the Gel/PCR DNA Fragments Extraction Kit (AVE Gene, Korea) and sequenced on an ABI3730XL automated sequencer using the Macrogen sequencing service (Macrogen Inc., Seoul, Korea). Pherogram editing and contig assembly was done manually.
Addition and analysis of GenBank sequence data
We additionally took rbcL and ndhF sequences (see Additional file 1, Table S1) for relevant taxa from GenBank, and in a separate set of analyses combined them with our three marker dataset. Taxon sampling of these four- and five-region datasets was adapted to include only taxa with all regions present.
Because the position of Hydrostachys remained inconsistent in previous studies, all sequences from that genus existing in GenBank were blasted against the entire data of GenBank via blastn [61]. Additionally, trnK/matK, rps16 and trnL-F sequences for Hydrostachys from a collection independent from those previously used [31, 33, 62, 63] were generated; all sequences used, including voucher information, are given in Table 1. The newly generated Hydrostachys matK sequence was aligned to an existing angiosperm matK alignment [35] and subjected to parsimony analysis.
Alignment and indel coding
DNA sequences were manually aligned in PhyDE [64], taking microstructural changes into account as outlined elsewhere [58, 65]. Regions of uncertain homology were excluded from phylogenetic analyses. For maximum parsimony (MP) analyses and Bayesian Inference of Phylogeny (BI), indels were coded according to simple indel coding (SIC) [66] using the program SeqState [67].
Parsimony analyses
Searches for the shortest tree were performed using the parsimony ratchet approach implemented in PRAP2 [68] using the following settings: 10 random addition cycles with 200 ratchet replicates, setting the weight for 25% of the characters to 2. The files generated were executed in PAUP* v4.0b10 [69]. Bootstrapping was performed with 10,000 replicates, each using TBR branch swapping and holding only one tree [70]. We measured the additional information provided by SIC-coded indels by the difference in decay indices (computed with PRAP2) for each node, comparing analyses with and without indels.
Bayesian Inference of Phylogeny
Bayesian inference (BI) of phylogeny was done with help of MrBayes v3.1.2 [71]. The model of best fit for the combined dataset as well as for each of the three partitions (trnK/matK, rps16 and trnL-F) was found to be GTR+G+I model was found as the optimal one using jModelTest v.0.1.1 [72]. The indel partition was co-analyzed together with the DNA partition, with the restriction site (binary) model applied to the gap characters and the ascertainment (coding) bias set to "variable". Default priors were used, i.e. flat dirichlets (1.0, 1.0) for state frequencies and instantaneous substitution rates, a uniform prior (0.0, 50.0) for the shape parameter of the gamma distribution, a uniform prior (0.0, 1.0) for the proportion of invariable sites, a uniform topological prior, an exponential prior Exp (10.0) for branch lengths. Four categories were used to approximate the gamma distribution. Two runs with 5 million generations each were run, and four chains were run in parallel for each run, with the temperature set to 0.2. The chains were sampled every 100th generation, and the burnin was set to 5000. To check for convergence of the independent runs under a given model, it was ensured that the plots of both runs indicated that the stationary phase was reached, that the potential scale reduction factor approached 1 for all parameters, and that no supported conflicting nodes were found among the consensus trees generated from each run. Convergence and effective sampling sizes (ESS) of all parameters were assessed with halp of Tracer v1.5 [73].
Maximum likelihood analyses
For maximum likelihood (ML) analyses RAxML v7.0.0 [74] was used. During the search for the best tree, the GTRGAMMA model was used, while the slightly simpler GTRCAT model was employed by RAxML during the 500 bootstrap replicates. Support values from all types of analysis were mapped on the tree topology from the ML analysis and conflicting nodes were identified with help of TreeGraph2 [75].
Topological tests
Topological tests were used to see whether alternative topologies could be rejected with confidence. Specifically it was tested whether evidence against Byblidaceae being sister to Lentibulariaceae was strong. Under parsimony, the Templeton and Winning-sites (sign) tests were used ("NonparamTest" option in Paup*), while under the likelihood criterion, the Approximately Unbiased test (AU-Test) [76] along with the more classical Shimodaira-Hasegawa test (SH-test [77]), as implemented in consel 0.1j [78], were employed.
Ancestral state reconstruction
We inferred ancestral states for ten selected morphological characters. Information on character states was compiled from different sources [79, 1, 27, 80] and is given in Table 3. We took the fully resolved best tree from the RAxML search, and traced the evolution of these characters on that topology via maximum likelihood, using the "multistate" command in BayesTraits [81].
Results
Sequence statistics and results from tree searches
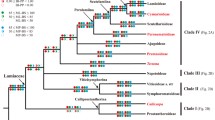
Sequences of trnK/matK, trnL-F and rps16 yielded an alignment of 7809 characters, of which 1739 were excluded from subsequent analysis because of uncertain homology. The alignment is available from TreeBase (http://purl.org/phylo/treebase/phylows/study/TB2:S10963); detailed sequence statistics are given in Table 4. Consensus trees from parsimony analyses were well resolved and supported. The MP trees from substitutions only were 13118 steps long (CI 0.419, RI 0.504,), those based on substitution and indel characters had a length of 14719 steps (CI 0.453, RI 0.507,). Comparison of decay values of substitution data versus substitutions plus SIC-coded indels showed higher decay values for most nodes when indel information was included (see Additional file 2, Figure S1). Trees from coding rbcL and ndhF seqences were far less resolved than those from our three marker combined analysis (Additional file 3 Figure S2 and Additional file 4, Figure S3). The tree topology from the ML analysis is shown in Figure 2, collapsing nodes support by less than 50% in at least one of the tree methodological approaches. BI and ML trees generally showed slightly higher resolution and statistical support than trees from MP searches. Effective sampling sizes (ESS) of all parameters from the Bayesian analysis were > 150. A phylogram from BI with branch lengths indicating relative substitution rates is given in Figure 3.

Phylogeny of Lamiales inferred from parsimony, likelihood and Bayesian analysis of sequences from plastid trnK / matK , trnL-F and rps 16. Topology from the maximum likelihood tree depicted, collapsing nodes not supported by > = 50% in at least one of the three analyses. Bold numbers above branches are posterior probabilities from Bayesian inferences, italic numbers above branches are MP bootstrap values, number below branches indicate ML bootstrap proportions. Numbers in brackets indicate that the respective node was not supported by all three methodological approaches. The bracketed number then indicates the strongest support found for any node that contradicts the shown node [69]. Familial annotation according to APG III [28]. For Phrymaceae monophyly is not confirmed, so subfamilies are annotated; Rehmannia is currently not assigned to a family.

Phylogram from Bayesian Inference of phylogeny with branch lengths giving the relative substitution rates using the GTR+G+I model.
Resolution of the backbone of the Lamiales phylogeny
The precise branching pattern of the nine first-branching families in the Lamiales tree (Plocospermataceae, Carlemanniaceae, Oleaceae, Tetrachondraceae, Calceolariaceae, Gesneriaceae, Plantaginaceae (incl. Gratioleae), Scrophulariaceae) is inferred with very high or maximum (most cases) support (Figure 2). A total of 16 nodes determining this branching pattern among families along the spine of the basal Lamiales grade receive very high or maximum support by all (most cases) or at least two out of three inference methods. An additional 19 of the nodes indicating delimitation and relative position of the remaining 15 more derived families receive very high or maximum support by at least one out of three analytic approaches.
Phylogenetic position of Hydrostachys
In our blastn searches, all sequences (rbcL, atpB, 18s rDNA, 26s rDNA, ndhF, matK) reached highest similarity scores to other Hydrostachys sequences, followed by sequences from Cornales taxa (Hydrangeaceae, Cornaceae, Loasaceae), with the exception of the matK sequence of Hydrostachys multifida (AY254547) of Hufford et al. [82] used in the study of Burleigh et al. [31]. This sequence showed highest similarity with Hydrangea hirta and a number of sequences from Avicennia. When included in the present trnK/matK alignment, the high similarity of sequence AY254547 to Avicennia is obvious. A blast search of the newly generated matK sequence of Hydrostachys [EMBL: FN8112689] resulted in best matches with taxa from Cornales. Aligning and analyzing the newly generated trnK/matK, rps16 and trnL-F sequences, Hydrostachys is resolved outside Lamiales. Parsimony analysis of the newly generated matK sequence in the context of the angiosperm matK data set [35] evidently places the newly generated matK sequence of Hydrostachys outside Lamiales, although its precise position within asterids remains unresolved in the 50%-majority-rule-bootstrap tree (Additional file 5, Figure S4).
Position of carnivorous lineages
In neither the Bayesian nor the maximum likelihood analysis Byblidaceae were found closely related to Lentibulariaceae. In MP analyses, the position of Byblidaceae receives no bootstrap support; interestingly, however, the strict consensus from all shortest trees depicts Byblidaceae as sister to Lentibulariaceae, regardless of the inclusion of indels. Because of this incongruence, albeit unsupported, topological tests were employed to further investigate the position of Byblidaceae. Under a parsimony framework, the Templeton and sign tests find the ML topology (Byblidaceae not closely related to Lentibulariaceae) not to be significantly less parsimonious than the shortest tree (Table 5), indicating that even under parsimony there is no significant evidence against the ML position of Byblidaceae or for its sister-group relationship to Lentibulariaceae. The AU-Test and SH-Test indicate that a sister-group relationship of Byblidaceae and Lentibulariaceae is significantly less likely than the maximum likelihood and Bayesian consensus topology.
Results from ancestral state reconstruction
Ancestral state reconstruction indicated the probabilities of the individual character states to be expected along branches as shown in Figure 4.

Evolution of selected morphological characters in Lamiales. ML ancestral state reconstruction on the ML topology (Figure 2) simplified to represent families by only one OTU and collapsing nodes not supported by > = 50% in at least one of the analyses. Pie charts give probabilities of character states; white indicates absence in case of binary (presence-absence) characters, while color indicates presence. Otherwise, colors indicate states as shown in legend.
Discussion
Lamiales sensu APGIII [28] (including Carlemanniaceae and Plocospermataceae) receive maximal support in the present study which is the first to sample taxa from these two families in a multigene study; a single gene study [36] did not provide support for the branching order of the early branching lamialean families.
The phylogenetic position of Hydrostachys
Hydrostachys as a rheophyte with tuber-like rhizomes, fibrous roots, and no stomata is a morphologically highly aberrant genus [32], which has always hampered inference of its phylogenetic affinities based on morphology. Embryological characters such as endosperm development and the apical septum in the ovary [83] might be interpreted as supporting a placement of Hydrostachys in Lamiales [31]. The first molecular study, however, placed it within Cornales [34]. In all previous phylogenetic studies, the genus was found on a long branch, indicating strongly elevated substitutional rates - a fact that could have misled previous phylogenetic inferences [33]. Burleigh et al. [31] recently used a 5-gene data matrix to infer an angiosperm phylogeny, and resolved Hydrostachys as nested in Lamiales, branching right after Oleaceae. Results from our re-sequencing and re-analysis, along with a blast screening of existing GenBank sequences, strongly suggest that this placement most likely was due to an erroneous matK sequence used in their study. That sequence was first published by Hufford et al. [82] but is identical to one published earlier by Hufford et al. [62], although citing a different voucher. Interestingly, Burleigh et al. [31] report that the 3-gene matrix (rbcL, atpB, 18S) places Hydrostachys in Cornales, while in the 5-gene matrix (additional matK and 26S data), Hydrostachys is found in Lamiales. The authors suggest the matK sequence to be the driving force for this result. Indeed, the most likely incorrect matK sequence misinforms phylogenetic inference, even though only one out of five genes provides the erroneous signal. If nothing else, this demonstrates the strong phylogenetic signal and potential of matK for phylogenetic analyses at the given phylogenetic depth. Phylogenetic reconstruction using our newly generated sequences in the context of the three-marker matrix compiled here and in the context of the angiosperm matK alignment clearly places Hydrostachys outside Lamiales, which is consistent with earlier findings [36, 84, 85] and with the analysis of two unpublished matK sequences by Kita and Kato (AB038179, AB038180).
A robust hypothesis on the basal grade in Lamiales
The Central American Plocospermataceae branch first in Lamiales (Figure 2), a scenario also found earlier in all studies that sampled this monotypic family [26, 35, 36]. A clade consisting of Carlemanniaceae plus Oleaceae branches second. A close relationship between these two families was found weakly supported (64% BS) previously [36] based on rbcL sequences, and was also observed in a study dealing with plastome rearrangements in Oleaceae [35], when Carlemanniaceae appeared sister to Oleaceae despite being set to as outgroup. We find the sister group relationship between Carlemanniaceae and Oleaceae with maximum support.
Tetrachondraceae are recovered with maximum support in all three analyses as third branch in Lamiales. While this relationship has been observed previously [36, 26], statistical support for it has increased significantly in our study (59% MP BS support in Savolainen et al. [36] versus PP 1.00, 100% ML BS, 94% MP BS, support in our tree). The family comprises two genera, Tetrachondra and Polypremum, both of which were sampled here. The genus Tetrachondra has a disjunct distribution (New Zealand/South America) and comprises the two aquatic or semi-aquatic species, while the monotypic Polypremum is found from southern U.S. to the northern part of South America.
Relationships within core Lamiales
The core Lamiales (sensu [35], all Lamiales excluding Carlemanniaceae, Oleaceae, Plocospermataceae, and Tetrachondraceae; Figure 2) are unambiguously recovered by our analysis. As first branch within this core group a maximally supported clade composed of Calceolariaceae and Gesneriaceae (Figure 1f, g) is found. The phylogenetic affinities of both families had remained unclear so far [45, 38, 2] but both share the presence of cornoside and absence of iridoids [86]. Gesneriaceae are a large (ca. 3200 species), predominantly pantropical family of herbaceous perennials (rarely woody shrubs and small trees), about one fifth of them growing as epiphytes [87]. In contrast to many other lamialean families, molecular phylogenetics confirmed their traditional circumscription, as proposed by Bentham in 1876 [88].
Plantaginaceae
Next in the basal grade of core Lamiales is a clade comprising Plantaginaceae as currently defined [28] (PP 1.00, 100% ML BS, 84% MP BS), in which a major split separates two groups from each other. All former studies focusing on Plantaginaceae relationships found a major dichotomy within this family [38, 22, 39, 89]. Rahmanzadeh et al. [2] argued that the finding of a well supported clade including genera from Gratioleae together with unclear relationships of this group to other families is handled best with the recognition of a separate family. Thus, Gratiolaceae were resurrected [2]. Current phylogenies allow both the recognition of two families, as well as the treatment of Plantaginaceae with two major subfamilies. Since the taxon sampling is still far from being complete, and clear morphological characters for either of the groups are lacking, we solely accept Plantaginaceae throughout this manuscript. Rahmanzadeh et al. [2] tentatively assigned 36 genera to their Gratiolaceae, 13 of which were included in our phylogenetic study. Among the genera proposed to be part of Gratiolaceae, the widespread genus Limosella was found in Scrophulariaceae [22, 39], and the present analysis confirms placement of Limosella in Scrophulariaceae. Stemodiopsis is found in Linderniaceae, while Lindenbergia is sister to the remaining Orobanchaceae. According to Olmstead et al. [38] and Rahmanzadeh et al. [2], Angelonieae (two genera: Angelonia and Monopera) appears closely related to Gratioleae. Gratioleae have an integument 3-6 cells across, with large, transversely elongated endothelial cells in vertical rows; this causes its seeds to have longitudinal ridges. The exotestal cells have hook-like thickenings [1]. Stevens et al. [1] suggest Angelonieae (integument 5-12 cells across) should also be included in Gratioleae. However, a denser taxon sampling will be needed to further test what belongs in this clade-regardless of the taxonomic level on which it might be recognized.
Scrophulariaceae
Scrophulariaceae in their new circumscription, including former Buddlejaceae and Myoporaceae, are the sister to all other higher core Lamiales (PP 1.00, 100% ML BS, 79% MP BS). This was already indicated by previous studies [2, 39] and is confirmed here with high confidence. A vastly expanded circumscription of Scrophulariaceae that was presented as a possibility in APGIII [28] would thus mean that all higher core Lamiales would have to be included in order to respect the principle of monophyletic families. Such a classification would have to include a morphologically very heterogeneous assemblage of lineages with more than 17.000 species and does therefore not appear as very helpful.
Higher core Lamiales (HCL) and the evolution of carnivory
The remaining families Acanthaceae, Bignoniaceae, Byblidaceae, Lamiaceae, Lentibulariaceae, Linderniaceae, Orobanchaceae, Paulowniaceae, Pedaliaceae, Phrymaceae, Schlegeliaceae, Stilbaceae, Thomandersiaceae, and Verbenaceae form a clade strongly supported by BI (PP 1.00) and ML (100% ML BS) analysis, but only moderately supported (76% MP BS) in MP trees (referred to as "higher core Lamiales", or HCL clade, in the following). There is no morphological synapomorphy known for this clade.
A monophyletic origin of carnivory in Lamiales has been discussed since the introduction of molecular phylogenetics to the field of angiosperm systematics (see chapter on Lamiales in [90]). In the earliest analyses of rbcL sequences, the genus Byblis was found sister to Lentibulariaceae, but this placement gained only weak statistical support [19]. Later, an analysis of three coding plus three non-coding chloroplast markers [26] found Byblidaceae as sister to Lentibulariaceae with 65% jackknife support. This is the highest statistical support ever reported for this relationship, but only one Byblis species and one Pinguicula species were sampled in that study.
Based on our data, a close relationship of carnivorous Byblidaceae and Lentibulariaceae is extremely unlikely. The placement of Byblidaceae next to Lentibulariaceae, as found in previous studies and even in single MP tree topologies of the current study, has been rejected at highest significance levels by our topological tests and is contradicted with substantial statistical support by our ML and BI trees. It might be due to long branch attraction, to which MP is much more susceptible than the other two approaches [91].
Accordingly, carnivory evolved at least twice within Lamiales, in congruence with Müller et al. [13]. Our data still do not provide enough resolution to identify the immediate sister group of Lentibulariaceae. The family appears in a weakly supported group together with Acanthaceae, Thomandersiaceae and Martyniaceae/Schlegeliaceae and Bignoniaceae, Pedaliaceae and Verbenaceae. An earlier study, sampling only one species from Lentibulariaceae (Pinguicula), found Elytraria (Acanthaceae) as sister to Lentibulariaceae [39] with 52% parsimony BS. In contrast, the monophyly of Acanthaceae, including Elytraria, was strongly supported in a more recent study sampling 85 taxa from Acanthaceae [92]. In congruence with that, we find Elytraria sister to remaining Acanthaceae.
The lack of resolution in higher core Lamiales still hampers a clear identification of the precise degree of relatedness to Martyniaceae, two strongly glandular members of which (Ibicella and Proboscidea) have been reported to attract and catch numerous arthropods, and thus have been classified as "protocarnivorous". Recent tests for protease activity of glands of the two respective genera were negative [93]; however, putatively mutualistic arthropods have been reported to be associated with each genus [94], from which the plant might benefit in a manner similar to the symbiosis observed in the African Roridula (Roridulaceae, Ericales) [93].
Next relatives to the supposedly carnivorous or "protocarnivorous" genus Philcoxia are found in Gratioleae, as previously suggested [21]. Without any doubt, Gratioleae have no close connection to Lentibulariaceae, despite some morphological similarity. Should further tests identify Philcoxia as a truly carnivorous plant, this would be the third independent origin of the syndrome within the order.
Further insights into the family circumscriptions in higher core Lamiales
Linderniaceae
The exact position of Linderniaceae within higher core Lamiales remains unclear. It is found unresolved in tritomy together with Byblidaceae and a clade including Acanthaceae, Bignoniaceae, Lamiaceae, Lentibulariaceae, Martyniaceae, Orobanchaceae, Paulowniaceae, Pedaliaceae, Phrymaceae, Schlegeliaceae, Stilbaceae, Thomandersiaceae, and Verbenaceae. Only the maximum likelihood tree depicts Linderniaceae and Byblidaceae forming a poorly supported clade. The centers of diversity of this family are in Southeast Asia and tropical Africa. Among them, desiccation tolerant plants like Craterostigma are found.
Stilbaceae and remaining families
Within the remaining families, the African Stilbaceae branch first; this scenario gains convincing support from Bayesian Inference (PP 0.93), weak support from ML bootstrapping (62% ML BS), and lacks parsimony bootstrap support. Molecular phylogenetic studies had expanded the traditional circumscription of Stilbaceae [38, 39, 95, 96] to 11 genera (3 of which we sampled here) with a predominantly South African distribution. Only Nuxia extends to tropical Africa and the Arabian Peninsula.
One of two major clades in this assembly comprises Lamiaceae, Phrymaceae, Paulowniaceae, Rehmannia, and Orobanchaceae. Although this clade also was recovered previously [39], this is the first time it receives support from BI and ML. Within that group, Lamiaceae are sister to the remaining taxa, supported by 50% ML BS (our study), and PP 0.92 and 58% MP BS value [39]. We find subfamily Mazoideae of Phrymaceae sister to a clade including Paulownia, Phrymaceae subfamily Phrymoideae, Rehmannia and Orobanchaceae. Herein, Rehmannia is weakly linked to Orobanchaceae, while the relationship between Paulownia and Phrymoideae remains unresolved. Previous studies dealing with the next relatives of Orobanchaceae found either Paulownia [38], or Phryma and Paulownia together, but as unresolved tritomy [26], or Mimulus and Paulownia as successive sisters to Orobanchaceae [2] but did not include Rehmannia and/or Triaenophora.
With regard to Orobanchaceae relationships, the most extensive sampling in terms of both taxa and character number are that of Xia et al. [43] and Albach et al. [50]. The authors found Rehmannia and Triaenophora together as sister clade to Orobanchaceae, which should either be included in Orobanchaceae, as suggested by Albach et al. [50], or be recognized as a new family. As a morphological synapomorphy, Orobanchaceae, Rehmannia and Triaenophora share alveolated seeds [43]. Although a well resolved phylogeny of Orobanchaceae exists, it still remains to be tested using plastid sequence data whether the non-parasitic Lindenbergia alone is sister to the remaining Orobanchaceae, or if Lindenbergia plus the hemiparasitic genera Siphonostegia, Schwalbea, Monochasma, Cymbaria and Bungea are in the respective position [49].
Including taxa from both subfamilies of Phrymaceae in a context of putative relatives, no evidence for the monophyly of Phrymaceae was found [37, 39]. Only Beardsley and Olmstead [51] found weak support for a monophyletic Phrymaceae, but this result is probably due to the specific sampling used. In that study [51], chloroplast data alone did not support this clade, while nuclear data and the combined analysis did so. The incongruence might be caused by a plastid-nuclear genome incongruity, which must be confirmed by additional data.
The two subfamilies of Phrymaceae, Phrymoideae and Mazoideae, do not form a clade in any of the trees in Xia et al. [43] or Albach et al. [50], and the branching order of Mazoideae, Phrymoideae and Paulownia is inconsistent in different analyses of these studies. Hence, the authors abstain from assigning these groups to families. In the light of our data we suggest to segregate Mazoideae from Phrymaceae and elevate it to family rank.
The position of Lamiaceae distinct from Verbenaceae (Figure 2) is an important and noteworthy finding. It ends a century-old discussion on close relationships of a Lamiaceae-Verbenaceae complex [88, 97, 98]. Molecular phylogenetic analysis rather concluded that Lamiaceae may not be monophyletic with respect to Verbenaceae [99]. However, analyses of rbcL [100, 99] were not conclusive about their relationships and even a combined matK/trnK analysis [2] did not provide sufficient support for Lamiaceae and Verbenaceae.
The families Acanthaceae, Bignoniaceae, Lentibulariaceae, Martyniaceae, Pedaliaceae, Schlegeliaceae, Thomandersiaceae, and Verbenaceae form a clade in our Bayesian and ML analyses (PP 1.00, ML BS 48%). For all families for which more than one taxon was sampled, monophyly is confirmed, but there is only little resolution of intra-familial relationships in that clade, especially in MP trees. In the work of Oxelman et al. [39], a corresponding clade was found, including the families mentioned above, except Pedaliaceae. We find weak support for Schlegeliaceae to be sister to Martyniaceae, while Oxelman et al. [39] found Martyniaceae, Verbenaceae and Schlegeliaceae in a clade (PP 0.82). Wortley et al. [42] found Thomandersia weakly linked to Schlegeliaceae, however, our data do not exhibit evidence for support such a relationship. A close examination of the floral anatomy of Thomandersia [101] could not improve the knowledge on its relationships.
Implications for the evolution of floral symmetry and other characters
Within Lamiales, both polysymmetric and monosymmetric (zygomorphic) flowers occur. Next to the typical pentamerous flowers, some groups exhibit tetramerous morphology. With the most highly resolved phylogeny of Lamiales to date, the evolution of floral symmetry and flower merosity within the order can be studied in more detail than previously possible. Assuming the ancestral asterid flower to be pentamerous and polysymmetric, Plocospermataceae as the most basal family of Lamiales, share this plesiomorphic character state (Figure 4). Regarding the evolution of tetramery, there are two possible scenarios. In the first, tetramery evolved once after the branching of Plocospermataceae in Lamiales, with two reversals to pentamery in both Gesneriaceae and then independently in all Lamiales branching after the Calceolariaceae/Gesneriaceae clade, this possibility is the one which is favoured by our ML ancestral state reconstruction. In the second scenario, tetramery evolved three times independently in (i) Oleaceae/Carlemanniaceae clade, (ii) Tetrachondraceae, and (iii) Calceolariaceae. Both options require three changes in flower merosity, and thus are equally parsimonious. However, there are details in floral development that differ among the tetramerous families. In Oleaceae, sepals are initiated in orthogonal positions, and petals are in diagonal position, whereas in Tetrachondraceae, sepals are initiated in diagonal, and petals in orthogonal position [102]. Initiation in Calceolariaceae follows that in Oleaceae; data for Carlemanniaceae are missing. Because tetramery in the early branching lineages of Lamiales is different for each group on more detailed level, independent gains seem more likely than a general shift towards tetramery and two independent reversals to pentamery. Tetramerous flowers are also found in the more derived Gratioleae, Veroniceae and Plantagineae (Plantaginaceae). Based on mixed evidence for fusion and loss of flower parts in these groups, multiple origins of tetramery within Plantaginaceae have been assumed. For the Plantaginaceae, Bello et al. [103] hypothesize two shifts from pentamery to tetramery: (i) in Amphianthus, which has recently been shown to be nested in Gratiola [89], and (ii) in a clade consisting of Aragoa, Plantago and Veronica. An independent shift to tetramery has been suggested by Albach et al. [104] based on loss of a sepal in Veroniceae and fusion in Plantago and Aragoa. But in these taxa the upper lip is composed out of two petals. Evidence for this is vascularization with two midribs, teratologic, pentamerous flowers, and an evolutionary row from pentamerous to tetramerous flowers within this tribe [98, 82]. The evolution of flower symmetry can be easily reconstructed. Lamiales descended from a polysymmetric ancestor, and early branching lineages in Lamiales share this character state. After branching of Tetrachondraceae, the ancestor of the following taxa once acquired monosymmetric flowers, accompanied by a reduction from five stamens to four stamens plus one staminode. There are multiple transitions back to actinomorphic flowers in Lamiales, e.g. in the case of Plantago (Plantaginaceae) [103, 105], in some taxa in Lamiaceae, Scrophulariaceae, Gesneriaceae, and in all Byblidaceae. The corolla of Byblidaceae is treated here as actinomorphic, although the curved stamens introduce a slight element of zygomorphy.
Further morphological characters
Several morphological or biochemical characters lend further support to some of our hypothesized phylogenetic relationships in Lamiales. Carlemanniaceae and Oleaceae share the characteristic of having only two stamens, while the first-branching Plocospermataceae have five stamens, and the lineages branching later in the evolution of Lamiales generally have four stamens. The sister-group relationship between Calceolariaceae and Gesneriaceae is further confirmed by two morphological characters shared by these families (see Figure 4): (i) the thyrsic inflorescence with pair flowered cymes, and (ii) aulacospermous alveolated seeds [102]. Aulacospermous seeds are otherwise only found in Linderniaceae (Crepidorhopalon, Hartliella). However, an aberrant type of aulacospermous seeds is found in some genera of Scrophulariaceae s.str.. Here not all cells of the endothelium protrude into the endosperm and the ontogeny is different from Calceolariaceae, Gesneriaceae and Linderniaceae [44, 106]. With regard to chemical compounds, Plocospermataceae, Oleaceae and Carlemanniaceae have no anthraquinones from the shikimic acid metabolism, Tetrachondraceae have not been examined for the occurrence of these compounds, and all other lineages in Lamiales possess them. Consequently, these anthraquinones have evolved immediately before or immediately after branching of Tetrachondraceae. Group II decarboxylated iridoids most likely evolved once after the branching of Calceolariaceae + Gesneriaceae, since they are shared by all taxa branching after this clade [1]. The close relationship between Rehmannia and Orobanchaceae is supported by the shared occurrence of alveolated seeds.
Divergence ages in Lamiales
There have been several attempts to estimate Asterid divergence ages, using fossil calibration points outside Lamiales. By means of the earliest relaxed clock dating method NPRS [107], Wikström et al. [108] provided estimates for Lamiales stem group (sga) and crown group ages (cga) of 74 mya and 64 mya, respectively. Using a more sophisticated approach (PL, [107]), the later results of Bremer at al. [109] and Janssens et al. [110] were quite congruent, estimating the stem group age at 106 and 104 mya, and the crown group age at 97 and 95 mya, respectively. The recent study of Magallon and Castillo [111] presents a diversification hypothesis for all angiosperms derived from constraining minimal ages of 49 nodes with fossil data. This setup resulted in a sga of 80 mya and a cga of 63 mya for Lamiales, maybe because of the strongly reduced taxon sampling among Lamiales compared to Bremer et al. [109]. Furthermore, the highest diversification rates among angiosperms were found in Lamiales [112]. This rapid radiation could be a reason for the difficulty in untangling the relationships in Lamiales, as previously supposed [2]. The very short branches among the representatives of Higher Core Lamiales (see Figure 3) are putatively indicative of a rapid radiation. So far, reliable relaxed-clock estimates for the age of major Lamiales lineages have been lacking for two reasons, one of which is the scantiness of useful fossil calibration points. Only few fossils, sometimes with questionable assignment [113], are known from Lamiales. They include a mummified Byblis seed (middle Eocene[114]), a fruit from Bignoniaceae (middle Eocene, [115]), Justicia-like pollen (Neogene, [116]), and vegetative parts from Hippuris (Hippuridaceae), Fraxinus (Oleaceae), and Chilopsis (Bignoniaceae) from Oligocene [117]. The second reason for the absence of dating attempts in Lamiales has been the uncertainty with respect to the phylogenetic position of the families within Lamiales. We believe that our study represents good progress with regard to this second problem. Nevertheless, we refrain from trying to obtain divergence age estimated based on our data at this point, because (i) the sparseness of reliable and useful fossil calibration points would force us to either use an insufficient number of calibration points or use calibration points that themselves are molecular-clock based estimates with a substantial error margin, and (ii) because the remaining uncertainties in the branching order within Lamiales would translate into inferring clade ages with unsatisfyingly wide confidence intervals.
Conclusions
Utility of chloroplast markers for Lamiales phylogenetics
Phylogenetic analysis of combined trnK/matK, trnL-F and rps16 intron sequences enhanced both resolution and statistical support compared to previous studies. Addition of the more slowly evolving protein coding rbcL and ndhF genes to our three-marker dataset did not increase resolution and support values of trees to the slightest degree (Additional file 6, Figure S5), and analyses of each of the coding markers alone yield highly unresolved topologies.
Despite the step forward reported here, more data need to be compiled to clarify the affinities within the derived Lamiales, especially for finding the next relatives of carnivorous lineages and a better understanding of the path to carnivory in the order. A recent simulation study argued for accumulating many more characters from slow evolving markers, and recommends 10,000-20,000 characters for Lamiales [40]. Apart from the much greater effort required by this strategy, the simulation approach taken by the authors does not allow a rejection of the utility of non coding markers. This is because the distribution of rates and homoplasy at individual sites, which seems to be a very important factor determining phylogenetic utility [57], was not taken into account by the authors. Moreover, simulations were exclusively based on substitutional patterns derived from functionally highly constrained ndhF and rbcL data sets with a scarce taxon sampling and a very rough estimation of phylogeny by neighbor-joining. A currently popular approach in large scale angiosperm phylogenetics takes this idea one step further and uses concatenated coding sequences extracted from complete cp genome sequences (e.g. [118]).
However, regardless of the markers and number of characters used, it has emerged as highly crucial to maintain a high taxon sampling density while accumulating more characters [40, 112, 119]. Although the cost for complete cp genome sequences have dropped dramatically in the past years, in particular when only protein coding regions are targeted and no assembly is aimed at, the cost/benefit ratio so far has prevented researchers from taking this avenue for resolving the Lamiales phylogeny. For such an approach, it is currently unclear whether an appropriate number of taxa could be upheld while keeping costs at a reasonable level, and whether the information content in even a large number of slowly evolving protein coding genes would significantly exceed that in just a few more quickly evolving cp genome regions. In view of the substantial progress made here with this kind of marker, adding further data from non-protein coding chloroplast regions seems a promising strategy that, alone or in combination with phylogenomic approaches, might finally provide us with a clear picture of Lamiales evolution.
References
Stevens PF: Angiosperm Phylogeny Website. 2001, Version 7, May 2006, [http://www.mobot.org/MOBOT/research/APweb/]
Rahmanzadeh R, Müller KF, Fischer E, Bartels D, Borsch T: Linderniaceae and Gratiolaceae (Lamiales) are further lineages distinct from Scrophulariaceae. Plant Biology. 2005, 7: 67-78. 10.1055/s-2004-830444.
Bartels D: Desiccation tolerance studied in the resurrection plant Craterostigma plantagineum. Integr Comp Biol. 2005, 45: 696-701. 10.1093/icb/45.5.696.
Young ND, Steiner KE, dePamphilis CW: The evolution of prasitism in Scrophulariaceae/Orobanchaceae: plastid gene sequences refute an evolutionary transition series. Ann MO Bot Gard. 1999, 86: 876-893. 10.2307/2666173.
Richies CR, Parker C: Parasitic plants as weeds. Parasitic plants. 1995, London, UK: Chapman & Hall, 226-255.
Lloyd FE: Carnivorous plants. 1942, Massachusetts: Waltham
Juniper BE, Robins RJ, Joel DM: The carnivorous plants. 1989, London: Academic press
Brugger J, Rutishauser R: Bau und Entwicklung landbewohnender Utricularia-Arten. Bot Helv. 1989, 99: 91-146.
Rutishauser R, Sattler R: Complementarity and heuristic value of contrasting models in structural botany: 3. Case study on shoot-like "leaves" and leaf-like "shoots" in Utricularia macrorhiza and U. purpurea (Lentibulariaceae). Bot Jahrb. 1989, 111: 121-137.
Rutishauser R, Isler B: Developmental genetics and morphological evolution of flowering plants, especially Bladderworts (Utricularia): Fuzzy arberian morphology complements classical morphology. Ann Bot. 2001, 88: 1173-1202. 10.1006/anbo.2001.1498.
Barthlott W, Porembski S, Fischer E, Gemmel B: First protozoa-trapping plant found. Nature. 1998, 392: 447-10.1038/33037.
Greilhuber J, Borsch T, Müller KF, Worberg A, Porembski S, Barthlott W: Smallest angiosperm genomes found in Lentibulariaceae, with chromosomes of bacterial size. Plant Biology. 2006, 8: 770-777. 10.1055/s-2006-924101.
Müller KF, Borsch T, Legendre L, Porembski S, Theisen I, Barthlott W: Evolution of carnivory in Lentibulariaceae and the Lamiales. Plant Biology. 2004, 6: 477-490. 10.1055/s-2004-817909.
Müller KF, Borsch T, Legendre L, Porembski S, Barthlott W: Recent progress in understanding the evolution of Lentibulariaceae. Plant Biology. 2006, 8: 748-757. 10.1055/s-2006-924706.
Hartmeyer S: Carnivory of Byblis revisited-A simple method for enzyme testing on carnivorous plants. Carniv Pl Newslett. 1997, 26: 34-38.
Plachno BJ, Adamec L, Lichtscheidl IK, Peroutka M, Adlassnig W, Vrba J: Fluorescence labelling of phosphatase activity in digestive glands of carnivorous plants. Plant Biology. 2006, 8: 813-820. 10.1055/s-2006-924177.
Lang F: Untersuchungen über Morphologie, Anatomie und Samenentwicklung von Polypompholyx und Byblis gigantea. Flora. 1901, 149-206.
Conran JG: The embryology and relationships of the Byblidaceae. Aust Sys Bot. 1996, 9: 243-254. 10.1071/SB9960243.
Albert VA, Williams SE, Chase MW: Carnivorous plants: Phylogeny and structural evolution. Science. 1992, 257: 1491-1495. 10.1126/science.1523408.
Taylor P, Souza VC, Giulietti AM, Harley RM: Philcoxia: A new genus of Scrophulariaceae with three new species from Eastern Brazil. Kew Bulletin. 2000, 55: 155-163. 10.2307/4117770.
Fritsch P, Almeda F, Martins AB, Cruz BC, Estes D: Rediscovery and phylogenetic placement of Philcoxia minensis (Plantaginaceae), with a test of carnivory. Proc Calif Acad Sci. 2007, 58: 447-467.
Albach DC, Meudt HM, Oxelman B: Piecing together the "new" Plantaginaceae. Am J Bot. 2005, 92: 297-315. 10.3732/ajb.92.2.297.
McDade LA, Moody ML: Phylogenetic relationships among Acanthaceae: evidence from noncoding trnL-trnF chloroplast DNA sequences. Am J Bot. 1999, 86: 70-80. 10.2307/2656956.
Olmstead RG, Bremer B, Scott KM, Palmer JD: A parsimony analysis of the Asteridae s.l. based on rbcL sequences. Ann MO Bot Gard. 1993, 80: 700-722. 10.2307/2399855.
Olmstead RG, Kim KJ, Jansen RK, Wagstaff SJ: The phylogeny of the Asteridae sensu lato based on chloroplast ndhF gene sequences. Mol Phylogenet Evol. 2000, 16: 96-112. 10.1006/mpev.1999.0769.
Bremer B, Bremer K, Heidari N, Erixon P, Olmstead RG, Anderberg AA, Källersjö M, Barkhordarian E: Phylogenetics of asterids based on 3 coding and 3 non-coding chloroplast DNA markers and the utility of non-coding DNA at higher taxonomic levels. Mol Phylogenet Evol. 2002, 24: 274-301. 10.1016/S1055-7903(02)00240-3.
Soltis DE, Soltis PS, Endress PK, Chase MW: Phylogeny and evolution of angiosperms. 2005, Sunderland, Massachusetts, USA: Sinauer Associates
APG III: An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot J Linn Soc. 2009, 161: 105-121. 10.1111/j.1095-8339.2009.00996.x.
Dahlgren G, (ed): Systematische Botanik. 1987, Berlin Heidelberg New York: Springer
Takhtajan A: Diversity and Classification of Flowering Plants. 1997, New York: Columbia University Press
Burleigh JG, Hilu K, Soltis D: Inferring phylogenies with incomplete data sets: a 5-gene, 567-taxon analysis of angiosperms. BMC Evol Biol. 2009, 9: 61-10.1186/1471-2148-9-61.
Xiang Q, Moody ML, Soltis DE, Fan CZ, Soltis PS: Relationships within Cornales and circumscription of Cornaceae--matK and rbcL sequence data and effects of outgroups and long branches. Mol Phylogenet Evol. 2002, 24: 35-57. 10.1016/S1055-7903(02)00267-1.
Fan C, Xiang Q: Phylogenetic analyses of Cornales based on 26S rRNA and combined 26S rDNA-matK-rbcL sequence data. Am J Bot. 2003, 90: 1357-1372. 10.3732/ajb.90.9.1357.
Hempel A, Reeves PA, Olmstead R, Jansen RK: Implications of rbcL sequence data for higher order relationships of the Loasaceae and the anomalous aquatic plant Hydrostachys (Hydrostachyaceae). Plant Syst Evol. 1994, 194: 25-37. 10.1007/BF00983214.
Hilu KW, Borsch T, Müller KF, Soltis DE, Soltis PS, Savolainen V, Chase M, Powell M, Alice LA, Evans R, Sauquet H, Neinhuis C, Slotta TA, Rohwer JG, Campbell CS, Chatrou L: Angiosperm phylogeny based on matK sequence information. Am J Bot. 2003, 90: 1758-1776. 10.3732/ajb.90.12.1758.
Savolainen V, Fay MF, Albach DC, Backlund A, Van der Bank M, Cameron KM, Johnson LA, Lledó MD, Pintaud J-, Powell M, Sheaham MC, Soltis DE, Soltis PS, Weston P, Whitten WM, Wurdack KJ, Chase MW: Phylogeny of the eudicots: a nearly complete familial analysis based on rbcL gene sequences. Kew Bulletin. 2000, 55: 257-309. 10.2307/4115644.
Tank DC, Beardsley PM, Kelchner SA, Olmstead RG: L. A. S. JOHNSON REVIEW No. 7. Review of the systematics of Scrophulariaceae s.l. and their current disposition. Aust J Bot. 2006, 19: 289-307. 10.1071/SB05009.
Olmstead RG, DePamphilis CW, Wolfe AD, Young ND, Elisons WJ, Reeves PA: Disintegration of the Scrophulariaceae. Am J Bot. 2001, 88: 348-361. 10.2307/2657024.
Oxelman B, Kornhall P, Olmstead RG, Bremer B: Further disintegration of Scrophulariaceae. Taxon. 2005, 54: 411-425. 10.2307/25065369.
Wortley AH, Rudall PJ, Harris DJ, Scotland RW: How much data are needed to resolve a difficult phylogeny? Case study in Lamiales. Syst Biol. 2005, 54: 697-709. 10.1080/10635150500221028.
Andersson S: On the phylogeny of the genus Calceolaria (Calceolariaceae) as inferred from ITS and plastid matK sequences. Taxon. 2006, 55: 125-137. 10.2307/25065534.
Wortley AH, Harris DJ, Scotland RW: On the Taxonomy and Phylogenetic Position of Thomandersia. Syst Botany. 2007, 32: 415-444. 10.1600/036364407781179716.
Xia Z, Wang Y, Smith JF: Familial placement and relations of Rehmannia and Triaenophora (Scrophulariaceae s.l.) inferred from five gene regions. Am J Bot. 2009, 96: 519-530. 10.3732/ajb.0800195.
Fischer E: Scrophulariaceae. The Families and Genera of Vascular Plants. Edited by: Kubitzki K. 2004, Berlin: Springer, 333-432.
Olmstead RG, Reeves PA: Evidence for the polyphyly of the Scrophulariaceae based on chloroplast rbcL and ndhF sequences. Ann MO Bot Gard. 1995, 82: 176-193. 10.2307/2399876.
APG2: An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot J Linn Soc. 2003, 141: 399-436. 10.1046/j.1095-8339.2003.t01-1-00158.x.
dePamphilis CW, Young ND, Wolfe AD: Evolution of plastid gene rps2 in a lineage of hemiparasitic and holoparasitic plants: many losses of photosynthesis and complex patterns of rate variation. Proc Natl Acad Sci USA. 1997, 94: 7367-7372. 10.1073/pnas.94.14.7367.
Wolfe A, dePamphilis C: The effect of relaxed functional constraints on the photosynthetic gene rbcL in photosynthetic and nonphotosynthetic parasitic plants. Mol Biol Evol. 1998, 15: 1243-1258.
Bennett JR, Mathews S: Phylogeny of the parasitic plant family Orobanchaceae inferred from phytochrome A. Am J Bot. 2006, 93: 1039-1051. 10.3732/ajb.93.7.1039.
Albach DC, Yan K, Jensen SR, Li H: Phylogenetic placement of Triaenophora (formerly Scrophulariaceae) with some implications for the phylogeny of Lamiales. Taxon. 2009, 58: 749-756.
Beardsley PM, Olmstead RG: Redefining Phrymaceae: The placement of Mimulus, tribe Mimuleae, and Phryma. Am J Bot. 2002, 89: 1093-1102. 10.3732/ajb.89.7.1093.
Magin N, Classen R, Gack C: The morphology of false anthers in Craterostigma plantagineum and Torenia polygonioides (Scrophulariaceae). Can J Bot. 1989, 67: 1931-1937. 10.1139/b89-245.
Fischer E: Systematik der afrikanischen Lindernieae (Scrophulariaceae). Trop Subtrop Pflanzenwelt. 1992, 82: 1-365.
Spangler RE, Olmstead RG: Phylogenetic analysis of Bignoniaceae based on the cpDNA gene sequences rbcL and ndhF. Ann MO Bot Gard. 1999, 86: 33-46. 10.2307/2666216.
Nakai T: Classes, Ordines, Familiae, Subfamiliae, Tribus, Genera nov quae attinet ad plantas Koreanas. J Jap Bot. 1949, 24: 8-14.
Reveal J: Newly required suprageneric names in vascular plants. Phytologia. 1995, 79: 68-76.
Müller KF, Borsch T, Hilu KW: Phylogenetic utility of rapidly evolving DNA at high taxonomical levels: Contrasting matK, trnT-F, and rbcL in basal angiosperms. Mol Phylogenet Evol. 2006, 41: 99-117. 10.1016/j.ympev.2006.06.017.
Borsch T, Hilu KW, Quandt D, Wilde V, Neinhuis C, Barthlott W: Non-coding plastid trnT-trnF sequences reveal a well resolved phylogeny of basal angiosperms. J Evol Biol. 2003, 16: 558-576. 10.1046/j.1420-9101.2003.00577.x.
Worberg A, Quandt D, Barniske A-, Löhne C, Hilu KW, Borsch T: Phylogeny of basal eudicots: Insights from non-coding and rapidly evolving DNA. Org Divers Evol. 2007, 7: 55-77. 10.1016/j.ode.2006.08.001.
Borsch T, Quandt D: Mutational dynamics and phylogenetic utility of noncoding chloroplast DNA. Plant Syst Evol. 2009, 282: 169-199. 10.1007/s00606-009-0210-8.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Hufford L, Moody ML, Soltis DE: A phylogenetic analysis of Hydrangeaceae based on sequences of the plastid gene matk and their combination with rbcl and morphological data. Int J Plant Sci. 2001, 162: 835-846. 10.1086/320789.
Albach DC, Soltis PS, Soltis DE, Olmstead RG: Phylogenetic analysis of asterids based on sequences of four genes. Ann MO Bot Gard. 2001, 88: 163-212. 10.2307/2666224.
Müller J, Müller KF, Neinhuis C, Quandt D: PhyDE - Phylogenetic Data Editor. 2006, [http://www.phyde.de]
Kelchner SA: The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann MO Bot Gard. 2000, 87: 482-498. 10.2307/2666142.
Simmons MP, Ochoterena H: Gaps as characters in sequence-based phylogenetic analyses. Syst Biol. 2000, 49: 369-381. 10.1093/sysbio/49.2.369.
Müller KF: SeqState: Primer design and sequence statistics for phylogenetic DNA datasets. Appl Bioinformatics. 2005, 4: 65-69. 10.2165/00822942-200504010-00008.
Müller KF: PRAP - computation of Bremer support for large data sets. Mol Phylogenet Evol. 2004, 31: 780-782. 10.1016/j.ympev.2003.12.006.
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (* and other Methods). 1998, Sinauer Associates, Sunderland, Massachussets
Müller KF: The efficiency of different search strategies in estimating parsimony jackknife, bootstrap, and Bremer support. BMC Evol Biol. 2005, 5: 58-10.1186/1471-2148-5-58.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
Posada D: jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008, 25: 1253-1256. 10.1093/molbev/msn083.
Andrew Rambaut, Alexei Drummond: Tracer. 2009, Edinburgh: Institute for Evolutionary Biology
Stamatakis A: RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006, 22: 2688-2690. 10.1093/bioinformatics/btl446.
Stöver B, Müller KF: TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics. 2010, 11: 7-10.1186/1471-2105-11-7.
Shimodaira H: An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002, 51: 492-508. 10.1080/10635150290069913.
Shimodaira H, Hasegawa M: Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999, 16: 1114-1116.
Shimodaira H, Hasegawa M: CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001, 17: 1246-1247. 10.1093/bioinformatics/17.12.1246.
Watson L, Dallwitz M: The families of flowering plants: descriptions, illustrations, identification, and information retrieval. 1992
Mabberley D: The plant-book: a portable dictionary of the vascular plants. 2008, Cambridge: Cambridge University Press
Pagel M, Meade A, Barker D: Bayesian Estimation of Ancestral Character States on Phylogenies. Systematic Biology. 2004, 53: 673-684. 10.1080/10635150490522232.
Hufford L, McMahon MM, Sherwood AM, Reeves G, Chase MW: The major clades of Loasaceae: Phylogenetic analysis using the plastid matK and trnL-trnF regions. Am J Bot. 2003, 90: 1215-1228. 10.3732/ajb.90.8.1215.
Jäger-Zürn I: Zur Frage der systematischen Stellung der Hydrostachyaceae auf Grund ihrer Embryologie, Blüten-und Infloreszenzmorphologie. Plant Syst Evol. 1965, 112: 621-639. 10.1007/BF01373191.
Soltis DE, Soltis PS, Chase MW, Mort ME, Albsch DC, Zanis M, Savolainen V, Hahn WH, Hoot SB, Fay MF, Axtell M, Swensen SM, Prince LM, Kress WJ, Nixon KC, Farris JS: Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot J Linn Soc. 2000, 133 (81): 381--461.
Albach D: Phylogenetic placement of the enigmatic angiosperm Hydrostachys. Taxon. 2001, 50: 781-805. 10.2307/1223707.
Jensen SR, Albach DC, Ohno T, Grayer RJ: Veronica: Iridoids and cornoside as chemosystematic markers. Biochem Syst Ecol. 2005, 33: 1031-1047. 10.1016/j.bse.2005.03.001.
Smith JF, Wolfram JC, Brown KD, Carroll CL, Denton DS: Tribal Relationships in the Gesneriaceae: evidence from DNA sequences of the chloroplast gene ndhF. Ann MO Bot Gard. 1997, 84: 50-66. 10.2307/2399953.
Bentham G: Gesneriaceae. Genera Plantarum 2. Edited by: Bentham G, Hooker JD. 1876, 990-1025. Reeve
Estes D, Small RL: Phylogenetic relationships of the monotypic genus Amphianthus (Plantaginaceae tribe Gratioleae) inferred from chloroplast DNA sequences. Syst Botany. 2008, 33: 176-182. 10.1600/036364408783887375.
Soltis DE, Soltis PS, Endress PK, Chase MW: Phylogeny and Evolution of Angiosperms. 2005
Felsenstein J: Cases in which parsimony or compatibility methods will be positively misleading. Syst Biol. 1978, 27: 401-410.
McDade LA, Daniel TF, Kiel CA: Toward a comprehensive understanding of phylogenetic relationships among lineages of Acanthaceae s.l. (Lamiales). Am J Bot. 2008, 95: 1136-1152. 10.3732/ajb.0800096.
Plachno BJ, Adamec L, Huet H: Mineral nutrient uptake from prey and glandular phosphatase activity as a dual test of carnivory in semi-desert plants with glandular leaves suspected of carnivory. Ann Bot-London. 2009, 104: 649-654. 10.1093/aob/mcp155.
Rice B: Reassessing commensal-enabled carnivory in Proboscidea and Ibicella?. Carniv Pl Newslett. 2008, 15-19.
Bremer B, Olmstead RG, Struwe L, Sweere JA: rbcL sequences support exclusion of Retzia, Desfontainia, and Nicodemia from the Gentianales. Plant Syst Evol. 1994, 190: 213-230. 10.1007/BF00986194.
Oxelman B, Backlund M, Bremer B: Relationships of the Buddlejaceae s.l. investigated using parsimony jackknife and branch support analysis of chloroplast ndhF and rbcL sequence data. Syst Botany. 1999, 24: 164-182. 10.2307/2419547.
Cronquist A: The Evolution and Classification of Flowering Plants. 1988, New York: The New York Botanical Garden, 2
Briquet J: Labiatae. Die natürlichen Pflanzenfamilien. Edited by: Engler A, Prantl K. 1895, Leipzig: Engelmann, 4/3a: 132-182.
Cantino PD: Evidence for a polyphyletic origin of the Labiatae. Ann MO Bot Gard. 1992, 361-379. 10.2307/2399774.
Wagstaff SJ, Hickerson L, Spangler R, Reeves PA, Olmstead RG: Phylogeny in Labiatae s.l., inferred from cpDNA sequences. Plant Syst Evol. 1998, 209: 265-274. 10.1007/BF00985232.
Wortley AH, Scotland RW, Rudall PJ: Floral anatomy of Thomandersia (Lamiales), with particular reference to the nature of the retinaculum and extranuptial nectaries. Bot J Linn Soc. 2005, 149: 469-10.1111/j.1095-8339.2005.00507.x.
Mayr EM, Weber A: Calceolariaceae: floral development and systematic implications. Am J Bot. 2006, 93: 327-343. 10.3732/ajb.93.3.327.
Bello MA, Rudall PJ, González F, Fernández-Alonso JL: Floral morphology and development in Aragoa (Plantaginaceae) and related members of the order Lamiales. Int J Plant Sci. 2004, 165: 723-738. 10.1086/422046.
Albach DC, Martinez-Ortega MM, Fischer MA, Chase MW: Evolution of Veroniceae: A phylogenetic perspective. Ann MO Bot Gard. 2004, 91: 275-302.
Endress P: Symmetry in flowers: diversity and evolution. Int J Plant Sci. 1999, 160: S3-S23. 10.1086/314211.
Hartl D: Das alveolierte Endosperm bei Scrophulariaceen, seine Entstehung, Anatomie und taxonomische Bedeutung. Beiträge zur Biologie der Pflanzen. 1959, 35: 95-110.
Sanderson MJ: Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Mol Biol Evol. 2002, 19: 101-109.
Wikström N, Savolainen V, Chase MW: Evolution of the angiosperms: calibrating the family tree. Proc R Soc Lond [Biol]. 2001, 268: 2211-2220. 10.1098/rspb.2001.1782.
Bremer K, Friis EM, Bremer B: Molecular phylogenetic dating of asterid flowering plants shows early Cretaceous diversification. Syst Biol. 2004, 53: 496-505. 10.1080/10635150490445913.
Janssens SB, Knox EB, Huysmans S, Smets EF, Merckx VS: Rapid radiation of Impatiens (Balsaminaceae) during Pliocene and Pleistocene: Result of a global climate change. Mol Phylogenet Evol. 2009, 52: 806-824. 10.1016/j.ympev.2009.04.013.
Magallon S, Castillo A: Angiosperm diversification through time. Am J Bot. 2009, 96: 349-365. 10.3732/ajb.0800060.
Zwickl DJ, Hillis DM: Increased taxon sampling greatly reduces phylogenetic error. Syst Biol. 2002, 51: 588-598. 10.1080/10635150290102339.
Nie Z, Sun H, Beardsley PM, Olmstead RG, Wen J: Evolution of biogeographic disjunction between eastern Asia and eastern North America in Phryma (Phrymaceae). American Journal of Botany. 2006, 93: 1343-1356. 10.3732/ajb.93.9.1343.
Conran JG, Christophel DC: A fossil Byblidaceae seed from Eocene South Australia. Int J Plant Sci. 2004, 165: 691-694. 10.1086/386555.
Pigg KB, Wehr WC: Tertiary Flowers, Fruits, and Seeds of Washington State and Adjacent Areas-Part III. Wash Geol. 2002, 30: 3-16.
Germeraad J, Hopping C, Muller J: Palynology of tertiary sediments from tropical areas. Review of Palaeobotany and Palynology. 1968, 6: 189-198. 10.1016/0034-6667(68)90051-1. 200-210, 212-228, 230-259, 261, 263-348
Axelrod DI: The Late Oligocene Creede Flora, Colorado. 1987, Berkeley, Los Angeles, London: University of California Press, 130:
Jansen RK, Cai Z, Raubeson LA, Daniell H, dePamphilis CW, Leebens-Mack J, Müller KF, Guisinger-Bellian M, Haberle RC, Hansen AK, Chumley TW, Lee S, Peery R, McNeal JR, Kuehl JV, Boore JL: Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci USA. 2007, 104: 19369-19374. 10.1073/pnas.0709121104.
Qiu Y, Li L, Wang B, Chen Z, Knoop V, Groth-Malonek M, Dombrovska O, Lee J, Kent L, Rest J, Estabrook GF, Hendry TA, Taylor DW, Testa CM, Ambros M, Crandall-Stotler B, Duff RJ, Stech M, Frey W, Quandt D, Davis CC: The deepest divergences in land plants inferred from phylogenomic evidence. Proc Natl Acad Sci USA. 2006, 103: 15511-15516. 10.1073/pnas.0603335103.
Johnson LA, Soltis DE: Phylogenetic inference in Saxifragaceae s.str. and Gilia (Polemoniaceae) using matK sequences. Ann MO Bot Gard. 1995, 82: 149-175. 10.2307/2399875.
Müller KF, Borsch T: Phylogenetics of Amaranthaceae based on matK/trnK sequence data evidence from parsimony, likelihood, and Bayesian analyses. Ann MO Bot Gard. 2005, 92: 66-102.
Taberlet P, Gielly L, Pautou G, Bouvet J: Universal primers for amplification of 3 noncoding regions of chloroplast DNA. Plant Mol Biol. 1991, 17: 1105-1109. 10.1007/BF00037152.
Oxelman B, Lidén M, Berglund D: Chloroplast rps16 intron phylogeny of the tribe Sileneae (Caryophyllaceae). Plant Syst Evol. 1997, 206: 393-410. 10.1007/BF00987959.
Acknowledgements
This study was funded by DFG grant "Carnivory in Lamiales: understanding character evolution, substitution rate plasticity, and genome miniaturization", MU2875/2, to K.F.M. Additional funding was obtained from the DFG project "Mutational dynamics of non-coding genomic regions and their potential for reconstructing evolutionary relationships in eudicots" (BO1815/2-1 and/-2; QU153/2-1 and/2-2) to T.B. and Dietmar Quandt. Thanks to Nadja Korotkova for a photograph used in Figure 1. The authors would like to thank the staff of the Bonn Botanical Gardens for cultivating plants analyzed in the present study, and the curators of the respective herbaria (BONN, M) for providing material for DNA extraction. We also want to thank Richard Olmstead and two anonymous reviewers for very helpful comments that helped to improve the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
B.S. generated data and drafted the manuscript. K.F.M. was responsible for the conception of the study and helped writing the manuscript. D.C.A. provided data and improved the manuscript. A.F. and T.B. provided plant material. T.B. contributed during manuscript preparation. A.F., E.F. and G.H. improved the manuscript. T.B., E.F., and D.C.A. contributed to the conception of the study during its initial phase, G.H. in its final phase. A.F. contributed during manuscript preparation. All authors have given final approval of the version to be published.
Electronic supplementary material
12862_2010_1567_MOESM1_ESM.DOC
Additional file 1: Table S1: Taxa, specimens and GenBank acession numbers for sequences used in the 5 gene analysis. Voucher information. (DOC 100 KB)
12862_2010_1567_MOESM2_ESM.PDF
Additional file 2: Figure S1: A comparison of decay values. Numbers above branches give decay values from nucleotide data matrix; numbers below branches that from nucleotides plus coded indels. (PDF 607 KB)
12862_2010_1567_MOESM3_ESM.PDF
Additional file 3: Figure S2: Tree from rbcLanalysis. Strict consensus of 100 MP bootstrap replicates performed. (PDF 376 KB)
12862_2010_1567_MOESM4_ESM.PDF
Additional file 4: Figure S3: Tree from ndhFanalysis. Strict consensus of 100 MP bootstrap replicates performed. (PDF 389 KB)
12862_2010_1567_MOESM5_ESM.PDF
Additional file 5: Figure S4: Tree from angiosperm-wide matKanalysis of the Hilu et al. 2003 dataset plus our newly generated Hydrostachyssequence. Strict consensus of 100 MP bootstrap replicates performed. (PDF 65 KB)
12862_2010_1567_MOESM6_ESM.PDF
Additional file 6: Figure S5: Tree from combined trnK/matK, trnL-F, rps16, rbcL, ndhFanalysis. 100 bootstrap replicates performed. (PDF 521 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Schäferhoff, B., Fleischmann, A., Fischer, E. et al. Towards resolving Lamiales relationships: insights from rapidly evolving chloroplast sequences. BMC Evol Biol 10, 352 (2010). https://doi.org/10.1186/1471-2148-10-352
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-10-352