
Abstract
Background
The Wnt signal transduction pathway is important in a wide variety of developmental processes as well as in the genesis of human cancer. Vertebrate Wnt pathways can be functionally separated into two classes, the canonical Wnt/beta-catenin pathway and the non-canonical Wnt/Ca2+ pathway. Supporting differences in Wnt signaling, gain of function of Wnt-1 in C57mg mouse mammary epithelial cells leads to their morphological transformation while loss of function of Wnt-5a leads to the same transformation. Many downstream target genes of the Wnt/beta-catenin pathway have been identified. In contrast, little is known about the Wnt/Ca2+ pathway and whether it regulates gene expression.
Results
To test the hypothesis that a specific cell line can respond to distinct Wnts with different patterns of gene expression, we over-expressed Wnt-5a and Rfz-2 in C57mg mammary epithelial cells and compared this cell line to C57mg cells over-expressing Wnt-1. These Wnts were chosen since previous studies suggest that C57mg cells respond differently to these Wnts, and since these Wnts can activate different signaling pathways in other systems. Using DNA microarray analysis, we identified several genes that are regulated by Wnt-5a and Rfz-2 as well as by Wnt-1. We then focused on two genes previously linked to various cancers, mesothelin and stromelysin-1, which are respectively up-regulated by Wnt-1 and Wnt-5a in C57mg cells.
Conclusion
Different Wnts have distinct effects on gene expression in a single cell line.
Similar content being viewed by others


Background
Wnt ligands are secreted glycoproteins that function in intracellular signaling pathways to regulate a variety of developmental processes including cell growth, cell differentiation, cell polarity, and apoptosis. Mis-regulation of Wnt signaling leads to the development of several human cancers, including colon carcinomas and melanoma [1]. Canonical Wnt signaling initiates in the binding of a Wnt ligand to its cell surface receptor, members of the frizzled gene family, along with the co-receptor LRP5/6 [2, 3], leading to changes in the activities of cytoplasmic effectors resulting in the stabilization of beta-catenin protein. Beta-catenin then accumulates in the nucleus where it interacts with its transcriptional co-activators, members of the Lymphoid Enhancer Factor/T Cell Factor (LEF/TCF) family of high mobility group DNA binding proteins to regulate gene expression [1]. Many downstream gene targets of the canonical Wnt/beta-catenin pathway have been identified http://www.stke.org.
There is a large family of Wnt ligands and not all Wnts are thought to function in the same pathway. Wnts can be operationally separated into two classes; the first being a class that transforms C57mg mouse mammary epithelial cells and also promotes duplication of the dorso-ventral axis when over-expressed in Xenopus embryos. Wnts in this functional class activate the canonical Wnt/beta-catenin pathway (Wnt-1, -3A, and -8). The second functional group of Wnts does not transform C57mg cells nor promote axis duplication. Instead, when over-expressed in frog or fish embryos, these Wnts perturb the movements of gastrulation. Wnts in this class have been shown to signal in a non-canonical Wnt signaling pathway, the Wnt/Ca2+ pathway (Wnt-5a, -4, -11). The Wnt/Ca2+ pathway has been shown to increase intracellular Ca2+ levels in zebrafish embryos [4] and to activate PKC and CamKII in Xenopus embryos [5, 6] as well as in human melanomas [7]. Moreover, it may activate Cdc42 [8] and JNK [9]. It is important to emphasize that whether Wnts activate distinct responses and likely pathways is context-dependent, and thus in other contexts a given Wnt may activate another pathway.
Vertebrate Wnts signal through frizzled receptors that in some experimental contexts (e.g., without co-expressing LRP5/6) preferentially activate the Wnt/beta-catenin or Wnt/Ca2+ pathways [5]. Interestingly, Wnt-5a may couple to either pathway depending on which frizzled receptor is present. When co-expressed with human frizzled-5, Wnt-5a can induce a secondary axis in Xenopus [10]. In other assays, however, Wnt-5a along with rat frizzled 2 (Rfz-2) increases intracellular Ca2+ levels and activates PKC and CamKII [4–6]. The activation of Wnt/Ca2+ signaling by Wnt-5a and Rfz-2 seems to be antagonistic to the canonical Wnt signaling pathway. Specifically, Wnt-5a expression inhibits ectopic Wnt-8 induction of secondary axis formation in Xenopus embryos [11]. Supportingly, the endogenous expression of Wnt-5a maintains C57mg cells in a normal growth state since anti-sense Wnt-5a mimics Wnt-1 transformation of C57mg cells [12]. This data suggests that Wnt-1 and Wnt-5a work in an opposing manner in some cellular contexts. This potential role of Wnt-5a as a tumor suppressor is further supported by the evidence that ectopic Wnt-5a in human uroepithelial cells prevents tumorigenesis when injected into athymic nude mice [13].
Much is still unknown about how Wnt-5a signals and, importantly, whether it regulates any genes. The ability of dnWnt-11 to block expression of some genes in Xenopus [6] and the ability of Wnt-5a to activate the Ca2+ sensitive transcription factor nuclear factor of activated T cells (NFAT) [14], support the likelihood that Wnt-5a can regulate gene expression through a beta-catenin-independent mechanism. We sought to identify genes regulated by Wnt-5a and compare whether these were different from canonical Wnt-1 gene targets using DNA microarray analysis.
Results
To identify downstream genes regulated by Wnt-5a we created C57mg cells that stably express both Wnt-5a and Rfz-2, a potential ligand and receptor pair that in some assays function in the same signaling pathway. We overexpressed Rfz-2 in addition to Wnt-5a in C57mg cells in order to augment the activation of noncanonical Wnt signaling. Both have been shown to elevate intracellular Ca2+ levels and to activate PKC and CamKII [4–6]. In addition Wnt-5a and Rfz-2 have been shown to synergize in their induction of CamKII [6]. Wnt-1/C57mg cells were also used for comparison, in an effort to identify genes preferentially regulated by Wnt-5a. C57mg cells appeared to respond to ectopic expression of both Wnt-5a and Rfz-2 in C57mg cells since the cells exhibited a moderate reduction in growth rate in comparison to wildtype (wt) C57mg cells as well as Wnt-1/C57mg cells (data not shown). Phenotypically the Wnt-5a/Rfz-2/C57mg cells were more spread out and the overall size of the cells increased compared to wt C57mg cells or Wnt-1/C57mg cells (data not shown). In contrast, ectopic expression of Wnt-1 induced the expected morphological transformation of C57mg cells (data not shown). Previous reports of ectopic expression of different Wnts in C57mg cells demonstrated Wnt-1 to be highly transforming whereas Wnt-5a failed to induce transformation [15, 16]. By identifying downstream targets of Wnt-5a and Rfz-2, we wanted to determine which genes are responsible for these phenotypic changes as well as which genes might be targets of noncanonical Wnt signaling.
RNA isolated from wt C57mg cells, C57mg cells stably transformed by Wnt-5a and Rfz-2 retroviruses, and C57mg cells stably transformed by Wnt-1 retrovirus were used for DNA microarray analysis. Biotinylated cRNA probes synthesized from the RNA were hybridized to Affymetrix mouse GeneChips Mu11k subA and subB arrays. Over 11,000 oligonucleotide probe sets were analysed including known genes and ESTs. Two independent sets of experiments were performed that initiated from the growth of cells. Probe sets that underwent a 2-fold or greater change in the Wnt-ligand expressing cells compared to control cells were selected for further analysis. Over 90 genes that repeated in both experiments appeared to change in the Wnt-expressing cells. Of those, 50 genes were chosen for validation by an independent method (quantitative RT-PCR). A list of some of these genes, with the observed fold-changes determined by DNA microarray analysis and quantitative RT-PCR, is shown (Table 1; list of primers shown in Table 2). Genes are separated by functional categories with the greatest number of genes having roles in cell adhesion, transcriptional regulation, and signaling. The list of genes shows those that are specific to Wnt-5a/Rfz-2, differential between Wnt-5a/Rfz-2 and Wnt-1, specific to Wnt-1, and common to both Wnt-1 and Wnt-5a/Rfz-2. Previously identified genes regulated by Wnt signaling in this list include fibronectin [17], IL-6 [18], BMP-4 [19], groucho related protein [20], and Krox-20 [21, 22]. Since Wnt-5a may couple to more than one frizzled receptor, different Wnt signaling pathways may be activated by Wnt-5a depending on the endogenous expression of various frizzled receptors [6, 10]. Thus, it is possible that the gene expression pattern we identified may be due to the activation of both canonical and noncanonical Wnt signaling pathways. We compared the Wnt-5a/Rfz-2 expression profile to the Wnt-1 expression profile to rule out genes common in both pathways and to identify those genes specific to Wnt-5a and Rfz-2.
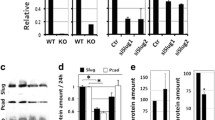
We next wanted to examine whether any of these genes were regulated in a similar manner if we used alternative methods for expressing Wnt-1 or Wnt-5a. In particular, we wanted to activate the function of these Wnts for short durations rather than relying on stable expression. The first approach was to co-culture C57mg cells with cells secreting Wnt ligand. For the co-culture experiments, C57mg cells were grown together with Wnt-1 or Wnt-5a expressing 293T cells for 24 hours. RNA was isolated and quantitative RT-PCR was performed using primers to the list of genes from Table 1. The majority of genes had little if any background in the PCR reactions from the human 293T cell RNA (data not shown). One gene, mesothelin, exhibited a 3-fold induction in C57mg cells treated with the Wnt-1 but not the Wnt-5a secreting 293T cells (Figure 1, panel A). This fold change was similar to the data obtained from stable expression with Wnt-1 in C57mg cells (see Table 1). Mesothelin is a cell-surface antigen of unknown function that is expressed in the mesothelium. It is synthesized as a 69 kDa precursor that is proteolytically processed into a 40 kDa membrane bound form that is glycosylphosphatidylinositol (GPI)-linked and a 32 kDa secreted form. The secreted form has been referred to as megakaryocyte potentiating factor (MPF) for its ability to stimulate the megakaryocyte colony forming activity of murine IL-3 in mouse bone marrow cell culture [23, 24]. Mesothelin expression is induced in a variety of cancers including ovarian, pancreatic, lung, and cervical cancer [25, 26]. The soluble portion of mesothelin has drawn attention as a potential serum tumor marker since it is highly over-expressed in patients with ovarian carcinomas and pancreatic adenocarcinomas [27, 28].

Induction of mesothelin and SL-1 expression by Wnt-1 or Wnt-5a signaling. A. RT-PCR was performed on RNA obtained from 3 separate experiments in which C57mg cells were co-cultured with 293T cells expressing Wnt-1, Wnt-5a, or CS2+ as control. The amounts of mesothelin and SL-1 mRNAs were normalized to HPRT and plotted as fold increase relative to C57mg cells co-culture with 293T cells expressing CS2+. B. C57mg cells (wt) or C57mg cells stably expressing the Rfz-2/β2AR chimeric receptor were treated with the β2AR agonist isoproterenol or the antagonist propranolol as control for 8 hours prior to harvesting for RNA extraction and RT-PCR analysis. The amounts of mesothelin and SL-1 mRNAs were normalized to HPRT and plotted as fold increase relative to propranolol treated cells. Mean levels were determined from 3 separate experiments.
Stromelysin-1 (SL-1) was induced almost 2-fold in C57mg cells co-cultured with Wnt-5a secreting 293T cells but not with Wnt-1 secreting 293T cells (Figure 1, panel A). A similar level of induction was also observed with Wnt-5a/293T cells co-cultured with Rfz-2/C57mg cells (data not shown), suggesting that the induction of SL-1 is due to expression of Wnt-5a and not only to Rfz-2. These data are similar to the ~4 fold induction of SL-1 in Wnt-5a/Rfz-2/C57mg cells (Table 1). SL-1 is a matrix metalloproteinase that can alter ECM-mediated signaling events during development [29–31]. Both SL-1 and Wnt-5a have roles in mammary gland development and are elevated in certain breast cancers [32–35], although Wnt-5a is reduced in other types of breast cancer [36].
Since Wnt-5a may activate both vertebrate Wnt pathways we turned to an approach that more specifically would transiently activate the Wnt/Ca2+ signaling pathway. This approach involves the use of a drug-inducible chimeric receptor consisting of the extracellular and transmembrane spanning portions derived from the hamster β2 adrenergic receptor (β2AR) and the intracellular domains derived from Rfz-2. This receptor has previously been shown to elevate intracellular Ca2+ and activate CamKII in a ligand-dependent manner [6, 37, 38]. C57mg cells stably transformed with Rfz-2/β2AR retrovirus or wt C57mg cells were treated overnight with propranolol, a β2AR antagonist, to maintain the chimeric receptors in an inactivated state. Subsequently, the β2AR agonist isoproterenol was added for 8 hours at which point RNA was isolated and quantitative RT-PCR was performed. Supporting the co-culture data discussed above, SL-1 exhibited almost a 3-fold induction with isoproterenol treated Rfz-2/β2AR expressing C57mg cells but not wt C57mg cells treated with isoproterenol (Figure 1, panel B).
SL-1 appears to be up-regulated by Wnt-5a and not Wnt-1 (Table 1 and Figure 1, panel A). Conversely, mesothelin appears to be up-regulated specifically by Wnt-1 and not Wnt-5a since mesothelin was not induced by co-culturing with Wnt-5a secreting cells or by activation of the chimeric Rfz-2/β2AR receptor (Figure 1, panels A and B). To our knowledge this is the first report to show that two genes are differentially regulated by two different Wnts within the same cell type.
In order to examine the mechanism by which Wnt-1 or Wnt-5a regulates mesothelin or SL-1, respectively, C57mg cells were treated with either LiCl or phorbol myristic acetate (PMA). LiCl has previously been shown to mimic activation of Wnt-1 signaling by stabilizing beta-catenin through its inhibition of glycogen synthase kinase-3β (GSK-3β), a kinase that phosphorylates beta-catenin and promotes its degradation via the ubiquitin proteasome pathway [39]. C57mg cells treated with LiCl exhibited elevated levels of mesothelin but not SL-1 (Figure 2, left panel). On the other hand, PMA treatment of C57mg cells elevated SL-1 transcript levels 6-fold but not mesothelin (Figure 2, right panel). PMA, a phorbol ester, activates PKC and has previously been shown to activate SL-1 [40]. It appears that mesothelin is activated by Wnt-1, which can be mimicked by LiCl, and that SL-1 is activated by Wnt-5a in a beta-catenin-independent manner, which can be mimicked by activating PKC.

Effect of Li+ or PMA on mesothelin and SL-1 expression in C57mg cells. For Li+ treatment, C57mg cells were treated with 10 mM LiCl or 10 mM KCl as control for 6 hours. For PMA treatment, C57mg cells were treated with 1 μM PMA or ethanol as control for 6 hours. RNA was extracted and RT-PCR analysis was performed using primers specific to mesothelin or SL-1 mRNAs. mRNA levels were normalized to HPRT and fold increase shown is relative to KCl treatment in the case of Li+ and to ethanol treatment in the case of PMA. Mean levels were determined from 3 independent experiments.
Discussion
We used DNA microarray analysis to identify genes regulated by Wnt-1 and Wnt-5a in C57mg cells. C57mg cells were chosen since they become transformed by ectopic expression of Wnt-1 and by loss of function of Wnt-5a [12, 15, 16]. In addition, Wnt-5a/Rfz-2 C57mg cells exhibited phenotypic differences from wt C57mg cells (see results). We focused on two genes that showed differential regulation by Wnt-1 and Wnt-5a since their regulation was reproducible using several methods to activate the Wnt signaling pathways. The first, mesothelin, was up-regulated by Wnt-1 both by stable expression of Wnt-1 in C57mg cells and by co-culturing C57mg cells with Wnt-1 secreting cells. Similarly, mesothelin expression was induced by Li+, an inhibitor of GSK-3β, that mimics Wnt-1. Mesothelin is markedly over-expressed in a variety of cancers derived from tissues of the mesothelium, including lung, cervix, and pancreas. Interestingly, mesothelin expression is induced in lung, ovarian, and pancreatic carcinomas where constitutive activation of Wnt signaling is caused by stabilizing mutations in beta-catenin [27, 28, 41–44]. The induction of mesothelin in these carcinomas may be due to misregulation of Wnt signaling as are other targets of the Wnt pathway, c-myc and cyclin D1. Mutations in different effectors of Wnt signaling have been identified in a variety of cancers. Interestingly, Wnt-5a down-regulated mesothelin expression, perhaps through antagonism of endogenous Wnt/beta-catenin signaling.
Second, SL-1 was up-regulated by Wnt-5a in C57mg cells. Three lines of evidence demonstrate that SL-1 induction is associated with activation of Wnt-5a signaling. First, C57mg cells stably expressing Wnt-5a and Rfz-2 elevate SL-1 levels. Second C57mg cells co-cultured with Wnt-5a secreting cells up-regulate SL-1. Third, activation of chimeric Rfz-2 receptors stably expressed in C57mg cells augment SL-1 expression. To further examine how SL-1 is up-regulated by Wnt-5a, we tested whether SL-1 induction could be mediated by downstream effectors of Wnt/Ca2+ signaling such as PKC. Activation of PKC by PMA treatment of C57mg cells did indeed elevate SL-1 levels significantly. SL-1 is an important modulator of cell adhesion through its function as a matrix metalloproteinase that degrades the ECM. SL-1 induction in p2S cells, an untransformed mouse mammary epithelial cell line, triggered the disappearance of E-cadherin and beta-catenin from cell-cell junctions and reduced the expression of beta-catenin [35]. Wnt-5a and SL-1 may play similar roles in inhibiting cadherin-mediated cell-adhesion, as Wnt-5a decreases Ca2+-dependent cell adhesion similar to a dominant negative cadherin [11]. The regulation of SL-1 by Wnt-5a may be important in developmental processes such as gastrulation where Wnt-5a affects the morphogenetic movement of tissues [45].
Whether mesothelin and SL-1 are direct targets of Wnt-1 and Wnt-5a, respectively, and whether alterations in their expression are responsible for the observed phenotypic changes in Wnt-5a/Rfz-2/C57mg cells, has yet to be determined. Whether they are direct or indirect does not alter the observation that the expression of both change in an unexpected manner in response to these two Wnts. Much less is understood about Wnt-5a signaling pathway(s) and how Wnt-5a signals are transmitted to the nucleus to affect gene expression compared to the canonical Wnt/beta-catenin signaling pathway. A recent report demonstrated that Wnt-5a and Rfz-2 can induce the nuclear localization of the transcription factor NF-AT [14]. Their results show that NF-AT is a downstream target of the Wnt/Ca2+ pathway and antagonizes the Wnt/beta-catenin pathway in Xenopus embryos. The SL-1 promoter has putative NF-AT binding sites but whether these are important for Wnt-5a regulation requires further testing. Nevertheless, our data clearly demonstrate that different Wnts have distinct effects on gene expression in a single cell line.
Conclusions
Using DNA microarray analysis we identified several genes that are differentially regulated by Wnt-1 and Wnt-5a signaling within the same cell type, C57mg. We focused on two genes, mesothelin and SL-1, whose regulation by Wnts was reproducible using a variety of methods to activate the Wnt signaling pathways. Mesothelin, a potential tumor serum marker, was up-regulated by Wnt-1 and down-regulated by Wnt-5a. SL-1, a matrix metalloproteinase, was up-regulated by Wnt-5a only, showing that Wnt-5a can indeed affect gene expression. Our data indicate that different Wnts can stimulate distinct sets of genes.
Methods
Cell Culture, retroviral infections, and DNA transfections
C57mg cells were grown in DMEM supplemented with 10% FBS and 10 μg/ml of insulin. 293T cells were grown in DMEM supplemented with 10% FBS. The Wnt-5a/ Rfz-2 expressing C57mg cell line and the Rfz-2/β2AR expressing C57mg cell line were established by retroviral infections. The coding region for Xenopus Wnt-5a including the myc epitope tag in pSP64T [45] was digested with BglII, treated with klenow enzyme and ligated into the retroviral vector PMI-hCD5 [46] digested with NotI and treated with Klenow enzyme. The Rfz-2 coding sequence from plasmid CS2+ [47] was amplified by PCR using the following primers: 5'-AAAAGCGGCCGCGAGTGGGGGGCGGCGGCC-3' and 5'-AAAAGTCGACGCGCCCAGCAGCGAGACCGC-3'. The PCR product was cloned into the NotI and SalI sites in PMI-hCD2 [46]. The coding sequence for the Rfz-2/β2AR chimeric receptor in CS2+ [6, 37] was digested with EcoRI and XhoI and cloned into PMI-hCD2 at the EcoRI and SalI sites. Generation of the retrovirus, infection of the C57mg cell line, and enrichment of infected cells was performed as described [46, 48]. Cell populations enriched to >95% were used for RNA isolation. Total RNA was extracted using the RNeasy Mini Kit (Qiagen) as directed by the manufacturer. Wnt-1 expressing C57mg cells were a kind gift from G. Shackleford. For co-culture experiments, 293T cells were transfected with either Xenopus Wnt-5a [45], mouse Wnt-1 [49], or CS2+ as control using the lipofectamine PLUS reagents (GIBCO BRL) as directed by the manufacturer. 24 hours after transfection the Wnt-expressing 293T cells were transferred to culture dishes seeded with C57mg cells. Both cell types were plated at a 1:1 ratio. Cells were co-cultured for 24 hours and RNA was isolated. For induction of the Rfz-2/β2AR chimeric receptor, chimeric receptor expressing C57mg cells or wt C57mg cells were treated with 10 μM propranolol overnight and then with 10 μM isoproterenol for 8 hours. Control cells were treated with 10 μM propranolol only. For Li+ and PMA stimulation experiments, C57mg cells were incubated with 10 mM LiCl, 10 mM KCl as a control, or 1 μM PMA for 6 hours. All treatments were followed by RNA extraction.
Oligonucleotide Array
Total RNA was extracted using the RNeasy kit (Qiagen) according to the manufacturer's protocols. Transcript profiling with Affymetrix GeneChips (Affymetrix, Santa Clara, CA) was performed using the Mu11KsubA and subB chips containing ~11,000 probe sets. Briefly, 15 μg of total RNA were reverse-transcribed with an oligo(dT) primer coupled to a T7 RNA polymerase binding site. Biotinylated complementary RNA (cRNA) was then synthesized from the resulting complementary DNA (cDNA) with the use of T7 polymerase. 30 μg of biotinylated cRNA were then randomly sheared (to an approximate length of 50 nucleotides). Hybridization to the arrays and first-pass analysis of the scanned data were performed at the PAN Facility (Stanford University, Palo Alto, California).
Reverse Transcriptase-PCR (RT-PCR) Analysis
Confirmation of gene expression was performed using quantitative RT-PCR. cDNA was created from total RNA using the ThermoScript RT-PCR System (Invitrogen) with an oligo(dT)20 primer. Minus RT controls were also prepared similarly. 2 μg RNA were included in each reaction in a total volume of 20 μl. The reaction was performed at 55°C for 1 hour and terminated by incubating at 85°C for 5 minutes. The reaction was diluted 7.5-fold in water and 2.5 μl were added to the PCR reaction to yield a total volume of 25 μl. Since nonspecific amplification was obtained in testing some of the primers sets with the -RT controls, optimization of the primer concentration was performed as per the SYBR green PCR protocol (Applied Biosystems). Three concentrations of each of the forward and reverse primers were used, 50 nM, 300 nM, and 900 nM, to obtain nine different combinations. The amplification reaction was performed using the following thermocycler conditions: 94°C for 5 min followed by 40 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C 30 s. +RT, -RT, and no template controls were tested for the primer concentration optimization and samples were taken out at 20, 25, 30 and 35 cycles to run on 2% agarose gels. The combination of forward and reverse primer concentrations was selected based on the presence of a single band only in the +RT sample. Using the optimized primer concentrations, the PCR reaction was performed with the SYBR Green PCR Master Mix (Applied Biosystems) and the ABI Prism 7700 Sequence Detector (Applied Biosystems). The amplification reaction consisted of the following: 50°C for 2 min, denaturation at 95°C for 10 minutes, and 40 thermal cycles of 95°C for 15 s and 60°C for 1 min. PCR quantification was performed in triplicate. Fold induction was obtained by using the ΔΔCt method in which all samples are first normalized to the level of mouse hypoxanthine phosphoribosyltransferase (HPRT) in each sample. Relative normalized units were then compared between the experimental sample and its control. The primer sequences are shown in Table 2. To determine the specificity of the primer sets to the murine mesothelin and SL-1 targets genes, we performed PCR using the primers sets with cDNA from human 293T cells, and found no amplification.
References
Miller JR, Hocking AM, Brown JD, Moon RT: Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene. 1999, 18: 7860-7872. 10.1038/sj.onc.1203245.
Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R: A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996, 382: 225-230. 10.1038/382225a0.
Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S: arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000, 407: 527-530. 10.1038/35035110.
Slusarski DC, Corces VG, Moon RT: Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature. 1997, 390: 410-413. 10.1038/37138.
Sheldahl LC, Park M, Malbon CC, Moon RT: Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr Biol. 1999, 9: 695-698. 10.1016/S0960-9822(99)80310-8.
Kuhl M, Sheldahl LC, Malbon CC, Moon RT: Ca(2+)/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzled homologs and promotes ventral cell fates in Xenopus. J Biol Chem. 2000, 275: 12701-12711. 10.1074/jbc.275.17.12701.
Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM: Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell. 2002, 1: 279-288. 10.1016/S1535-6108(02)00045-4.
Choi SC, Han JK: Xenopus Cdc42 regulates convergent extension movements during gastrulation through Wnt/Ca2+ signaling pathway. Dev Biol. 2002, 244: 342-357. 10.1006/dbio.2002.0602.
Yamanaka H, Moriguchi T, Masuyama N, Kusakabe M, Hanafusa H, Takada R, Takada S, Nishida E: JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep. 2002, 3: 69-75. 10.1093/embo-reports/kvf008.
He X, Saint-Jeannet JP, Wang Y, Nathans J, Dawid I, Varmus H: A member of the Frizzled protein family mediating axis induction by Wnt- 5A. Science. 1997, 275: 1652-1654. 10.1126/science.275.5306.1652.
Torres MA, Yang-Snyder JA, Purcell SM, DeMarais AA, McGrew LL, Moon RT: Activities of the Wnt-1 class of secreted signaling factors are antagonized by the Wnt-5A class and by a dominant negative cadherin in early Xenopus development. J Cell Biol. 1996, 133: 1123-1137. 10.1083/jcb.133.5.1123.
Olson DJ, Gibo DM: Antisense wnt-5a mimics wnt-1-mediated C57MG mammary epithelial cell transformation. Exp Cell Res. 1998, 241: 134-141. 10.1006/excr.1998.4030.
Olson DJ, Gibo DM, Saggers G, Debinski W, Kumar R: Reversion of uroepithelial cell tumorigenesis by the ectopic expression of human wnt-5a. Cell Growth Differ. 1997, 8: 417-423.
Saneyoshi T, Kume S, Amasaki Y, Mikoshiba K: The Wnt/calcium pathway activates NF-AT and promotes ventral cell fate in Xenopus embryos. Nature. 2002, 417: 295-299. 10.1038/417295a.
Wong GT, Gavin BJ, McMahon AP: Differential transformation of mammary epithelial cells by Wnt genes. Mol Cell Biol. 1994, 14: 6278-6286.
Olson DJ, Papkoff J: Regulated expression of Wnt family members during proliferation of C57mg mammary cells. Cell Growth Differ. 1994, 5: 197-206.
Gradl D, Kuhl M, Wedlich D: The Wnt/Wg signal transducer beta-catenin controls fibronectin expression. Mol Cell Biol. 1999, 19: 5576-5587.
Sen M, Lauterbach K, Firestein GS, Corr M, Carson DA: Expression and function of wingless and frizzled homologs in rheumatiod arthritis. Proc Natl Acad Sci USA. 2000, 97: 2791-6. 10.1073/pnas.050574297.
Baker JC, Beddington RS, Harland RM: Wnt signaling in Xenopus embryos inhibits bmp4 expression and activates neural development. Genes Dev. 1999, 13: 3149-59. 10.1101/gad.13.23.3149.
Tice DA, Szeto W, Soloviev I, Rubinfeld B, Fong SE, Dugger DL, Winer J, Williams PM, Wieand D, Smith V, Schwall RH, Pennica D, Polakis P: Synergistic induction of tumor antigens by Wnt-1 signaling and retinoic acid revealed by gene expression profiling. J Biol Chem. 2002, 277: 14329-14335. 10.1074/jbc.M200334200.
McGrew LL, Hoppler S, Moon RT: Wnt and FGF pathways cooperatively pattern anteroposterior neural ectoderm in Xenopus. Mech Dev. 1997, 69: 105-114. 10.1016/S0925-4773(97)00160-3.
Saint-Jeannet JP, He X, Varmus HE, Dawid IB: Regulation of dorsal fate in the neuraxis by Wnt-1 and Wnt-3a. Proc Natl Acad Sci U S A. 1997, 94: 13713-13718. 10.1073/pnas.94.25.13713.
Kojima T, Oh-eda M, Hattori K, Taniguchi Y, Tamura M, Ochi N, Yamaguchi N: Molecular cloning and expression of megakaryocyte potentiating factor cDNA. J Biol Chem. 1995, 270: 21984-21990. 10.1074/jbc.270.37.21984.
Yamaguchi N, Hattori K, Oh-eda M, Kojima T, Imai N, Ochi N: A novel cytokine exhibiting megakaryocyte potentiating activity from a human pancreatic tumor cell line HPC-Y5. J Biol Chem. 1994, 269: 805-3.
Chowdhury PS, Viner JL, Beers R, Pastan I: Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc Natl Acad Sci U S A. 1998, 95: 669-674. 10.1073/pnas.95.2.669.
Hassan R, Viner JL, Wang QC, Margulies I, Kreitman RJ, Pastan I: Anti-tumor activity of K1-LysPE38QQR, an immunotoxin targeting mesothelin, a cell-surface antigen overexpressed in ovarian cancer and malignant mesothelioma. J Immunother. 2000, 23: 473-479. 10.1097/00002371-200007000-00011.
Scholler N, Fu N, Yang Y, Ye Z, Goodman GE, Hellstrom KE, Hellstrom I: Soluble member(s) of the mesothelin/megakaryocyte potentiating factor family are detectable in sera from patients with ovarian carcinoma. Proc Natl Acad Sci U S A. 1999, 96: 11531-11536. 10.1073/pnas.96.20.11531.
Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, Murugesan SR, Leach SD, Jaffee E, Yeo CJ, Cameron JL, Kern SE, Hruban RH: Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. 2001, 7: 3862-3868.
Ganser GL, Stricklin GP, Matrisian LM: EGF and TGF alpha influence in vitro lung development by the induction of matrix-degrading metalloproteinases. Int J Dev Biol. 1991, 35: 453-461.
Nordstrom LA, Lochner J, Yeung W, Ciment G: The metalloproteinase stromelysin-1 (transin) mediates PC12 cell growth cone invasiveness through basal laminae. Mol Cell Neurosci. 1995, 6: 56-68. 10.1006/mcne.1995.1006.
Lelongt B, Trugnan G, Murphy G, Ronco PM: Matrix metalloproteinases MMP2 and MMP9 are produced in early stages of kidney morphogenesis but only MMP9 is required for renal organogenesis in vitro. J Cell Biol. 1997, 136: 1363-1373. 10.1083/jcb.136.6.1363.
McDonnell S, Matrisian LM: Stromelysin in tumor progression and metastasis. Cancer Metastasis Rev. 1990, 9: 305-319. 10.1007/BF00049521.
Lejeune S, Huguet EL, Hamby A, Poulsom R, Harris AL: Wnt5a cloning, expression, and up-regulation in human primary breast cancers. Clin Cancer Res. 1995, 1: 215-222.
Bui TD, Tortora G, Ciardiello F, Harris AL: Expression of Wnt5a is downregulated by extracellular matrix and mutated c-Ha-ras in the human mammary epithelial cell line MCF-10A. Biochem Biophys Res Commun. 1997, 239: 911-917. 10.1006/bbrc.1997.7530.
Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ: Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997, 139: 1861-1872. 10.1083/jcb.139.7.1861.
Jonsson M, Dejmek J, Bendahl PO, Andersson T: Loss of Wnt-5a protein is associated with early relapse in invasive ductal breast carcinomas. Cancer Res. 2002, 62: 409-416.
Liu X, Liu T, Slusarski DC, Yang-Snyder J, Malbon CC, Moon RT, Wang H: Activation of a frizzled-2/beta-adrenergic receptor chimera promotes Wnt signaling and differentiation of mouse F9 teratocarcinoma cells via Galphao and Galphat. Proc Natl Acad Sci U S A. 1999, 96: 14383-14388. 10.1073/pnas.96.25.14383.
Ishitani T, Kishida S, Hyodo-Miura J, Ueno N, Yasuda J, Waterman M, Shibuya H, Moon RT, Ninomiya-Tsuji J, Matsumoto K: The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol Cell Biol. 2003, 23: 131-139. 10.1128/MCB.23.1.131-139.2003.
Klein PS, Melton DA: A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996, 93: 8455-8459. 10.1073/pnas.93.16.8455.
Benbow U, Brinckerhoff CE: The AP-1 site and MMP gene regulation: what is all the fuss about?. Matrix Biol. 1997, 15: 519-526. 10.1016/S0945-053X(97)90026-3.
Miettinen M, Sarlomo-Rikala M: Expression of calretinin, thrombomodulin, keratin 5, and mesothelin in lung carcinomas of different types: an immunohistochemical analysis of 596 tumors in comparison with epithelioid mesotheliomas of the pleura. Am J Surg Pathol. 2003, 27: 150-158. 10.1097/00000478-200302000-00002.
Sunaga N, Kohno T, Kolligs FT, Fearon ER, Saito R, Yokota J: Constitutive activation of the Wnt signaling pathway by CTNNB1 (beta-catenin) mutations in a subset of human lung adenocarcinoma. Genes Chromosomes Cancer. 2001, 30: 316-321. 10.1002/1098-2264(2000)9999:9999<::AID-GCC1097>3.0.CO;2-9.
Zhai Y, Wu R, Schwartz DR, Darrah D, Reed H, Kolligs FT, Nieman MT, Fearon ER, Cho KR: Role of beta-catenin/T-cell factor-regulated genes in ovarian endometrioid adenocarcinomas. Am J Pathol. 2002, 160: 1229-1238.
Tanaka Y, Kato K, Notohara K, Hojo H, Ijiri R, Miyake T, Nagahara N, Sasaki F, Kitagawa N, Nakatani Y, Kobayashi Y: Frequent beta-catenin mutation and cytoplasmic/nuclear accumulation in pancreatic solid-pseudopapillary neoplasm. Cancer Res. 2001, 61: 8401-8404.
Moon RT, Christian JL, Campbell RM, McGrew LL, DeMarais AA, Torres M, Lai CJ, Olson DJ, Kelly GM: Dissecting Wnt signalling pathways and Wnt-sensitive developmental processes through transient misexpression analyses in embryos of Xenopus laevis. Dev Suppl. 1993, 85-94.
Deftos ML, He YW, Ojala EW, Bevan MJ: Correlating notch signaling with thymocyte maturation. Immunity. 1998, 9: 777-786. 10.1016/S1074-7613(00)80643-3.
Yang-Snyder J, Miller JR, Brown JD, Lai CJ, Moon RT: A frizzled homolog functions in a vertebrate Wnt signaling pathway. Curr Biol. 1996, 6: 1302-1306. 10.1016/S0960-9822(02)70716-1.
He YW, Deftos ML, Ojala EW, Bevan MJ: RORgamma t, a novel isoform of an orphan receptor, negatively regulates Fas ligand expression and IL-2 production in T cells. Immunity. 1998, 9: 797-806. 10.1016/S1074-7613(00)80645-7.
Papkoff J, Schryver B: Secreted int-1 protein is associated with the cell surface. Mol Cell Biol. 1990, 10: 2723-2730.
Acknowledgements
MP is an Associate, and RTM an Investigator, of the HHMI.
Author information
Authors and Affiliations
Corresponding author
Additional information
Author's contributions
MP performed the experiments. RTM supervised the work. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Prieve, M.G., Moon, R.T. Stromelysin-1 and mesothelin are differentially regulated by Wnt-5a and Wnt-1 in C57mg mouse mammary epithelial cells. BMC Dev Biol 3, 2 (2003). https://doi.org/10.1186/1471-213X-3-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-213X-3-2