
Abstract
Background
Natural antisense transcripts (NATs) are endogenous RNA molecules that exhibit partial or complete complementarity to other RNAs, and that may contribute to the regulation of molecular functions at various levels. In recent years, large-scale NAT screens in several model organisms have produced much data, but there is no database to assemble all these data. AntiCODE intends to function as an integrated NAT database for this purpose.
Results
This release of antiCODE contains more than 30,000 non-redundant natural sense-antisense transcript pairs from 12 eukaryotic model organisms. In order to provide an integrated NAT research platform, efficient browser, search and Blast functions have been included to enable users to easily access information through parameters such as species, accession number, overlapping patterns, coding potential etc. In addition to the collected information, antiCODE also introduces a simple classification system to facilitate the study of natural antisense transcripts.
Conclusion
Though a few similar databases also dealing with NATs have appeared lately, antiCODE is the most comprehensive among these, comprising almost all currently detected NAT pairs.
Similar content being viewed by others


Background
Natural antisense transcripts (NATs) are endogenous RNA molecules that exhibit partial or complete complementarity to other transcripts, through which they may contribute to the regulation of molecular expression at various levels. Though many natural antisense transcripts were discovered through their regulatory function on the expression of mRNAs [1, 2], some global predictions of NATs in several species have also been published [3–10]. The first of these used mRNA data to predict natural antisense transcripts [4]. With the appearance of more draft genomes and full length cDNA data, the scale of NATs predictions has been extended. Several datasets, mainly based on full length cDNAs, have been published for mouse [8, 11], rice [12] and Arabidopsis thaliana [7]. Since 2006, the trend in NATs prediction has turned to multi-species comparisons [6, 13]. A number of published NATs have been validated by various experimental approaches, such as RT-PCR [10] and microarray [5], further confirming that antisense transcript is a common occurrence in eukaryote transcriptomes.
The background for the emergence of so much NAT data in recent years, is on the one hand the availability of more genomic and full length cDNA data, and on the other hand a growing realization of the important functions of natural antisense transcripts. Antisense RNAs may contribute regulatory activity at various levels, such as post-transcription [14, 15], splicing [16, 17], transport [18], and genomic imprinting[19, 20], and have been shown to be involved in the control of developmental processes [21], adaptation to various stresses [22], and viral infection [23, 24] through annealing to complementary sequences.
To facilitate research, previous publications have suggested a few classification systems for NATs. The most basic of these is the cis/trans system [4] in which an antisense transcript from the same genomic loci as the sense transcript is labelled a cis-NAT, whereas a trans-NAT is an antisense transcript expressed from a genomic locus different from that of the sense transcript. A second classification system is based on the overlapping position of the complementary pair, which will be divided into 5–6 categories according to their patterns of gene structure, e.g. depending on whether the pair overlaps at their 5' ends, 3' ends, completely, or in the introns [6, 7, 10, 11]. A third classification system considers the respective coding potential of the complementary pair, and includes the categories coding-coding, coding-noncoding and noncoding-noncoding [8, 13].
Up to present, a number of large-scale NAT data have been published and several functional studies of NATs have been carried out, however, thus far no database has been set up to collect and order all these transcripts. In order to serve the need of the NAT research, we have over the past two years built the antiCODE database. The purpose of the database is to collect the existing NAT data, and to provide a useful browsing and search platform for these data. This release of antiCODE contains more than 30,000 natural sense-antisense transcript pairs from the 12 model organisms Homo sapiens (human), Mus musculus (mouse), Rattus norvegicus (rat), Xenopus tropicalis (western clawed frog), Drosophila melanogaster (fruit fly), Caenorhabditis elegans (nematode), Ciona intestinalis (seasquirt), Gallus gallus (chicken), Danio rerio (zebrafish), Bos taurus (cow), Oryza sativa (rice) and Arabidopsis thaliana (thale cress).
Construction and content
All NATs in the database have been collected from recent articles [4–8, 10–13]. The original datasets used for construction of the database are listed in Table 1, which include 11,287 human NAT pairs [4, 5, 10], 14,199 mouse NAT pairs [8, 11], 1,339 A. thaliana NAT pairs [7], 687 rice NAT pairs [12] and more than 5,000 NAT pairs from other species [6, 13].
Classification
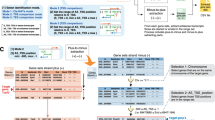
After collecting the NAT pairs, there was a need for uniform criteria to organize the data. Based on the previous classifications, we developed a classification system that includes three complementary aspects for which we use the terms "5/3/c/o", "cis/trans" and "coding/noncoding". The "5/3/c/o" system represents a simplification of the existing classification based on gene structure [6, 11], and indicates which parts of the two sequences overlap, i.e. the 5' ends (5' overlapping), the 3' ends (3' overlapping), or one transcript completely covered by the other (complete; see Figure 1). If neither applies, the NAT pair will be marked "o" (other), for instance if only partial overlap between the two transcripts. The "cis/trans" scheme tells whether or not the two sequences of a NAT pair are located at the same chromosomal loci, i.e. if both of them are located at the same genomic position they will be named a cis-NAT pair, otherwise a trans-NAT pair. The "coding/noncoding" scheme indicates whether the two overlapping RNAs are (protein) coding RNAs or noncoding RNAs. We have not adopted the system [6, 11] that divided NAT pairs according to their exon-intron structures, because we wish to provide more compact and practical information and thus enable quick retrieval of the most useful bits from the abundance of available information. For more detailed information on particular NAT pairs, users may visit other relevant databases through the provided links.

The "5/3/c/o" classification system. The arrows indicate the transcriptional orientation of the NAT pair. A solid line indicates an exon and a broken line an intron.
Database Construction
We obtained accession numbers and clone IDs for the NAT pairs from the supplementary material of published articles and downloaded the annotation information and sequences from the NCBI and FANTOM websites. In the first step, we divided the NAT pairs to cis/trans classes according to information in referenced papers. The second step was to classify the NAT pairs according to the coding/noncoding system, thus, all NAT pairs were sorted as coding-coding, coding-noncoding and noncoding-noncoding. In the third step, Blat [25] was used to classify the NAT pairs according to the 5/3/c/o system. Finally, we have removed redundant NAT pairs derived from different datasets.
Website Features
The three core functions of antiCODE database are browse, search and sequence alignment with Blast. Under the browse option, there are five sub-options – Pair ID, cis/trans, overlap, coding/noncoding, and species – by which users can browse all NAT pairs by pair ID, or NAT pair classes.
More specific lookups can be executed by the search function. Users can enter the exact gene accession number or clone ID to see whether a sequence of interest has a possible complementary transcript. If one is interested in NAT pairs relating to some particular condition, e.g. cancer, a relevant key word can be entered in the Text search frame under the search option.
If a sequence of interest cannot be found in the database or a user want to investigate whether some novel sequence possibly overlap with known NAT pairs, the Blast option will be very useful. Users just needs to paste her sequence in the sequence window, or load them into the Blast web page, and select the appropriate choices, such as expected number of hits (Figure 2), and then the Blast result will be returned.

The Blast options. In the database frame, 12 genomes could be selected as Blast databases. More detailed options could be found below which allow users to personalize the Blast results according to complexity, expect value and graphical overview options.
After a NAT pairs of interest have been found, all information pertaining to the NAT pair, including annotation and map view links to other databases, affiliated classes, a simple description and references, will appear. More detailed annotations and comments can be obtained through the links to other relevant databases.
Utility and discussion
Recently, new technologies, such as microarray, SAGE, and MPSS have played prominent roles in the identification of NAT pairs. Before 2005 only EST (UniGene) and mRNAs had been used for NAT prediction. Later large scale full-length cDNA data emerged, based on which more than 1,000 rice NATs[12] were first reported, closely followed by mouse [8, 11] and Arabidopsis [7] NATs. For NAT prediction in Arabidopsis [7] also MPSS data has been used, and in 2005, a new NAT dataset based on SAGE was reported in mouse [26]. In 2007, data [27] from whole-genome arrays was employed for NAT prediction in Arabidopsis. It is expected that along with the improvement in array technology, more transcripts from tilling microarrays will be used for future NAT predictions, hopefully resulting in an accurate and exhaustive set of NAT data.
Conclusion
The most recently released NAT datasets [9, 26–28] have yet not been included in antiCODE, but will be included in the next release of the database. However, compared with other existing databases [29], antiCODE is presently the most comprehensive and integrated database for NAT pairs. The most distinctive features of antiCODE are as follows; (i) antiCODE includes almost all known natural antisense transcript (NAT) pairs from 12 eukaryotic model organisms, (ii) antiCODE provides substantial and compact information relating to NATs (e.g. accession number, clone ID, species, classification etc.), (iii) we have introduced a classification system based on the previous notions which should give users an immediate impression of the basic features of each NAT pair, (iv) a Blast service is provided, and (v) antiCODE provides a user-friendly interface and a convenient search option, allowing efficient investigation and verification of natural antisense pairs from different species.
Availability and requirements
The antiCODE database and related resources can be freely accessed at its websites http://bioinfo.ibp.ac.cn/ANTICODE or http://www.anticode.org
References
Billy E, Brondani V, Zhang H, Muller U, Filipowicz W: Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc Natl Acad Sci U S A 2001, 98(25):14428–14433. 10.1073/pnas.261562698
Faghihi MA, Wahlestedt C: RNA interference is not involved in natural antisense mediated regulation of gene expression in mammals. Genome Biol 2006, 7(5):R38. 10.1186/gb-2006-7-5-r38
Engstrom PG, Suzuki H, Ninomiya N, Akalin A, Sessa L, Lavorgna G, Brozzi A, Luzi L, Tan SL, Yang L, Kunarso G, Ng EL, Batalov S, Wahlestedt C, Kai C, Kawai J, Carninci P, Hayashizaki Y, Wells C, Bajic VB, Orlando V, Reid JF, Lenhard B, Lipovich L: Complex Loci in human and mouse genomes. PLoS Genet 2006, 2(4):e47. 10.1371/journal.pgen.0020047
Lehner B, Williams G, Campbell RD, Sanderson CM: Antisense transcripts in the human genome. Trends Genet 2002, 18(2):63–65. 10.1016/S0168-9525(02)02598-2
Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, Nemzer S, Pinner E, Walach S, Bernstein J, Savitsky K, Rotman G: Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol 2003, 21(4):379–386. 10.1038/nbt808
Zhang Y, Liu XS, Liu QR, Wei L: Genome-wide in silico identification and analysis of cis natural antisense transcripts (cis-NATs) in ten species. Nucleic Acids Res 2006, 34(12):3465–3475. 10.1093/nar/gkl473
Wang XJ, Gaasterland T, Chua NH: Genome-wide prediction and identification of cis-natural antisense transcripts in Arabidopsis thaliana. Genome Biol 2005, 6(4):R30. 10.1186/gb-2005-6-4-r30
RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group) and the FANTOM Consortium, Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, Suzuki H, Carninci P, Hayashizaki Y, Wells C, Frith M, Ravasi T, Pang KC, Hallinan J, Mattick J, Hume DA, Lipovich L, Batalov S, Engstrom PG, Mizuno Y, Faghihi MA, Sandelin A, Chalk AM, Mottagui-Tabar S, Liang Z, Lenhard B, Wahlestedt C: Antisense Transcription in the Mammalian Transcriptome10.1126/science.1112009. Science 2005, 309(5740):1564–1566. 10.1126/science.1112009
Li YY, Qin L, Guo ZM, Liu L, Xu H, Hao P, Su J, Shi Y, He WZ, Li YX: In silico discovery of human natural antisense transcripts. BMC Bioinformatics 2006, 7: 18. 10.1186/1471-2105-7-18
Chen J, Sun M, Kent WJ, Huang X, Xie H, Wang W, Zhou G, Shi RZ, Rowley JD: Over 20% of human transcripts might form sense-antisense pairs. Nucleic Acids Res 2004, 32(16):4812–4820. 10.1093/nar/gkh818
Kiyosawa H, Yamanaka I, Osato N, Kondo S, Hayashizaki Y: Antisense transcripts with FANTOM2 clone set and their implications for gene regulation. Genome Res 2003, 13(6B):1324–1334. 10.1101/gr.982903
Osato N, Yamada H, Satoh K, Ooka H, Yamamoto M, Suzuki K, Kawai J, Carninci P, Ohtomo Y, Murakami K, Matsubara K, Kikuchi S, Hayashizaki Y: Antisense transcripts with rice full-length cDNAs. Genome Biol 2003, 5(1):R5. 10.1186/gb-2003-5-1-r5
Sun M, Hurst LD, Carmichael GG, Chen J: Evidence for variation in abundance of antisense transcripts between multicellular animals but no relationship between antisense transcriptionand organismic complexity. Genome Res 2006, 16(7):922–933. 10.1101/gr.5210006
Luther HP: Role of endogenous antisense RNA in cardiac gene regulation. J Mol Med 2005, 83(1):26–32. 10.1007/s00109-004-0613-5
Hastings ML, Ingle HA, Lazar MA, Munroe SH: Post-transcriptional regulation of thyroid hormone receptor expression by cis-acting sequences and a naturally occurring antisense RNA. J Biol Chem 2000, 275(15):11507–11513. 10.1074/jbc.275.15.11507
Enerly E, Sheng Z, Li KB: Natural antisense as potential regulator of alternative initiation, splicing and termination. In Silico Biol 2005, 5(4):367–377.
Munroe SH: Antisense RNA inhibits splicing of pre-mRNA in vitro. Embo J 1988, 7(8):2523–2532.
Werner A, Preston-Fayers K, Dehmelt L, Nalbant P: Regulation of the NPT gene by a naturally occurring antisense transcript. Cell Biochem Biophys 2002, 36(2–3):241–252. 10.1385/CBB:36:2-3:241
Sleutels F, Barlow DP, Lyle R: The uniqueness of the imprinting mechanism. Curr Opin Genet Dev 2000, 10(2):229–233. 10.1016/S0959-437X(00)00062-9
Rougeulle C, Heard E: Antisense RNA in imprinting: spreading silence through Air. Trends Genet 2002, 18(9):434–437. 10.1016/S0168-9525(02)02749-X
Coudert AE, Pibouin L, Vi-Fane B, Thomas BL, Macdougall M, Choudhury A, Robert B, Sharpe PT, Berdal A, Lezot F: Expression and regulation of the Msx1 natural antisense transcript during development. Nucleic Acids Res 2005, 33(16):5208–5218. 10.1093/nar/gki831
Borsani O, Zhu J, Verslues PE, Sunkar R, Zhu JK: Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 2005, 123(7):1279–1291. 10.1016/j.cell.2005.11.035
Michael NL, Vahey MT, d'Arcy L, Ehrenberg PK, Mosca JD, Rappaport J, Redfield RR: Negative-strand RNA transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by Tat. J Virol 1994, 68(2):979–987.
Briquet S, Richardson J, Vanhee-Brossollet C, Vaquero C: Natural antisense transcripts are detected in different cell lines and tissues of cats infected with feline immunodeficiency virus. Gene 2001, 267(2):157–164. 10.1016/S0378-1119(01)00404-8
Kent WJ: BLAT--the BLAST-like alignment tool. Genome Res 2002, 12(4):656–664. 10.1101/gr.229202. Article published online before March 2002
Siddiqui AS, Khattra J, Delaney AD, Zhao Y, Astell C, Asano J, Babakaiff R, Barber S, Beland J, Bohacec S, Brown-John M, Chand S, Charest D, Charters AM, Cullum R, Dhalla N, Featherstone R, Gerhard DS, Hoffman B, Holt RA, Hou J, Kuo BY, Lee LL, Lee S, Leung D, Ma K, Matsuo C, Mayo M, McDonald H, Prabhu AL, Pandoh P, Riggins GJ, de Algara TR, Rupert JL, Smailus D, Stott J, Tsai M, Varhol R, Vrljicak P, Wong D, Wu MK, Xie YY, Yang G, Zhang I, Hirst M, Jones SJ, Helgason CD, Simpson EM, Hoodless PA, Marra MA: A mouse atlas of gene expression: large-scale digital gene-expression profiles from precisely defined developing C57BL/6J mouse tissues and cells. Proc Natl Acad Sci U S A 2005, 102(51):18485–18490. 10.1073/pnas.0509455102
Yamada K, Lim J, Dale JM, Chen H, Shinn P, Palm CJ, Southwick AM, Wu HC, Kim C, Nguyen M, Pham P, Cheuk R, Karlin-Newmann G, Liu SX, Lam B, Sakano H, Wu T, Yu G, Miranda M, Quach HL, Tripp M, Chang CH, Lee JM, Toriumi M, Chan MM, Tang CC, Onodera CS, Deng JM, Akiyama K, Ansari Y, Arakawa T, Banh J, Banno F, Bowser L, Brooks S, Carninci P, Chao Q, Choy N, Enju A, Goldsmith AD, Gurjal M, Hansen NF, Hayashizaki Y, Johnson-Hopson C, Hsuan VW, Iida K, Karnes M, Khan S, Koesema E, Ishida J, Jiang PX, Jones T, Kawai J, Kamiya A, Meyers C, Nakajima M, Narusaka M, Seki M, Sakurai T, Satou M, Tamse R, Vaysberg M, Wallender EK, Wong C, Yamamura Y, Yuan S, Shinozaki K, Davis RW, Theologis A, Ecker JR: Empirical analysis of transcriptional activity in the Arabidopsis genome. Science 2003, 302(5646):842–846. 10.1126/science.1088305
Wang H, Chua NH, Wang XJ: Prediction of trans-antisense transcripts in Arabidopsis thaliana. Genome Biol 2006, 7(10):R92. 10.1186/gb-2006-7-10-r92
Zhang Y, Li J, Kong L, Gao G, Liu QR, Wei L: NATsDB: Natural Antisense Transcripts DataBase. Nucleic Acids Res 2006.
Acknowledgements
This work was supported by grants from Youth Foundation of Peking Union Medical College Hospital (No. 2005 37A), National Natural Science Foundation of China (No. 30570393 and No. 30600729) and China Medical Board in New York (No.06837).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Authors' contributions
Yifei Yin and Yi Zhao carried out the design and the collection of data. Jie Wang carried for building the database. Changning Liu participated in the design of the study. Shuguang Chen helped to draft the manuscript. Runsheng Chen and Haitao Zhao participated in the design and coordination. All authors read and approved the final manuscript.
Yifei Yin, Yi Zhao contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yin, Y., Zhao, Y., Wang, J. et al. antiCODE: a natural sense-antisense transcripts database. BMC Bioinformatics 8, 319 (2007). https://doi.org/10.1186/1471-2105-8-319
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-8-319