
Abstract
Background
Although closely related, the alpha-proteobacteria Wolbachia and the Rickettsiacae (Rickettsia and Ehrlichia), employ different evolutionary life history strategies. Wolbachia are obligate endocellular symbionts that infect an extraordinary host range and, in contrast to the infectious and pathogenic Rickettsia and Ehrlichia, profoundly influence host reproductive biology.
Results
Phylogenies of the Rickettsia, Ehrlichia, and Wolbachia were independently inferred from 16S rDNA sequences and GroEL amino acid sequences. Topologies inferred from both sets of sequence data were consistent with one another, and both indicate the genus Wolbachia shared a common ancestor most recently with Ehrlichia. These two genera are a sister group to the genus Rickettsia. Mapping biological properties onto this phylogeny reveals that manipulation of host reproduction, characteristic of Wolbachia strains, is a derived characteristic. This evolutionary novelty is accompanied by the loss of the ability to infect vertebrate hosts.
Conclusions
Because of the contrasting transmission strategies employed by each, Wolbachia is expected to maximize efficiency of vertical transmission, while Ehrlichia and Rickettsia will optimize horizontal transfer of infection. Wolbachia manipulation of host reproduction could thus be viewed as strategy employed by this bacterium to foster its own propagation via vertical transmission.
Similar content being viewed by others


Background
In recent years a great deal of excitement has been generated by the discovery that Wolbachia infections in Ecdyzoa (arthropods and nematodes) can manipulate reproduction of their hosts in a variety of ways; e.g., induced parthenogenesis, male killing, feminization, and cytoplasmic incompatibility (Cl) [1, 2]. Superimposing the biological properties of this lineage onto a phylogeny of Wolbachia and closely related genera indicates that this reproductive manipulation is an evolutionary novelty.
Initially identified as a rickettsial organism, Wolbachia pipientis and related bacterial strains have been extensively documented in arthropods and nematodes, and are now treated as a separate genus within the Rickettsiae [1]. Wolbachia is clearly part of a monophyletic lineage containing the genera Rickettsia and Ehrlichia, and all members of this clade share the defining characteristic of being obligate endosymbionts. Rickettsia and Ehrlichia species are commonly found in arthropods, which serve as reservoirs of infection for a number of diseases of vertebrates, e.g., typhus, caused by R. prowazekii, and various ehrlichioses [3–5]. By contrast, Wolbachia strains are restricted in nature to ecdyzoan hosts, and have never been linked to vertebrate disease. Mapping the traits which are peculiar to Wolbachia onto a phylogeny of representative taxa from the three bacterial genera indicates that these are derived characteristics. Therefore, the manipulation of host reproduction is an evolutionary novelty acquired by the Wolbachia lineage, which meanwhile has lost the ability, manifest in Ehrlichia and Rickettsia species, to infect vertebrate, and in particular, mammalian, hosts.
Results and Discussion
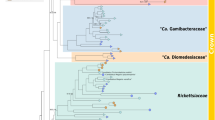
Previous phylogenetic analyses of Wolbachia sequence data focused upon relationships among Wolbachia strains, but relationships with other members of the Rickettsaceae have not been examined in any great detail [6, 7]. Other analyses have focused upon relationships within genus Rickettsia[8] or genus Ehrlichia[9]. In our analysis, we used 16S rDNA sequences from Rickettsia, Wolbachia, and Ehrlichia to specifically and extensively test whether Wolbachia is a sister taxon to genus Ehrlichia and/or Rickettsia. In addition, amino acid sequences from a subset or Rickettsia, Wolbachia, and Ehrlichia species were also analyzed. Our results confirm and augment previously published trees, which indicate that Wolbachia and the genus Ehrlichia together comprise a sister taxon to the genus Rickettsia. This clustering is extremely robust; analyses using different outgroups, and sequences from a variety of Ehrlichia, Rickettsia, and Wolbachia taxa converge upon the same topology, with good resolution of major branches (Figure 1). Different tree building algorithms (maximum likelihood, parsimony) likewise yield similar topologies. A phylogeny derived from amino acid sequences of GroEL proteins is consistent with the phylogeny inferred from rDNA sequences, although the high level of divergence manifest among these genes precludes unambiguous alignment of rickettsial GroEL sequences to homologous sequences from taxa outside the Rickettsiae. Instead, we aligned representative GroEL amino acid sequences from the three rickettsial genera, and constructed an unrooted tree (Figure 2). This tree is congruent with the tree derived from 16S rDNA sequences. Our analyses confirm monophyly for Wolbachia taxa, including representatives of Wolbachia strains infecting filarial nematodes. When Ehrlichia sennetsu is included in the analysis, this genus is shown to be paraphyletic, with Ehrlichia sennetsu basal to a node linking Wolbachia taxa and all other Ehrlichia taxa included in these analyses (Figure 3). These results indicate that Wolbachia and Ehrlichia lineages shared a common ancestor, and that this ancestral lineage diverged from the genus Rickettsia before the Wolbachia-Ehrlichia split. This conclusion has been implicit in previous studies [10–12]; here we make these relationships explicit, and examine the consequences for our understanding of Wolbachia evolution.

Phylogeny derived from 1.3 kb of 16S rDNA sequence. Parsimony tree (PAUP vers. 4.0b4a). All branches had 100% bootstrap support, except where indicated by numbers on branches. Bold line indicates the lineage which has specialized in manipulating host reproduction, and which has lost the ability to infect vertebrate hosts. Wolbachia host genera are shown in parentheses. Wolbachia sequences used in this analysis were from infections in coleopteran (Diabrotica), dipteran (Drosophila), hymenopteran (Muscidifurax) and filarial worm (Brugia) hosts. Crossed line indicates ancestral loss of the ability to infect vertebrates, and acquisition of reproductive specialization.

Shortest maximum parsimony trees derived from GroEL amino acid sequence (left) and 16S rDNA sequences (right), showing congruence of 16S rDNA and GroEL trees. Rickettsia and Ehrlichia sequences used to construct both trees are from the same species, Wolbachia sequences used in the GroEL analysis are from different taxa than those used in the 16S analysis. Host genera of Wolbachia strains are shown in brackets.

Maximum likelihood tree derived from 16S rDNA sequences. Taxa included are the same as those in Figure 1, with the addition of E. sennetsu.
These results have important ramifications for future work on members of this clade. As has been previously noted, phylogenetic analyses of diverse Wolbachia strains imply horizontal transfer of Wolbachia between hosts [1, 7, 10] and has been demonstrated under laboratory conditions [13, 14]. Because the infection frequently results in similar manifestations in taxonomically divergent hosts, this implies bacterial interaction with conserved aspects of host cell and reproductive biology [15]. The obligate endocellular existence is common to all three genera, and presumably represents the ancestral condition of all of these genera. However, the most parsimonious explanation for the evolution and maintenance of traits that alter reproduction, a defining characteristic of the Wolbachia lineage, is to assume that this ability appeared as an evolutionary novelty in the direct ancestor of extant Wolbachia strains. The alternative hypothesis requires numerous independent acquisitions by an endosymbiont of the capacity to induce Cl, alter sex ratios, suppress production of males, and/or feminize host embryos. Hence, the most likely explanation is that a Wolbachia ancestor acquired the ability to manipulate arthropod reproduction in a manner that facilitated bacterial transmission into the next generation, and this property persists in surviving lineages.
There are two reported instances of Rickettsia species which affect host reproduction, one which causes female-biased sex ratio in mites, and a second which causes male killing in ladybird beetles [16]. These two species are fairly distantly related within the genus Rickettsia[8], and intervening members of this genus have not to date been shown to influence or affect reproduction. Therefore, given current information, and in contrast with the situation in the Wolbachia clade, we conclude that these instances are most likely independent acquisitions of the capacity to influence reproduction in their respective hosts. Should further investigation of Rickettsia host reproduction provide evidence of extensive manipulation of host reproduction, we would have to revisit our conclusions. However, since Wolbachia would seem to be selected for efficient vertical transmission, while Rickettsia and Ehrlichia need to maximize the efficiency of horizontal transfer, this does not seem especially likely. This raises questions of a possible trade-offs in life history strategies – did Wolbachia specialization on reproductive manipulation in arthropod hosts cause a loss in the ability to infect vertebrate hosts? Or did loss of the ability to infect vertebrate hosts foster specialization in vertical transmission, and hence, reproductive manipulation? Answers to these questions may come from genetic and molecular studies of the underlying mechanisms involved in Wolbachia-mediated alterations in host reproductive biology.
Conclusions
We have used phylogenetic analysis of related rickettsial genera to track the evolution of life-history traits associated with these bacteria. These analyses expand upon and confirm previous phylogenies of this bacterial clade, in that the Wolbachia lineage is shown to be a sister taxon to genus Ehrlichia, and these genera together form the sister-group to genus Rickettsia. Furthermore, analysis of the phyletic distribution of life-history traits indicates that the propensity of Wolbachia to influence the reproductive biology of infected hosts is a derived state, an investigation of evolutionary space which apparently optimizes vertical transmission and fosters the spread of Wolbachia infection, absent the need for a second host, in contrast with other rickettsial genera.
Materials and Methods
Sequences used in these analyses were obtained from the NCBI database. Rickettsia 16S sequences from: pea aphid rickettsia (PAR), accession no. U42084; R. typhi, U12463; R. rickettsii, U11021; R. bellii, U11014. Ehrlichia 16S sequences from: E. canis. AF156785; E. chaffeensis, AF147752; E. muris, AB013008; E. risticii, AF179351; E. sennetsu, M73225. Wolbachia 16S sequences from Wolbachia endosymbiont in: Brugia pahangi, AJ012646; Diabrotica vergifera, U83098; Drosophila mauritiana, U 17060; Gryllus integer, U83096; Muscidifurax uniraptor, L02882; Sphaeroma hookeri, AJ001610. GroEL amino acid sequences were obtained for: R. rickettsii, U96733; E. canis, U96731; E. sennetsu, U88092; and Wolbachia endosymbionts in Ephestia kuehniella, AB002291; Ephestia cautella, AB002290; Drosophila simulans, AB002287.
Sequences were aligned with ClustalW [17], GroEL data incorporates 148 aligned amino acid residues, 16S data incorporates 1320 bp of aligned DNA sequence. Maximum parsimony phylogenetic analysis shown in Figures 1 and 2 was carried out using the Exhaustive Search option of PAUP vers. 4.0b4a [18], ignoring gaps. The maximum likelihood analysis was accomplished using the Branch and Bound search option of PAUP [18], ignoring gaps. Analysis assumes all sites evolve at an equal rate, a transition :transversion ratio of 2:1, molecular clock not enforced.
References
Werren JH: Biology of Wolbachia. Ann Rev Entomol. 1997, 42: 587-609. 10.1146/annurev.ento.42.1.587.
Stouthamer R, Breeuwer JAJ, Hurst GDD: Wolbachia pipientis: Microbial Manipulator of Reproduction. Ann Rev Microbiol. 1999, 53: 71-102. 10.1146/annurev.micro.53.1.71.
Hackstadt T: The Biology of Rickettsiae. Infectious Agents and Disease. 1996, 5: 127-143.
Hackstadt T: The diverse habitats of obligate intracellular parasites. Current Opinion in Microbiol. 1996, 1: 82-87. 10.1016/S1369-5274(98)80146-X.
Azad AF, Beard CF: Rickettsial pathogens and their arthropod vectors. Emerging Infectious Diseases. 1998, 4(2): 179-186.
Werren JH, Zhang W, Guo L: Evolution and phylogeny of Wolbachia: reproductive parasites of arthropods. Proc R Soc London Ser B. 1995, 251: 55-71.
Schulenburg JHG, Hurst GDD, Huigens TME, van Meer MMM: Molecular evolution and phylogenetic utility of Wolbachia ftsZ and wsp gene sequences with special reference to the origin of male-killing. Mol Biol Evol. 2000, 17: 584-600.
Chen DQ, Campbell BC, Purcell AH: A new Rickettsia from a herbivorous insect, the pea aphid Acyrthosiphon pisum (Harris). Current Microbiology. 1996, 33: 123-128. 10.1007/s002849900086.
Yu XJ, Zhang XF, McBride JW, Zhang Y, Walker DH: Phylogenetic relationships of Anaplasma marginale and Ehrlichia platys to other Ehrlichia species determined by GroEL amino acid sequences. IntI J Syst Evol Microbiol. 2001, 51: 1143-1146.
O'Neill S, Giordano R, Karr TL, Robertson HM: 16S rDNA analysis of the symbionts associated with cytoplasmic incompatibility in insects. Proc NatI Acad Sci (USA). 1992, 89: 2699-2702.
Breeuwer JAJ, Stouthamer R, Barns SM, Pelletier DA, Weisburg WG, Werren JH: Phylogeny of cytoplasmic incompatiblity microorganisms in the parasitoid wasp genus Nasonia (Hymenoptera, Pteromalidae) based on 16S ribosomal DNA sequences. Insect Mol Biol. 1992, 1: 25-36.
Stouthamer R, Breeuwer JA, Luck RF, Werren JH: Molecular identification of microorganisms associated with parthenogenesis. Nature. 1993, 361: 66-68. 10.1038/361066a0.
Boyle L, O'Neill SL, Robertson HM, Karr TL: Interspecific and intraspecific horizontal transfer of Wolbachia in Drosophila. Science. 1993, 260: 1796-1799.
Schilthuizen M, Stouthamer R: Horizontal transmission of parthenogenesis-inducing microbes in Trichogramma wasps. Proc Roy Soc Lond (B). 1997, 264: 361-366. 10.1098/rspb.1997.0052.
Karr TL: Giant steps sideways. Curr Biol. 1994, 4: 537-540.
Werren JH, Hurst GDD, Zhang W, Breeuwer JAJ, Stouthamer R, Majerus ME: Rickettsial relative associated with male-killing in the ladybird beetle (Adalia bipunctata.). J Bacteriol. 1994, 176: 388-394.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994, 22: 4673-4680.
Swofford DL: PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates,. 2000
Acknowledgements
Authors wish to thank M. dark, Y. Gottleib, and anonymous reviewer for suggesting improvements to the manuscript.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Anderson, C.L., Karr, T.L. Wolbachia: Evolutionary novelty in a rickettsial bacteria. BMC Evol Biol 1, 10 (2001). https://doi.org/10.1186/1471-2148-1-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-1-10