
Abstract
Background
Species in the varied geographic topology of Taiwan underwent obvious demographic changes during glacial periods. Cinnamomum kanehirae has been exploited for timber and to obtain medicinal fungi for the past 100 years. Understanding anthropogenic factors influencing the demography of this species after the last glacial maximum (LGM) is critically important for the conservation of this species.
Results
Populations of C. kanehirae were classified into four geographic regions: northwestern (NW), west-central (WC), southwestern (SW), and southeastern (SE). In total, 113 individuals from 19 localities were sampled, and variations in the chalcone synthase gene (Chs) intron and leafy (Lfy) intron-2 sequences of nuclear DNA were examined in order to assess phylogeographic patterns, the timescales of demographic and evolutionary events, and recent anthropogenic effects. In total, 210 Chs and 170 Lfy sequences, which respectively constituted 36 and 35 haplotypes, were used for the analyses. Estimates of the migration rate (M) through time revealed a pattern of frequent gene flow during previous and the present interglacials. The isolation-by-distance test showed that there generally was no significant correlation between genetic and geographic distances. The level of among-region genetic differentiation was significant when comparing eastern to western populations. However, no significant among-region genetic differentiation was found in comparisons among the four geographic regions. Moreover, essentially no genetic structuring was found for the three regions west of the CMR. A fit of spatial range expansion was found for pooled and regional samples according to the non-significant values of the sum of squared deviations. Using the Bayesian skyline plot (BSP) method, a recent bottleneck after the LGM expansion was detected in both regional and pooled samples.
Conclusions
Common haplotype distributions among geographic regions and the relatively shallow genetic structuring displayed are the result of historical gene flows. Southward dispersals in an earlier time frame from the NW region and in a later time frame from the SE region were inferred. The BSP analysis suggested a postglacial expansion event. Recent trends, however, refer to a bottleneck due to human interventions observed for both pooled and regional C. kanehirae samples.
Similar content being viewed by others


Background
Climate change is an important factor influencing organisms' evolutionary history, especially their demographic dynamics [1]. The Pleistocene was an era of temperature oscillations due to cyclic glacial/interglacial events since 0.9 million years ago (Mya) [2]. Many species experienced huge bottleneck events during glacial periods because their growth under the prevailing unpleasant cold and dry conditions was restricted to refugia [1, 2]. Contraction into refugia was reported for both animals [3–5] and plants [6–8]. Although glacial events occur on a global scale, the lowlands of southeastern Asia were not covered with ice but were drier and colder. Thus, the climatic changes were cyclic pluvial and interpluvial in Asia (respectively corresponding to interglacial and glacial periods) during the Pleistocene. According to Hewitt's [2] speculation on the climate of tropical mountains according to several faunal studies, stable moist habitats in areas not covered by ice provided suitable refugia for species survival and the generation of new lineages. Glacials changed climates in Asia and several refugia of different species are consistently being found [9–12], which shows that climate changes in the Pleistocene indeed affected the demographic history of Asian species.
Taiwan is a continental island situated off the coast of southern East Asia. The Central Mountain Ridge (CMR) is a continuous ridge trending north-south and divides Taiwan into eastern and western geographic areas [13]. There are more than 200 peaks exceeding 3000 m in elevation in the CMR. The varied geographical topologies of Taiwan allowed habitats and lineages to persist through altitudinal shifts during glacial/interglacial cycles [cf. [2]]. The remnants of glaciations at the top of some peaks indicate the former occurrence of glaciers [14, 15]. The floristic composition of Taiwan is characterized by high levels of endemism (40%) and species diversity [16]. Forests in middle to low elevations of Taiwan consist of evergreen broadleaf trees, including camphor (Cinnamomum spp.), banyan (Ficus spp.), beech (Fagus spp.), etc. During glacial periods, conifers which were distributed at high elevations in Taiwan spread to lower elevations to escape the ice-covered mountaintops [14, 17, 18], and those species originally distributed in middle and low elevations slowly migrated to the lowlands. However, the dry environments of the lowlands during glacial periods may have restricted the distributions of broadleaf trees and resulted in bottleneck effects. Following glacial periods, climates of interglacial periods were warm and moist in Taiwan, and populations would have recovered and expanded. Climate-dependent changes in demographics of trees in Taiwan are evidenced by the pollen record [19] and genetics, e.g., Michelia formosana (Magnoliaceae) [20], Cunninghamia konishii (Cupressaceae) [21], and Quercus (or Cyclobalanopsis) glauca (Fagaceae) [22].
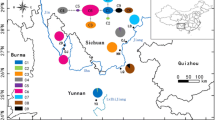
In this study, we chose Cinnamomum kanehirae Hayata (Lauraceae) as an example to explore the influence of the evolutionary history of middle-elevation broadleaf trees. Cinnamomum kanehirae belongs to the Cinnamomum group of the Cinnamomeae sensu Kostermans. The Cinnamomum group shows an amphi-Pacific disjunct distribution with its sister group, the Persea group, the disruption of boreotropical ranges of which is attributed to Eocene-Oligocene climatic cooling. The genus Cinnamomum is considered to have originated about 35 ± 6 or 138 ± 19 Mya by estimates of the relaxed molecular clock using outgroups with two different divergence times [23]. The genus Cinnamomum comprises approximate 250 species distributed in tropical and subtropical eastern Asia, Australia, and the Pacific islands, and 8 of 14 Cinnamomum species in Taiwan are endemic, including C. kanehirae. Cinnamomum kanehirae is distributed at 200~2000 m in elevation, and is mixed with C. micranthum in broadleaf forests throughout the island [24]. Cinnamomum kanehirae has a long lifespan, was abundant at the beginning of the 20th century and is one of the most valuable timber species in Taiwan [25]. The range distribution of C. kanehirae is contiguous throughout Taiwan except the northeastern part east of the CMR [25, 26] (Figure 1). A fungus called Antrodia cinnamomea Chang & Chou, parasitic inside the C. kanehirae trunk [27–29], is valued as a medicine due to its antioxidant [30–32], anticancer [33], antiviral [34], and antibiotic properties [33]. In total, 46,000 trees consisting of live, dead, and root stumps were recorded during an extensive survey of all areas, with the exception of most of the southeastern part of Taiwan between 1918 and 1922 [26], but it is now extremely difficult to find a standing live tree in the wild due to the intensive logging during 1920~1970 especially of populations of the northwestern and southwestern regions as well as selective cutting to obtain the medicinal fungus in recent years.

Geographical distribution and magnitudes of migration routes of Cinnamomum kanehirae. In the upper panel, the flow of the Kuroshio Current is shown. In the bottom left panel, four geographic regions defined by Lin et al. [35] together with the location of populations are indicated. The northwestern region (NW) contains populations TP, KW, TH, and NC; the west-central region (WC) contains populations WC, CT, WH, and ALS; the southwestern region (SW) contains populations KS, HNS, FKS, and TN; and the southeastern region (SE) contains populations FL, HKL, CL, YL, CP, YF, and TML. Abbreviations of population codes are listed in Table 1. The dark area indicates elevations above 2000 m, and the gray area indicates the distribution range of C. kanehirae outside the area above 2000 m [26, 35]. HR, Huseshan Range; CMR, Central Mountain Ridge. In the bottom right panel, directions and magnitudes of gene flow estimated by the Bayesian approach are shown. Arrows indicate the direction of migration and the thickness of the arrows indicates values of the migration parameter (M) calculated by the computer software, MIGRATE 3.0 [54].
According to an allozyme survey, C. kanehirae has a relatively higher genetic diversity in terms of heterozygosity and other parameters compared to those of other local tree species [35]. DNA is a more-sensitive genetic marker, and is very useful for studying the demographic history and/or migratory footprints of species. Phylogeographic studies of several species in Taiwan consistently point to a rapid postglacial expansion event. The suggestion of population expansion of most of those studies was based on the "star-like" phylogeny of chloroplast DNA (cpDNA) haplotypes due to the high frequencies of singletons [21, 36, 37]. Approaches based on the coalescence theory are used to trace the evolutionary history, e.g., the Bayesian skyline plot (BSP) analysis [38]. Plastid DNA is commonly used for making phylogeographic inferences; however, a strong selective sweep acting across the entire plastid genome can reduce the genetic variation of plastid DNA [cf. [39]]. The genetic variation of C. kanehirae estimated from cpDNA is very low (only six haplotypes in populations and π = 0.00016 ± 0.00007) [40], and the amounts of variation cannot reflect the long-term demographic history or the historical effective population size. Nuclear DNA is a suitable marker to obtain more information so that the evolutionary history of C. kanehirae can be traced in Taiwan.
In this study, the intron of chalcone synthase (Chs) and the second intron of the Leafy gene (Lfy) of nuclear DNA were sequenced to assess the phylogeographic structure of C. kanehirae in Taiwan. Population genetic analyses of C. kanehirae should provide more insights into geographic influences on migratory events (historical gene flow and recent dispersal). Furthermore, a coherent timescale for the geologic time and demographic events as well as climate change was established using the coalescent-based Bayesian framework. The ancestral effective population size of C. kanehirae was estimated in order to understand the trend of historical demography. Such a study is vital to our understanding of the process of demographic changes corresponding to how climate changes affected subtropical tree species in the Pleistocene.
Methods
Plant materials and DNA amplification
In total, 113 individuals originating from 19 populations of C. kanehirae of Taiwan were collected from a scion garden at the Liukuei Research Center, Taiwan Forestry Research Institute (TFRI), southwestern Taiwan (Table 1, Figure 1). This scion garden was set up in 1995 to house intraspecific variations of C. kanehirae. Sample sizes of each population range from 3 to 16 individuals. Young developed leaves were collected from this garden, preserved on silica gel, and carried back to the laboratory for DNA extraction. Total DNA was extracted from ground-up leaf powder according to a modified cetyltrimethyl ammonium bromide (CTAB) procedure [41]. DNA was precipitated with ethanol and after washing with 70% ethanol, was dissolved in 200 μL TE buffer (pH 8.0) and stored at -20°C. The DNA concentration was determined for each sample using the GeneQuant II RNA/DNA Calculator (Amersham Biosciences, Taipei, Taiwan).
Primers, polymerase chain reaction (PCR), and sequencing
PCRs were performed with universal primers for the Chs first intron (CHSx1F and CHSx2RN) [42] and the Lfy second intron (Mk.LFY-1n and Mk.LFY-2n) [43]. Amplifications were performed in a DNA programmable thermal cycler (PTC-100, MJ Research, Watertown, MA, USA) under the following protocol: initial denaturation at 94°C for 2 min followed by 35 cycles of 30 s at 94°C, 30 s of annealing at 52 or 48°C (Chs and Lfy, respectively), and 2 min at 72°C, followed by a subsequent 7-min final extension at 72°C. The PCR mixture (50 μL) contained 50 mM KCl, 1.5 mM MgCl2, 0.001% gelatin, 10 mM Tris-HCl (pH 8.3), 100 μM dNTPs, 0.2 μM of the primer, 20 ng of template DNA, 1 μg RNase, and 0.5 U Taq polymerase (Amersham Biosciences). Amplification products were run on 1% agarose gels, purified with a QiaGen purification kit (Taipei, Taiwan), and sequenced using both forward and reverse reactions with a Taq BigDye Terminator Cycle Sequencing Kit and a model ABI373A automated sequencer (Applied Biosystems, Foster City, CA, USA). This was applied to sequences of homozygotes or sequences containing one polymorphic site. Heterozygotes were visualized as "double peaks" at polymorphic sites in the chromatogram. The identity of the two haplotypes within a heterozygote was inferred through haplotype subtraction [44], which deduces haplotypes by a comparison of heterozygote sequences to haplotypes commonly observed in the global sample. When the chromatogram quality did not permit this procedure, PCR products were cloned using a yT&A vector (Yeastern Biotech, Taipei, Taiwan) according to the manufacturer's protocol. Three clones were sequenced for each individual when sequences contained two or more polymorphic sites and/or an unpaired indel (an indel of a different length). The two sequences of a heterozygote were separated by comparing sequences of the PCR product and cloned sequence. Fortunately, no sequence containing more than one indel was found in the length of Chs and Lfy used in this study. Because the Taq polymerase error was estimated to be as high as 0.1% [45] or even higher, sequences containing only one polymorphic site were considered to be produced by a Taq error and were excluded from the analysis. All haplotype sequences were deposited in the NCBI nucleotide sequence database under the following accession numbers: FJ858218~FJ858253 (Chs) and GQ260573~GQ260607 (Lfy).
DNA sequence analyses
Sequence alignments were performed with Clustal X [46] and manually edited using BioEdit vers. 7.0.9 [47]. After sequence alignment, indels were treated as the fifth character when calculating the indices of genetic diversity. The nucleotide diversity (π), haplotype diversity (Hd), and θ estimated by segregating sites (S) were calculated using DnaSP vers. 5.0 [48].
Population structure
Populations of C. kanehirae were formerly classified into four geographic regions, northwestern (NW), west-central (WC), southwestern (SW), and southeastern (SE), according to their geographic distributions (Figure 1) [35]. In addition, the CMR is considered to be a barrier to gene flow, and the structures of populations distributed in eastern and western Taiwan were also examined. The haplotype network was reconstructed with the algorithm of statistical parsimony using TCS vers. 1.21 [49]. The long-fragment indel of Chs sequences was treated as a one-mutation step and recoded. An analysis of molecular variance (AMOVA) was performed with Arlequin vers. 3.11 [50] in order to examine the components of genetic variation and the population structure.
Genetic differentiation between populations (pairwise FST) was calculated by Arlequin [50] based on Wright's [51] equation. To examine the influence of geographic barriers on levels of gene flow, the isolation-by-distance (IBD) model was used. If the stepping-stone dispersal model was tenable for C. kanehirae (and then the reinforcement of the geographic distance was necessary when estimating using MIGRATE), the IBD pattern was assumed. The IBD model was examined by the Mantel test (FST vs. geographic or altitudinal slope distance) to analyze the geographic effect on the genetic distribution. A reduced major axis (RMA) regression [52] was used to calculate the slope for the IBD test. Hellberg [52] indicated that the RAM regression is superior to the ordinary least squares regression for the IBD test based on simulation data. The IBD test considering both linear geographic distance and altitudinal slope was performed with the web-based computer program, IBDWS vers. 3.15 [53] (available at http://ibdws.sdsu.edu/~ibdws/).
Intralocus recombination
In the following analyses, all sites within studied loci were assumed to share the same genealogical history. However, recombination is believed to be common in nuclear loci and to influence the outcome of genealogically based analyses [54]. Therefore, we assessed recombination events with the "four-gamete test" using DnaSP vers. 5.0 [48]. Although this test only evaluates the minimum number of intralocus recombination events, it is sensitive to the presence of recombinations. When the test of the dataset of each locus (Chs and Lfy) suggested an intralocus recombination, we discarded the sites within the recombination block and retained the longest possible contiguous unrecombined sequence for subsequent analyses.
Migration rate through time
Because the incomplete migration-drift equilibrium was inferred based on the IBD results, the gene flow (Nm) estimated by the reciprocal of pairwise FST by Wright's [51] equation under the assumption of the migration-drift equilibrium may have been incorrect. Hence, varying degrees of the migration rate through time had to be considered. We jointly estimated all immigration and emigration rates by the Metropolis-coupled Markov chain Monte Carlo (MCMCMC) strategy in the MIGRATE program vers. 3.0 [55]. Because the Mantel test revealed essentially no correlations between genetic and geographic distances (see "Results"), the assumption of an identical migration potential was adopted in the MIGRATE algorithm. The prior distributions of uniform θ and equal migration rates were incorporated into the Bayesian approach in the MIGRATE algorithm. FST estimates were used as starting values for the initial analysis. For all other analyses, parameters at the end of the previous run were used as starting values for the next run until results converged at approximately the same values. We used settings of three long chains with 5 × 106 sampled and 5 × 103 recorded genealogies and discarded 25% of them at the beginning of a chain. Multiple long chains were combined for estimates. Adaptive heating was applied with temperature specifications of 1.0, 1.2, 1.5, and 3.0. Three replicates of the dataset were analyzed to calculate the average and confidence intervals. Because gene flow between populations estimated by pairwise M, which indicates the migration rate over the mutation rate, revealed confusing results (data not shown), four geographic regions based on the determination of Lin et al. [35] were used for the reanalysis.
Inferring the historical demography
Signatures of population demographic changes (bottlenecks or expansions) in C. kanehirae were first examined by Tajima's D [56] and Fu's Fs [57] statistics using DnaSP 5.0 [48] to see whether C. kanehirae data conformed to expectations of neutrality or departed from neutrality due to factors such as a population bottleneck or expansion. The examination of deviation from neutrality by both D and Fs indices was based on 1000 coalescent simulations with consideration of the recombination rate using DnaSP. Expectations of these statistics are nearly zero in a constant-size population; significant negative values indicate a sudden expansion in population size, whereas significant positive values indicate processes such as a population subdivision or recent population bottlenecks. Second, demographic changes in C. kanehirae were also examined by calculating the raggedness index of the observed mismatch distribution for each of the populations according to the population expansion model implemented in Arlequin 3.1 [50]. This measure quantifies the smoothness of the observed mismatch distribution. Small raggedness values represent a population which has experienced sudden expansion whereas higher values of the raggedness index suggest stationary or bottlenecked populations [58, 59]. Third, the demographic history of whether C. kanehirae underwent a range expansion was investigated by the spatial expansion model in Arlequin [50, 60–62]. We employed parametric bootstrapping (1000 replicates) as implemented in Arlequin to test the goodness-of-fit of the observed mismatch distribution to that expected under the spatial expansion model using the sum of squared deviations (SSD) statistic for regional and pooled samples. A significant SSD value is taken as evidence of departure from the estimated demographic model of spatial expansion in subdivided populations [60, 62] using Arlequin [50]. The model of spatial expansion assumes that subdivided populations expanded their distribution range and increased the total number of individuals [60, 61]. It estimates tau (τ), the demographic expansion factor, with 95% confidence intervals, and the mutation parameter, θ, as well as the number of migrants (M). This least-squares estimation of τ is reliable but was found to be unreliable for estimating the migration parameter, M [63]. Therefore, we took the M estimate of > 1 together with a non-significant SSD value as evidence of spatial expansion. The time at which the expansion event took place was dated following the expression, t = τ/2 μk, where τ is the estimated number of generations since the expansion, μ is the mutation rate per site per generation, and k is the sequence length. A mutation rate of 1.5% per 106 years per site was used for both Chs [64] and Lfy. We assumed a mean generation time of 20 years for C. kanehirae in converting the time to expansion to years.
Patterns of historical demography can also be inferred from estimates of the effective population size over time using the BSP method [38] as implemented in BEAST vers. 1.4.8 [65] for estimating fluctuations in the effective population size. This method estimates a posterior distribution of effective population sizes through time via MCMC procedures, by moving backward until the time of the most recent common ancestor is reached. The constant population size coalescent model was the basic assumption used for this approach. A HKY model together with among-site rate heterogeneity across all branches but assuming a strict molecular clock was used for this calculation. Markov chains were run for 2.5 × 107 generations and were sampled every 1000 generations, with the first 2500 samples discarded as burn-in. Three replicates were run and combined to separately analyze the Chs and Lfy datasets. Other parameters were set as default values. TRACER vers. 1.4 [66] was used to visualize the posterior probabilities of the Markov chain statistics and to calculate a statistical summary of the genetic parameters.
Results
Genetic diversity
In total, 82 of 113 samples of Chs (72.5%) and 41 of 94 samples of Lfy (43.6%) were heterozygotes. Although all 226 alleles of Chs and 188 alleles of Lfy were unambiguously identified, 16 and 18 of them, respectively, were probably derived from Taq errors and were thus excluded from further analysis. PCR amplification resulted in 637 and 657 bp of the Chs gene sequence spanning exon 1 (1~18 bp), intron 1 (19~594 bp or a 20-bp indel), and exon 2 (595~637 bp), and 741 bp of the Lfy gene sequence spanning exon 2 (1~75 bp), intron 2 (76~692 bp), and exon 3 (693~741 bp). All Chs variations occurred within the region of the intron. In aligned Lfy sequences, two variable sites were found within exon 2, 29 variable sites were found within intron 2, and three variable sites were found within exon 3 (Additional file 1). The aligned lengths of 210 Chs sequences were 657 bp and 170 Lfy sequences were 741 bp, which yielded 36 and 35 haplotypes, respectively (Table 1, Additional file 1). Details of the genetic diversity of populations and regions are presented in Table 2. Among 34 polymorphic sites of Chs, 14 were substitutions and 20 were indels; whereas only one indel was found among 35 polymorphic sites of Lfy. Among all of the populations examined, nucleotide diversity (π) was 0.00716 and ranged from 0.00372 (population CP in the SE region) to 0.00865 (population ALS in the WC region) in Chs and was 0.00479 and ranged from 0 (population YF in the SE region) to 0.00838 (population FL in the SE region) in Lfy. Haplotype diversity (Hd) was 0.841 and ranged from 0.515 (population CP in the SE region) to 1.000 (population HNS in the SW region) in Chs and was 0.895 and ranged from 0.000 (population YF in the SE region) to 1.000 (population WC in the WC region) in Lfy. Haplotype and nucleotide diversities of the four geographic regions were also calculated: haplotype diversities (Hd) ranged from 0.679 (SE region) to 0.906 (WC region) in Chs and from 0.674 (NW region) to 0.896 (SE region) in Lfy; nucleotide diversities ranged from 0.00511 (SE region) to 0.00784 (SW region) in Chs and from 0.00260 (WC region) to 0.00625 (SE region) in Lfy. When comparing the genetic diversity of regions to each other, the haplotype diversity and nucleotide diversity of the SE region estimated for Chs were significantly lower than those of the other three regions, but the nucleotide diversity of the SE region estimated for Lfy was significantly higher than those of the other regions. In addition, the haplotype diversity of the NW region estimated for Lfy was significantly lower than that of the SE region (Table 2).
Population structure and gene flow
Mean population pairwise FST values were 0.101 and 0.293 for the Chs and Lfy sequences, respectively (Additional file 2). The haplotype network depicted no clear patterns of population or regional genetic structuring (Additional file 3). Mean FST values for regional comparisons were 0.033 and 0.086 for the Chs and Lfy sequences, respectively (Table 3). Although the pairwise FST estimated from two nuclear DNA sequences showed discrepancies on the levels of genetic differentiation among regional comparisons, both Chs and Lfy pairwise FST matrices revealed higher genetic differentiation when SE was compared to the regions west of the CMR, but lower levels of genetic differentiations were found when only regions west of the CMR were compared (Table 3). The conspicuous level of genetic differentiation between regions on either side of the CMR indicates its barrier effect. A lack of migration-drift equilibrium can be assumed according to the result of the IBD tests (see below). Gene flow (Nm) estimated using Wright's model [51] which assumes migration-drift equilibrium was not appropriate for estimating magnitudes of gene flow. Therefore, gene flow varying over time was assumed when estimating the level of gene flow due to differential levels of common haplotype sharing among regions west of the CMR and across the CMR (Figure 2). We applied the Bayesian approach implemented in MIGRATE [55] to estimate the magnitudes and directions of gene flow between pairs of regional samples. The high number of migrants (M) calculated by the Bayesian approach indicated frequent gene flow, which was not hindered by geological interruption (Figure 1). The Bayesian analysis of migration showed that the SE region may have served as a dispersal source southwards in recent times (Figure 1).

Proportions of common and rare haplotypes of Cinnamomum kanehirae among the four geographic regions. The white bar indicates the proportion of haplotypes that specifically appeared in only one of the four regions; the dark bar is the proportion of haplotypes that also appeared in one of the three other regions; the intermediate-gray bar is the proportion of haplotypes that also appeared in two of the three other regions; and the light-gray bar indicates the proportion of haplotypes that appeared in all four regions.
At the entire population and regional scales, lack of correlations between genetic (FST) and geographic distances (both linear and altitudinal slope) was a common phenomenon according to the Mantel test, and only the SE-SW comparison showed a marginally significant correlation between genetic distance and linear geographic distance (r = 0.314, P = 0.049 for Chs sequences and r = 0.295, P = 0.047 for Lfy sequences, 1000 permutations, one-tailed test). If we consider the altitudinal slope, then the NW-SE and NW-WC comparisons showed significant correlations between genetic and geographic distances for Chs (r = 0.341, P = 0.011; r = 0.427, P = 0.044, respectively; 1000 permutations, one-tailed test), whereas no significant correlation was found based on Lfy sequences. In addition, we also estimated the most probable time at which gene flow occurred by the Bayesian approach performed using MIGRATE [55], and high probabilities of gene flow were found for each comparison during the last interglacial (Figure 3). This result indicates a trend of gene flow that occurred in the recent past or else a large, stable population size of this species in the past.

The number of migrants through time. Amplitudes of migrations through time are indicated by the parameter M multiplied by one thousand.
The AMOVA among the four geographic regions and between the east-west regions had similar results of a high proportion of the overall genetic variation being attributed to within-population levels (Table 4). On average, a relatively low percentage of the total genetic variation was attributable to among-region comparisons for both the four geographic regions and east-west regions. Nevertheless, the level of among-region genetic differentiation was significant when comparing eastern to western populations (ΦCT = 0.044, P < 0.01 for Chs and ΦCT = 0.170, P = 0.01 for Lfy). Significant genetic differentiation was also found in both sets of comparisons at the hierarchical level among populations within regions (ΦSC = 0.050 and ΦSC = 0.042 for Chs and ΦSC = 0.230 and ΦSC = 0.207 for Lfy, respectively, for comparisons of the four geographic regions and the eastern-western regions with P < 0.05). Moreover, genetic differentiation among populations was also significant for both sets of comparisons (ΦST = 0.067 and ΦST = 0.084 for Chs, and ΦST = 0.299 and ΦST = 0.341 for Lfy, respectively for comparisons of the four geographic regions and eastern-western regions with P < 0.01). When comparing the three geographic regions west of the CMR, both Chs and Lfy revealed no genetic structuring among regions, but a certain level of significant genetic structuring was found at the hierarchical levels among populations within regions and among populations (ΦSC = 0.080 and ΦST = 0.053 for Chs, and ΦSC = 0.163 and ΦST = 0.130 for Lfy, respectively). Although most comparisons of genetic structure were significant, the relatively shallow genetic structure on average especially when populations west of the CMR were compared represents a scenario of unhindered gene flow incompatible with the rugged geographic topology. The relatively shallow population structure was also supported by the mixed clustering relationships of populations (data not shown) and again reflects the large, contiguous distribution of this species in the past.
Demographic history
Neutrality tests of Tajima's D revealed non-significant negative values in many individual populations, pooled samples and the four regional samples for both the Chs and Lfy datasets (Table 2). Non-significant negative Fu's Fs values were also obtained for all populations, regional samples, and the pooled samples for Chs data. Similar results were also seen for most populations and regional samples with Lfy data; however, significant negative Fu's Fs values (P < 0.05) in the KS population, SW regions, and pooled samples were found (Table 2). Fu's Fs is considered to be more sensitive in detecting population expansion and suggesting possible past population growth, but the expansion may have been restricted to separate local areas that resulted in the non-significant negative Fu's Fs values for most populations and regions. Moreover, either population reduction, population subdivision, a recent bottleneck, or migration which resulted in secondary contact among previously differentiated lineages could have caused the discrepancy in detecting population expansion using the pooled samples with these two neutrality test statistics [67, 68]. Non-significant or small negative values of Tajima's D and Fu's Fs were found in many individual populations suggesting stationary in C. kanehirae populations. We also calculated the raggedness index under the demographic expansion model for each population and found that many individual populations had a high raggedness index (Table 2). We further analyzed the mismatch distribution under the spatial expansion model considering population subdivision [60–62]. Analysis of the mismatch distribution showed a multimodal pattern characteristic of population differentiation which corresponds to demographic growth and a subsequent bottleneck [[61], data not shown]. It is probable that loss of haplotypes due to the recent bottleneck would have caused the multimodal mismatch distribution. According to the mismatch distribution analysis, the hypothesis of spatial expansion could not be rejected in pooled samples (SSD = 0.021, P = 0.502 for Chs and SSD = 0.011, P = 0.38 for Lfy, Table 5). A good fit of the spatial expansion was also observed for regional samples according to the non-significant SSD values; however, different expansion times of regional samples estimated by τ were found due to different coalescent histories of Chs and Lfy sequences (Table 5). The relatively longer expansion time and smaller migration rate of the NW region for both sequence sets (τ = 8.95 and M = 0.02 for Lfy; τ = 25.8 and M = 0.89 for Chs) implies a far-distant spatial expansion for older lineages of NW regional samples. In contrast to NW samples, SE regional samples revealed a relatively shorter expansion time and larger migration rate (τ = 2.50 and M = 8.08 for Lfy; τ = 5.5 and M = 1.65 for Chs), which indicates a recent spatial expansion.
The BSP analysis suggested a past increase in the effective population size of C. kanehirae for both nuclear markers (Figure 4, Additional file 4). An increase in the effective population size inferred by Chs was smaller than that inferred by Lfy. Moreover, the time of population growth inferred by Chs was later than that inferred by Lfy. Slight differences in trends of the population size increase and expansion time are likely due to different coalescent histories of the two loci and uncertainty of molecular dating [69]. On the other hand, a clear recent bottleneck was revealed by the pooled samples of Chs sequences, but Lfy data showed only a slight decrease in population size after the last glacial maximum (LGM) expansion. In addition, regional samples of western, WC, and SE revealed a more-obvious population decline for Chs than for Lfy (Figure 4). Moreover, NW samples displayed a history of a prolonged population decline followed by a recent sudden expansion based on Lfy variations.

Bayesian skyline plot revealing the demographic trends of Chs and Lfy intron lineages of Cinnamomum kanehirae. The population size was measured as the effective population size per generation. MIS, marine isotope stage.
Discussion
Genetic diversity
Historical demographic events and population sizes of most natural populations fluctuated and left traces in patterns of their genetic diversity. In this study, C. kanehirae had higher or comparable nucleotide diversities for both Chs and Lfy or either one of them separately compared to the nucleotide diversity in population studies of nuclear genes such as Chs in the Hippophaë rhamnoides species complex [70], the glyceraldehyde-3-phosphate dehydrogenase (G3pdh) gene in Q. glauca [71] and the nucleotide diversity of G3pdh in Manihot esculenta [72], the PHYP and PHYO loci in Pinus sylvestris [73], and the fruit vacuolar invertase gene in Solanum pimpinellifolium [74]. Although the comparison of nucleotide diversity using nuclear DNA sequence data might not be complete, the high level of allozyme variation found also suggests a great potential for high genetic variation in nuclear DNA sequences of C. kanehirae [35]. High nucleotide diversity of nuclear genes may reflect a long evolutionary history of a large, stable population, or mixing of differential lineages via range expansion [75–77] and is consistent with field surveys of the contiguous range distribution of C. kanehirae in the past [25, 26].
Historical and recent demography of Cinnamomum kanehirae
Overall, it can be seen that the demographic history of C. kanehirae is far more complex than previously thought, with evidence of historical range expansion and a very recent bottleneck (Table 2, Figure 4). While population expansion was rejected by Tajima's D and Fu's Fs test statistics in most population and regional samples, the high raggedness index estimated for individual populations suggests the occurrence of bottleneck events [78]. Population bottlenecks were also evident when analyzed with the BSP (Figure 4). On the other hand, the mismatch analyses failed to reject a spatial expansion model for all datasets including pooled and regional samples (Table 5). Historical spatial expansion was also revealed by the BSP analysis (Figure 4). From these results the demographic history of C. kanehirae conformed to the hypothesis of past spatial range expansion followed by a very recent population bottleneck. However, the past spatial range expansion might not display a molecular signature of a unimodal distribution characteristic of sudden expansions in un-subdivided populations, even though populations had expanded by several orders of magnitude after the past expansion [61] and was evident through analysis with the MIGRATE software, which might reflect a historically large stable population of C. kanehirae (Figure 3). The level of nucleotide diversity of C. kanehirae might therefore be a species-associated feature, and also a result of its past evolutionary history as well as contemporary events.
The climate during interglacial periods in Taiwan was mostly humid and warm (in contrast to glacial periods) [79, 80], and therefore habitats at different elevations were appropriate for the growth of C. kanehirae. Population expansion after the LGM was inferred for several plant species including C. kanehirae based on cpDNA variations [21, 37, 40, 81]. Further information on the demographic history of C. kanehirae was revealed by nuclear DNA variations. It is likely that larger genetic variability that accumulated in nuclear markers coalesced into a longer genealogy relative to cpDNA, reflecting greater sensitivity in revealing insights into both recent and older events. Moreover, C. kanehirae is an insect-pollinated hermaphrodite with male and female flowers at different times possibly promoting outcrossing and therefore at least a 2-fold higher effective population size of nuclear loci relative to cpDNA makes it more sensitive to population oscillations. The vegetational history traced by pollen records illustrates rapid growth of Alnus, Liquidambar, Mallotus, Trema, and the Chenopodiaceae in subtropical forests of Taiwan [82]. Characteristics of a slow growth rate and long generation time (approximate 20 years) may be limiting factors in habitat establishment for C. kanehirae when competing with other plant species, which thus reinforced the present-day bottleneck.
Loss of genetic diversity due to anthropogenic factors might not be apparent because of the high genetic diversity that accumulated during past range expansions and resulted in only a slight decline in the recent effective population size as estimated by the BSP analysis. Nevertheless, this small decline in the recent population size reveals a commencing phase of a bottleneck in C. kanehirae inflicted by anthropogenic factors. The past increase in gene flow and spatial expansion should have assisted the dispersal of population- and region-specific haplotypes; however, human interventions including over-deforestation and habitat destruction caused declining of population size and fewer opportunities for colonization of these specific haplotypes.
Contemporary genetic structure and gene flow
Cinnamomum kanehirae is a plant species with bird-dispersed seeds. Forest birds in the warm period of the last interglacial [83] could have facilitated seed dispersal of Lauraceae species and accelerated gene flow between C. kanehirae populations. Cinnamomum kanehirae has a similar pattern of diversity distribution as that of Machilus thunbergii (Lauraceae) based on cpDNA variations with most populations possessing the ancestral haplotype [40, 84]. A shared ancestral polymorphism was the cause of the lack of genetic differentiation among populations in these species. In this study, relatively low levels of genetic differentiation (FST) and a high degree of gene flow were also found by estimates of M based on nuclear DNA variations. Moreover, the haplotype network depicted no clear patterns of geographic structuring, further supporting the regional migrations of haplotypes (Additional file 3). The overall spatial genetic differentiation estimated among all C. kanehirae populations was not purely stochastic, but instead probably reflects the effect of secondary contacts through migration due to range expansion after the LGM (Figure 3, Table 5).
Rates and patterns of dispersal and migration between populations will affect the genetic structure of species: the higher the dispersal ability, the lower the population structuring [85]. The AMOVA revealed a certain level of significant genetic structuring in C. kanehirae populations, but the level of the genetic structuring of the two nuclear DNA loci assayed was relatively low on average at all hierarchical levels. Relatively low levels of genetic differentiation in C. kanehirae, especially among populations west of the CMR (Table 3), can be attributed to two possibilities: first, gene exchange between long-term isolated populations and a second, recent fragmentation of a large population with recurrent gene flow. A similar consequence of a lack of genetic structuring and low population differentiation was observed for these two possibilities; however, there were slight differences in the outcomes. In the situation of long-term isolation, each separate population would have independently experienced genetic drift, and resulted in high frequencies of rare haplotypes in each population [86]. On the other hand, if the populations were recently separated, there would insufficient time for genetic drift to have occurred in each small fragmented population, and most populations would share common haplotypes [87]. Sharing of haplotypes was frequently found among populations and among regions (Table 1, Figure 2), although some haplotypes were region- and/or population-specific, however, with relatively low proportions. Therefore, the hypothesis of recent fragmentation of a large population with recurrent gene flow is highly probable. A general lack of IBD, at a regional scale to the west of the CMR, is typically considered to be the result of a large, stable population with historically recurring gene flow, which was further supported by the high migration rates among both populations and regions (Figure 3).
The implications of incomplete drift-migration equilibrium can further explain the contemporary genetic structure of C. kanehirae, which is the outcome of the interaction between inherent past spatial expansion with the barrier effect of the CMR and recent bottleneck events. Taiwan is a continental island which began to rise approximately 4 Mya by tectonic compression of the Eurasian and Philippine Sea Plates. The CMR geographically separates the landscape into eastern and western parts [13]. The barrier effect of gene flow by the CMR in Taiwan was observed in several plant and animal studies [37, 81, 88]. The AMOVA indicated some level of geographic structuring resulted in haplotypes specific to either the eastern or western populations of C. kanehirae, which might be related to the barrier effect of the CMR, and this was statistically supported (Table 4). The occurrence of population- or region-specific haplotypes may have resulted from a recent bottleneck event due to human interventions that caused the loss of haplotypes in other populations and regions; however, the possible failure to discover haplotypes due to the small sample size cannot be excluded. Historically, recurrent gene flow between populations, across geographic regions and across the CMR possibly diminished the level of genetic differentiation and the influence of the CMR barrier. Therefore, common haplotype distributions between geographic regions and relatively shallow genetic structuring are the result of historical gene flows. Under the drift-migration equilibrium, patterns of exclusivity are not expected, particularly for a large population size or a short isolation time. In this scenario, common haplotypes should be more abundant than private ones in an individual population. However, rare or population-specific haplotypes occurred in almost all of the populations examined (Table 1).
Dispersal after the LGM
Historical and extrinsic ecological events can leave signatures in the spatial genetic structure of a species and provide insights into understanding the species' evolutionary history [75]. In Taiwan, six subtropical tree species have been phylogeographically studied using cpDNA variations, and the SE part of Taiwan was inferred to be a major diversity center based on average FST divergences from populations of other regions [22, 37, 40, 71, 81, 84, 89], and that was further suggested by this study because a higher average FST divergence from other regions was found when the SE was compared to other regions (Table 3). However, the SE region being postulated as a diversity center in this way may only reflect its long isolation from other regions, which was suggested by the high level of regional-specific haplotypes and very recent expansion southward. In contrast, the southerly expansion event of the NW region to the west of the CMR was older according to the TMRCA estimates (Table 5). The trend of a temperature rise after cold climatic periods of the Pleistocene was inferred to have occurred from south to north assisted by the warm Kuroshio Current that flow northward offshore of the east coast of Taiwan [90]. Although the South China Sea Warm Current flowed through the Taiwan Strait after the LGM when sea levels again rose, the magnitude of the temperature rise might not have been significant, as the area west of the CMR faces cold fronts from the north as well as the barrier effect of the CMR in receiving warm temperatures from the south carried by the Kuroshio Current. Thus, a deferred timing of the temperature rise may have occurred in western populations of C. kanehirae (except populations in the SW region). The onset of a temperature rise after the LGM for populations located in the SE region preceded that of populations located in other regions, thereby facilitating the migration from SE to SW regions by C. kanehirae.
Conclusions
Distributions of genetic variations in wild populations provide insights into a species' evolutionary history and its future continuity [75, 91]. Cinnamomum kanehirae is confronted with conditions of being rare, fragmented, and threatened at the present time, consistent with the genetic survey that revealed a pattern of population growth followed by an ongoing bottleneck. Despite the declining effective population size of C. kanehirae, the genetic diversity of remnant individuals showed no dramatic reduction in genetic diversity. In C. kanehirae, a tree with a long generation time, the rapid population decline probably began decades ago due to anthropogenic interventions for two or more generations. Thus information on the loss of genetic diversity might not be spontaneously revealed, but will probably be seen within two or more generations if the populations are not restored. In light of the declining effective population size, the condition of the true population size of C. kanehirae at present may be far more severely threatened. In addition to the continuing illegal selective cutting for cultivating the medicinal fungus, A. cinnamomea, the loss of habitat is another serious issue contributing to population declines of C. kanehirae. Elevations where C. kanehirae grows range 200~2000 m, and overdevelopment for farming vegetables, tea, and fruit crops has destroyed the natural habitats of primeval forests, including habitats of C. kanehirae. Therefore, loss of the population size of C. kanehirae was mainly caused by human overexploitation. In consequence, it is essential that this valuable species be conserved through habitat restoration, which can advance the restoration of the effective population size of C. kanehirae. Further field surveys to locate plants and grafts for ex situ conservation are another requirement to conserve this species. Management strategies should also consider reinforcing local populations with individuals originating from populations showing the closest genetic relatedness to produce larger population with higher genetic variability. In summary, conservation strategies must therefore aim for habitat restoration and remnant diversity preservation, and avoid the occurrence of genetic inbreeding in populations of C. kanehirae.
References
Hewitt GM: Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc Lond. 1996, 58: 247-276.
Hewitt G: The genetic legacy of the Quaternary ice ages. Nature. 2000, 405: 907-913. 10.1038/35016000.
Lee-Yaw JA, Irwin JT, Green DM: Postglacial range expansion from northern refugia by the wood frog, Rana sylvatica. Mol Ecol. 2008, 17: 867-884.
Depraz A, Cordellier M, Hausser J, Pfenninger M: Postglacial recolonization at a snail's pace (Trochulus villosus): confronting competing refugia hypotheses using model selection. Mol Ecol. 2008, 17: 2449-2462. 10.1111/j.1365-294X.2008.03760.x.
Schmitt T, Haubrich K: The genetic structure of the mountain forest butterfly Erebia euryale unravels the late Pleistocene and postglacial history of the mountain coniferous forest biome in Europe. Mol Ecol. 2008, 17: 2194-2207. 10.1111/j.1365-294X.2007.03687.x.
Puscas M, Choler P, Tribsch A, Gielly L, Rioux D, Gaudeul M, Taberlet P: Post-glacial history of the dominant alpine sedge Carex curvula in the European Alpine System inferred from nuclear and chloroplast markers. Mol Ecol. 2008, 17: 2417-2429. 10.1111/j.1365-294X.2008.03751.x.
Ikeda H, Senni K, Fujii N, Setoguchi H: Consistent geographic structure among multiple nuclear sequences and cpDNA polymorphisms of Cardamine nipponica Franch. et Savat. (Brassicaceae). Mol Ecol. 2008, 17: 3178-3188. 10.1111/j.1365-294X.2008.03821.x.
Beck JB, Schmuths H, Schaal BA: Native range genetic variation in Arabidopsis thaliana is strongly geographically structured and reflects Pleistocene glacial dynamics. Mol Ecol. 2008, 17: 902-915. 10.1111/j.1365-294X.2008.03906.x.
Su YC, Chang YH, Lee SC, Tso IM: Phylogeography of the giant wood spider (Nephila pilipes, Araneae) from Asian-Australian regions. J Biogeogr. 2007, 34: 177-191. 10.1111/j.1365-2699.2006.01617.x.
Yasuda SP, Vogel P, Tsuchiya K, Han SH, Lin LK, Suzuki H: Phylogeographic patterning of mtDNA in the widely distributed harvest mouse (Micromys minutus) suggests dramatic cycles of range contraction and expansion during the mid- to late Pleistocene. Can J Zool. 2005, 83: 1411-1420. 10.1139/z05-139.
Brubaker LB, Anderson PM, Edwards ME, Lozhkin AV: Beringia as a glacial refugium for boreal trees and shrubs: new perspectives from mapped pollen data. J Biogeogr. 2005, 32: 833-848. 10.1111/j.1365-2699.2004.01203.x.
Bird MI, Taylor D, Hunt C: Environments of insular Southeast Asia during the Last Glacial Period: a savanna corridor in Sundaland?. Quat Sci Rev. 2005, 24: 2228-2242. 10.1016/j.quascirev.2005.04.004.
Shen CF: The biogeography of Taiwan: 2. Some preliminary thoughts and studies. National Taiwan Museum Annual Report. 1997, 40: 361-450.
Siame L, Chu HT, Carcaillet J, Bourles D, Braucher R, Lu WC, Angelier J, Dussouliez P: Glacial retreat history of Nanhuta Shan (north-east Taiwan) from preserved glacial features: the cosmic ray exposure perspective. Quat Sci Rev. 2007, 26: 2185-2200. 10.1016/j.quascirev.2007.04.016.
Böse M: Traces of glaciation in the high mountains of Taiwan. Quaternary glaciations-extent and chronology Part III: Asia, Latin America, Africa, Australasia, Antarctica. Edited by: Ehlers J, Gibbard PL. 2004, Amsterdam: Elsevier, 347-352. full_text.
Hsieh CF: Composition, endemism and phytogeographical affinities of the Taiwan flora. Taiwania. 2002, 47: 298-310.
Hebenstreit R, Bose M, Murray A: Late Pleistocene and early Holocene glaciations in Taiwanese mountains. Quat Int. 2006, 147: 76-88. 10.1016/j.quaint.2005.09.009.
Ono YG, Aoki T, Hasegawa H, Liu DL: Mountain glaciation in Japan and Taiwan at the global last glacial maximum. Quat Int. 2005, 138: 79-92. 10.1016/j.quaint.2005.02.007.
Liew PM, Huang SY, Kuo CM: Pollen stratigraphy, vegetation and environment of the last glacial and Holocene - a record from Toushe Basin, central Taiwan. Quat Int. 2006, 147: 16-33. 10.1016/j.quaint.2005.09.003.
Lin TP: Allozyme variations in Michelia formosana (Kanehira) Masamune (Magnoliaceae), and the inference of a glacial refugium in Taiwan. Theo Appl Genet. 2001, 102: 450-457. 10.1007/s001220051666.
Hwang SY, Lin TP, Ma CS, Lin CL, Chung JD, Yang JC: Postglacial population growth of Cunninghamia konishii (Cupressaceae) inferred from phylogeographical and mismatch analysis of chloroplast DNA variation. Mol Ecol. 2003, 12: 2689-2695. 10.1046/j.1365-294X.2003.01935.x.
Lee YJ, Hwang SY, Ho KC, Lin TP: Source populations of Quercus glauca in the last glacial age in Taiwan revealed by nuclear microsatellite markers. J Hered. 2006, 97: 261-269. 10.1093/jhered/esj030.
Chanderbali AS, Werff van der H, Renner SS: Phylogeny and historical biogeography of Lauraceae: evidence from the chloroplast and nuclear genomes. Annals of the Missouri Botanical Garden. 2001, 88: 104-134. 10.2307/2666133.
Liao JC: Lauraceae. Flora of Taiwan. Edited by: Boufford DE, Hsieh C-F, Huang T-C, Ohashi H, Yang Y-P, Lu S-Y. 1996, Taipei, Taiwan: Editorial Committee of Flora of Taiwan, 2: 433-499. 2
Lin TP: Cinnamomum kanehirae Hay. and Cinnamomum micranthum (Hay.) Hay. Bulletin of Taiwan Forest Research Institute. 1993, 8: 11-20.
Uka S: Report on the survey of Taiwan camphora trees. 1927, Taipei: Monopoly Bureau, Formosan Government
Chang TT, Chou WN: Antrodia cinnamomea sp. nov. on Cinnamomum kanehirai in Taiwan. Mycol Res. 1995, 99: 756-758. 10.1016/S0953-7562(09)80541-8.
Chang TT, Chou WN: Antrodia cinnamomea reconsidered and A. salmonea sp. nov. on Cunninghamia konishii in Taiwan. Bot Bull Acad Sin. 2004, 45: 347-352.
Wu SH, Ryvarden L, Chang TT: Antrodia camphorata ("niu-chang-chih"), new combination of a medicinal fungus in Taiwan. Bot Bull Acad Sin. 1997, 38: 273-275.
Song TY, Yen GC: Antioxidant properties of Antrodia camphorata in submerged culture. J Agric Food Chem. 2002, 50: 3322-3327. 10.1021/jf011671z.
Yang HL, Hseu YC, Chen JY, Yech YJ, Lu FJ, Wang HH, Lint PS, Wang BC: Antrodia camphorata in submerged culture protects low density lipoproteins against oxidative modification. Am J Chin Med. 2006, 34: 217-231. 10.1142/S0192415X06003783.
Hseu YC, Chang WC, Hseu YT, Lee CY, Yech YJ, Chen PC, Chen JY, Yang HL: Protection of oxidative damage by aqueous extract from Antrodia camphorata mycelia in normal human erythrocytes. Life Sci. 2002, 71: 469-482. 10.1016/S0024-3205(02)01686-7.
Chen CH, Yang SW, Shen YC: New steroid acids from Antrodia cinnamomea, a fungal parasite of Cinnamomum micranthum. J Nat Prod. 1995, 58: 1655-1661. 10.1021/np50125a002.
Lee IH, Huang RL, Chen CT, Chen HC, Hsu WC, Lu MK: Antrodia camphorata polysaccharides exhibit anti-hepatitis B virus effects. FEMS Microbiol Lett. 2002, 209: 63-67. 10.1111/j.1574-6968.2002.tb11110.x.
Lin TP, Cheng YP, Huang SG: Allozyme variation in four geographic areas of Cinnamomum kanehirae. J Hered. 1997, 88: 433-438.
Chung JD, Lin TP, Tan YC, Lin MY, Hwang SY: Genetic diversity and biogeography of Cunninghamia konishii (Cupressaceae), an island species in Taiwan: a comparison with Cunninghamia lanceolata, a mainland species in China. Mol Phylogenet Evol. 2004, 33: 791-801. 10.1016/j.ympev.2004.08.011.
Cheng YP, Hwang SY, Lin TP: Potential refugia in Taiwan revealed by the phylogeographical study of Castanopsis carlesii Hayata (Fagaceae). Mol Ecol. 2005, 14: 2075-2085. 10.1111/j.1365-294X.2005.02567.x.
Drummond AJ, Rambaut A, Shapiro B, Pybus OG: Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005, 22: 1185-1192. 10.1093/molbev/msi103.
Broughton RE, Harrison RG: Nuclear gene genealogies reveal historical, demographic and selective factors associated with speciation in field crickets. Genetics. 2003, 163: 1389-1401.
Kuo DC, Lin CC, Ho KC, Cheng YP, Hwang SY, Lin TP: Two genetic divergence centres revealed by chloroplastic DNA variation in populations of Cinnamomum kanehirae Hay. Conservation Genetics. 2009, doi: 10.1007/s10592-10009-19901-10595
Doyle JJ, Doyle JL: Isolation of plant DNA from fresh tissue. Focus. 1990, 12: 13-15.
Strand AE, LeebensMack J, Milligan BG: Nuclear DNA-based markers for plant evolutionary biology. Mol Ecol. 1997, 6: 113-118. 10.1046/j.1365-294X.1997.00153.x.
Frohlich MW, Meyerowitz EM: The search for flower homeotic gene homologs in basal angiosperms and gnetales: a potential new source of data on the evolutionary origin of flowers. Int J Plant Sci. 1997, 158: S131-S142. 10.1086/297513.
Clark AG: Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol. 1990, 7: 111-122.
Okuyama Y, Fujii N, Wakabayashi M, Kawakita A, Ito M, Watanabe M, Murakami N, Kato M: Nonuniform concerted evolution and chloroplast capture: heterogeneity of observed introgression patterns in three molecular data partition phylogenies of Asian Mitella (Saxifragaceae). Mol Biol Evol. 2005, 22: 285-296. 10.1093/molbev/msi016.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25: 4876-4882. 10.1093/nar/25.24.4876.
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symposium Series. 1999, 41: 95-98.
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452. 10.1093/bioinformatics/btp187.
Clement M, Posada D, Crandall KA: TCS, a computer program to estimate gene genealogies. Mol Ecol. 2000, 9: 1657-1659. 10.1046/j.1365-294x.2000.01020.x.
Excoffier L, Laval G, Schneider S: Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform Online. 2005, 1: 47-50.
Wright S: The genetical structure of populations. Ann Eugen. 1951, 15: 323-354.
Hellberg ME: Relationships between inferred levels of gene flow and geographic distance in a philopatric coral, Balanophyllia elegans. Evolution. 1994, 48: 1829-1854. 10.2307/2410512.
Jensen JL, Bohonak AJ, Kelley ST: Isolation by distance, web service. BMC Genetics. 2005, 6: 13-10.1186/1471-2156-6-13.
Wall JD: Estimating ancestral population sizes and divergence times. Genetics. 2003, 163: 395-404.
Beerli P: Migrate version 3.0 - a maximum likelihood and Bayesian estimator of gene flow using the coalescent. 2008, [http://popgen.sc.fsu.edu/Migrate-n.html]
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Fu YX: Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997, 147: 915-925.
Harpending HC: Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol. 1994, 66: 591-600.
Harpending HC, Sherry ST, Rogers AR, Stoneking M: The genetic structure of ancient human populations. Curr Anthropol. 1993, 34: 483-496. 10.1086/204195.
Excoffier L: Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol. 2004, 13: 853-864. 10.1046/j.1365-294X.2003.02004.x.
Ray N, Currat M, Excoffier L: Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol. 2003, 20: 76-86. 10.1093/molbev/msg009.
Schneider S, Excoffier L: Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates very among sites: application to human mitochondrial DNA. Genetics. 1999, 152: 1079-1089.
Hamilton G, Currat M, Ray N, Heckel G, Beaumont M, Excoffier L: Bayesian estimation of recent migration rates after a spatial expansion. Genetics. 2005, 170: 409-417. 10.1534/genetics.104.034199.
Koch MA, Haubold B, Mitchell-Olds T: Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol Biol Evol. 2000, 17: 1483-1498.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7: 214-10.1186/1471-2148-7-214.
Rambaut A, Drummond AJ: Tracer v1.4. 2007, [http://beast.bio.ed.ac.uk/Tracer]
Maruyama T, Fuerst PA: Population bottlenecks and non-equilibrium models in population genetics. I. Allele numbers when populations evolve from zero variability. Genetics. 1984, 108: 745-763.
Maruyama T, Fuerst PA: Population bottlenecks and non-equilibrium models in population genetics. II. Number of alleles in a small population that was formed by a recent bottleneck. Genetics. 1985, 111: 675-689.
Ho SY, Saarma U, Barnett R, Haile J, Shapiro B: The effect of inappropriate calibration: three case studies in molecular ecology. PLoS ONE. 2008, 3: e1615-10.1371/journal.pone.0001615.
Bartish IV, Kadereit JW, Comes HP: Late Quaternary history of Hippophae rhamnoides L. (Elaeagnaceae) inferred from chalcone synthase intron (Chsi) sequences and chloroplast DNA variation. Mol Ecol. 2006, 15: 4065-4083. 10.1111/j.1365-294X.2006.03079.x.
Shih FL, Cheng YP, Hwang SY, Lin TP: Partial concordance between nuclear and organelle DNA in revealing the genetic divergence among Quercus glauca (Fagaceae) populations in Taiwan. Int J Plant Sci. 2006, 167: 863-872. 10.1086/504923.
Olsen KM: Population history of Manihot esculenta (Euphorbiaceae) inferred from nuclear DNA sequences. Mol Ecol. 2002, 11: 901-911. 10.1046/j.1365-294X.2002.01493.x.
García-Gil MR, Mikkonen M, Savolainen O: Nucleotide diversity at two phytochrome loci along a latitudinal cline in Pinus sylvestris. Mol Ecol. 2003, 12: 1195-1206. 10.1046/j.1365-294X.2003.01826.x.
Caicedo AL, Schaal BA: Population structure and phylogeography of Solanum pimpinellifolium inferred from a nuclear gene. Mol Ecol. 2004, 13: 1871-1882. 10.1111/j.1365-294X.2004.02191.x.
Frankham R: Relationship of genetic variation to population size in wildlife. Conser Biol. 1996, 10: 1500-1508. 10.1046/j.1523-1739.1996.10061500.x.
Grant WS, Bowen BW: Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered. 1998, 89: 415-426. 10.1093/jhered/89.5.415.
Avise JC: Phylogeography: The History and Formation of Species. 2000, Cambridge, MA.: Harvard University Press
Rogers AR, Harpending H: Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992, 9: 552-569.
Wei KY: Environmental changes during the late quaternary in Taiwan and adjacent seas: an overview of recent results of the past decade (1990-2000). West Pac Earth Sci. 2002, 2: 149-160.
Liew PM, Kuo CM, Huang SY, Tseng MH: Vegetation change and terrestrial carbon storage of eastern Asia during the last glacial maximum as indicated by new pollen records of northern Taiwan. Glob Planet Change. 1998, 16: 85-94. 10.1016/S0921-8181(98)00014-9.
Huang SSF, Hwang SY, Lin TP: Spatial pattern of chloroplast DNA variation of Cyclobalanopsis glauca in Taiwan and east Asia. Mol Ecol. 2002, 11: 2349-2358. 10.1046/j.1365-294X.2002.01624.x.
Tsukada M: Late Pleistocene vegetation and climate in Taiwan (Formosa). PNAS. 1966, 55: 543-548. 10.1073/pnas.55.3.543.
Rhymer JM, McAuley DG, Ziel HL: Phylogeography of the American woodcock (Scolopax minor): Are management units based on band recovery data reflected in genetically based management units?. Auk. 2005, 122: 1149-1160. 10.1642/0004-8038(2005)122[1149:POTAWS]2.0.CO;2.
Wu SH, Hwang CY, Lin TP, Chung JD, Cheng YP, Hwang SY: Contrasting phylogeographical patterns of two closely related species, Machilus thunbergii and Machilus kusanoi (Lauraceae), in Taiwan. J Biogeogr. 2006, 33: 936-947. 10.1111/j.1365-2699.2006.01431.x.
Clegg SM, Degnan SM, Kikkawa J, Moritz C, Estoup A, Owens IPF: Genetic consequences of sequential founder events by an island-colonizing bird. PNAS. 2002, 99: 8127-8132. 10.1073/pnas.102583399.
Kimura M, Clegg SM, Lovette IJ, Holder KR, Girman DJ, Mila B, Wade P, Smith TB: Phylogeographical approaches to assessing demographic connectivity between breeding and overwintering regions in a Nearctic-Neotropical warbler (Wilsonia pusilla). Mol Ecol. 2002, 11: 1605-1616. 10.1046/j.1365-294X.2002.01551.x.
Printzen C, Ekman S, Tonsberg T: Phylogeography of Cavernularia hultenii: evidence of slow genetic drift in a widely disjunct lichen. Mol Ecol. 2003, 12: 1473-1486. 10.1046/j.1365-294X.2003.01812.x.
Toda M, Nishida M, Matsui M, Lue KY, Ota H: Genetic variation in the Indian rice frog, Rana limnocharis (Amphibia: Anura), in Taiwan, as revealed by allozyme data. Herpetologica. 1998, 54: 73-82.
Huang SF, Hwang SY, Wang JC, Lin TP: Phylogeography of Trochodendron aralioides (Trochodendraceae) in Taiwan and its adjacent areas. J Biogeogr. 2004, 31: 1251-1259. 10.1111/j.1365-2699.2004.01082.x.
Chen MP, Huang CK, Lo L, Wang CH: Late Pleistocene paleoceanography of the Kurosio Current in the area offshore southeast Taiwan. Terr Atm Ocean Sci. 1992, 3: 81-110.
Raybould AF, Clarke RT, Bond JM, Welters RE, Gliddon CJ: Inferring patterns of dispersal from allele frequency data. Dispersal Ecology. Edited by: Bullock JM, Kenward RE, Hails RS. 2002, Oxford: Blackwell Science, 89-110.
Acknowledgements
This investigation was funded by the Council of Agriculture (grant no. 96AS-7.2.2-FB-e2), Executive Yuan, Taiwan to Shih-Ying Hwang. The authors acknowledge the assistance of the Taiwan Forestry Research Institute in providing the materials of Cinnamomum kanehirae growing in a scion garden at the Liukuei Research Center, Kaohsiung, Taiwan.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
SYH and TPL conceived the research; DCK, CCL, and KCH collected the data; PCL analyzed the data and wrote the first draft; SYH led the writing. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2009_1340_MOESM1_ESM.DOC
Additional file 1: Polymorphic sites among haplotypes at Chs and Lfy. The table provides polymorphic information for both the Chs and Lfy loci for each haplotype. (DOC 333 KB)
12862_2009_1340_MOESM2_ESM.DOC
Additional file 2: Pairwise F ST of 19 Cinnamomum kanehirae populations. The table provides the pairwise genetic distance matrix obtained using Chs and Lfy DNA. (DOC 67 KB)
12862_2009_1340_MOESM3_ESM.DOC
Additional file 3: Phylogenetic network constructed by both Chs and Lfy sequences. The figure shows the haplotype networks constructed using a statistical parsimony approach. The long-fragment indel of Chs sequences was treated as one-mutation step and recoded. The limit of 90% connection was used to estimate the network. Four geographic regions were indicated proportionally with four colors. (DOC 328 KB)
12862_2009_1340_MOESM4_ESM.DOC
Additional file 4: Changes in effective population size in regional and pooled samples of Cinnamomum kanehirae. The figure shows the effective population size per generation for regional and pooled samples of Cinnamomum kanehirae based on Chs and Lfy datasets. The solid line represents mean values of the estimated effective population size with lower (0.05) and upper (0.95) bounds. (DOC 224 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liao, PC., Kuo, DC., Lin, CC. et al. Historical spatial range expansion and a very recent bottleneck of Cinnamomum kanehiraeHay. (Lauraceae) in Taiwan inferred from nuclear genes. BMC Evol Biol 10, 124 (2010). https://doi.org/10.1186/1471-2148-10-124
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-10-124