
Abstract
Background
In sequence analysis the multiple alignment builds the fundament of all proceeding analyses. Errors in an alignment could strongly influence all succeeding analyses and therefore could lead to wrong predictions. Hand-crafted and hand-improved alignments are necessary and meanwhile good common practice. For RNA sequences often the primary sequence as well as a secondary structure consensus is well known, e.g., the cloverleaf structure of the t-RNA. Recently, some alignment editors are proposed that are able to include and model both kinds of information. However, with the advent of a large amount of reliable RNA sequences together with their solved secondary structures (available from e.g. the ITS2 Database), we are faced with the problem to handle sequences and their associated secondary structures synchronously.
Results
4SALE fills this gap. The application allows a fast sequence and synchronous secondary structure alignment for large data sets and for the first time synchronous manual editing of aligned sequences and their secondary structures. This study describes an algorithm for the synchronous alignment of sequences and their associated secondary structures as well as the main features of 4SALE used for further analyses and editing. 4SALE builds an optimal and unique starting point for every RNA sequence and structure analysis.
Conclusion
4SALE, which provides an user-friendly and intuitive interface, is a comprehensive toolbox for RNA analysis based on sequence and secondary structure information. The program connects sequence and structure databases like the ITS2 Database to phylogeny programs as for example the CBCAnalyzer. 4SALE is written in JAVA and therefore platform independent. The software is freely available and distributed from the website at http://4sale.bioapps.biozentrum.uni-wuerzburg.de
Similar content being viewed by others

Background
Since multiple sequence alignments are the basis of many analyses, for example in phylogenetics or in analysing functional protein domains, there is a need for programs to create and improve those alignments. Currently, several programs are available to fulfil these necessities, e.g., CLUSTAL W [1], MUSCLE [2], DiAlign [3], T-Coffee [4] or DCA [5] all of which are able to align multiple sequences globally. The underlying methods have their strengths and weaknesses, and resulting alignments can diverge from the biologically correct ones. Editors like JalView [6], SEAVIEW [7], CINEMA [8] or Align [9] are needed to enhance the results by hand.
The just mentioned tools are based on sequence information only, but in RNA sequence analyses there is often also structural information available. Databases like the ITS2 Database [10–12] provide a growing number of sequences and their known secondary structures, as a prerequisite for constructing RNA alignments for inferring phylogenies, which of course is a precondition to understand the evolution of such RNA secondary structures [12].
All available methods that include structural information to build RNA sequence alignments have a very high complexity. Rfam [13] provides a method to compare a single nucleotide query sequence to handcurated alignments of non-coding RNA families with annotated consensus secondary structures. MARNA [14] and RNAforester [15] can be used to build global multiple alignments based on sequence and simultaneously on secondary structure information. However, the amount of sequences and/or sequence lengths is limited due to the complexity of their underlying algorithms, which is at least O(N3).
In current alignment editors like RALEE [16], DCSE [17] or jPHYDIT [18] secondary structure information support is very limited. While RALEE relys on the consensus structure only, jPHYDIT just shows the pairing information of the selected sequence. Another RNA alignment editor called SARSE has become available recently and focuses on detection and editing of structural groups in RNA families [19]. So there is no editor available to align both, sequence and secondary structure information of every single RNA sequence simultaneously.
Implementation
4SALE is entirely written in JAVA, which enables to execute the software on any platform with a JAVA 5.0 virtual machine available. The application consists of two parts, the alignment algorithm, which is based on standard protein alignment algorithms, and the graphical editor frontend. For sequence and secondary structure alignments running on the local machine 4SALE takes use of CLUSTAL W [1], so the binary is required to be installed.
Integration of different multiple alignment tools is realised by using SOAP based WebServices. Here, we take use of RNAforester [15], CLUSTAL W [1], DCA [5] or DiAlign [3]. The DiAlign and DCA WebServices currently support sequence alignments without secondary structure information only. The WebService technology enables the user to run the tasks on remote machines. Therefore, it is possible to use 4SALE without restriction of any kind during the calculation of the alignment. All WebServices require an internet connection.
Results
Supported data
For RNA sequence and secondary structure alignment and editing, 4SALE reads Vienna style DotBracket [20] formatted files. The ITS2 Database [10–12] represents a good source for these kind of data. In addition we provide direct access to the ITS2 Database from within 4SALE through the ITS2 SOAP interface [10]. While RNA sequence information only is supported via the standard FASTA format, alignment data can be loaded using the Clustal [1] importer. Furthermore 4SALE handles XML based RNA formats namely RNAStructML and RNAStructAlignmentML [21].
Standard features
In addition to the secondary structure based functionality, 4SALE integrates many useful features, that are known from other alignment editors/programs. This includes selecting multiple sections of an alignment to highlight interesting regions and temporarily hide sequences to focus on a subset of the alignment. Sequence-motifs, including those, which are based on sequence and secondary structure information, can easily be highlighted by pattern matching. Alignment column conservation based on sequence information is visualized by either sequence logos [22] or simply by bars on top of each column. Further importing, exporting and deleting sequences is possible by using the sequence names' context menu. Additionally sequences can be reordered with the help of the "Rearrange Sequences" window.
Algorithm
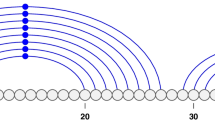
Beside the integration of RNAforester [15] we developed an algorithm that uses the secondary structure information of every single sequence to align multiple RNA sequences. This algorihm inherits the complexity of those based on sequence information only. We achieve this by mapping the sequence and secondary structure information of every single RNA sequence to artificial protein sequences. The algorithm can be described as string alignment on a 12 letter alphabet comprised of the 4 nucleotides in three structural states (unpaired, paired left, paired right). Horizontal dependencies given by the sequence bindings are not modeled by this approach. To align the string we use common alignment programs, like CLUSTAL W [1] with a suitable scoring matrix. There are several substitution models for this kind of scoring matrices discussed [23, 24], we used a model as described by [25, 26]. The model is based on subsitutions that were extracted from ITS2 sequence and secondary structure alignments (Fig. 1). Those sequences and their associated secondary structures were obtained from the ITS2 Database [10–12].

ITS2 sequence and secondary structure ratematrix. This figure shows the estimated sequence/secondary structure substitution rates (*105). Diagonal entries are by definition the negative sum of all row entries. Note, high rates depict frequent substitutions, and vice versa small rates depict rare substitutions, e.g., within a secondary structure Cs an Us are often replaced by each other.
Synchronous editing
One of the main features of 4SALE is synchronizing the sequence and secondary structure alignment, that is, every operation on the sequence alignment is also performed on the secondary structure alignment and vice versa. Alignment editing in general works like in most alignment editors by using the space key to insert and the backspace key to remove gaps.
Block editing
As an effect of not using horizontal dependencies in our greedy sequence and secondary structure alignment algorithm, there are often misaligned "blocks" in the result of CLUSTAL W [1]. As shown in Fig. 2e the alignment could be improved very fast, by using the block editing feature in 4SALE.

Overview. This figure shows a complete overview of the main features in 4SALE. All parts and their use are described in the boxes within the figure.
Cursors
To make editing as convenient as possible, we provide different edit cursors. Beside the standard cursor, that behaves as you expect from text editors for example, we have an exclusive cursor, which performs the edit operation on every sequence but the selected ones. The leftside cursor (c.f. [9]) allows edit operations only at the beginning of the sequence. As mentioned above all cursors perform synchronously on sequences and their secondary structures.
Working with secondary structures
As current predictions of secondary structure information is not highly reliable, performing changes to correct the secondary structures is often needed.
Adding secondary structure information
4SALE supports two methods to add secondary structure information to RNA sequences. First, by using the remote folding feature to call the RNAfold WebService provided by the University of Bielefeld (Germany), or manually adding secondary structures by using the secondary structure editing feature.
Secondary structure editing
The secondary structure editing mode available in 4SALE allows easy modification of the secondary structure information. It is context sensitive, which means it uses sequence and secondary structure information to validate whether a binding in this context is possible or not. Furthermore, it supports column based editing by holding the control key. This enables insertion and deletion of equivalent bindings in all sequences. In this case bindings are only added to sequences allowing this binding (same with deletions).
Secondary structure inspector
A secondary structure inspector allows to view and select specific helical regions in secondary structures of loaded sequences. The inspector consists of two parts; the upper part shows a consensus of all secondary structures, the lower part shows all secondary structures separately. The secondary structure consensus is calculated not only on column conservation, but also with respect of horizontal dependencies, so the result is a valid secondary structure. The conservation threshold can be modified using the slider above.
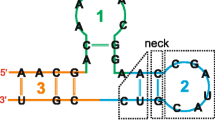
Selecting an element in the inspector highlights the corresponding part in the alignment view. As shown in Fig. 3, the inspector simplifies visualizing misaligned sequences. "Masking" the sequence alignment based on the current consensus structure is also possible. The result is shown in a new window, which contains the alignment based on sequence information only. The alignment can be processed like any other sequence alignment loaded in 4SALE. This is particularly useful for calculating phylogenetic trees based on the collective helical regions in the sequence alignment.

Synchronous editing. This figure illustrates the synchronous sequence and secondary structure handling in 4SALE. When selecting a helical region in the secondary structure alignment, as shown in this example, 4SALE synchronously selects its structural counterpart and its corresponding parts in the sequence alignment. The figure also shows very well, how easily an error in the alignment could be detected and corrected by using the selection and edit features in 4SALE.
Analyzing compensatory base changes
Compensatory base changes (CBC) occur when both nucleotides of a paired site mutate while the pairing itself stays stable. CBC analysis is important in detecting species that are discriminated by their sexual incompatibility [27–30]. We provide an easy-to-use CBC analysis mechanism with the ability to calculate CBC matrices on the current sequence and secondary structure based alignment. The numbers in the CBC matrix are the counts of compensatory base changes in a pairwise sequence structure alignment, which are naturally given in the considered multiple sequence structure alignment. A CBC-window in 4SALE (Fig. 2c) allows to select CBC counts between two sequences and highlights directly all CBCs within the alignment, giving an overview of all CBCs in the aligned sequences.
Output & connection to other tools
For further analyses we provide several output formats. Calculated CBC matrices can be saved as comma/tab seperated values to be used in CBCAnalyzer [27]. CBCTree (as implemented in CBCAnalyzer) can be used to calculate phylogenetic trees based on a CBC count matrix. At present, no program is available to handle alignment outputs that include sequences and their individual secondary structures. However, for viewing purposes and publication we support a MARNA-like [14] output. Sequence alignments optimized by structural information could, of course, be saved separately. For phylogenetic analyses here we support the PHYLIP [31, 32] formats. Other tools that rely on multiple alignments are supported by FASTA.
Discussion
4SALE is the first alignment editor which allows synchronous editing of sequences and their corresponding secondary structures. Since it is targeted on RNA sequence alignment and editing it contains many features using the secondary structure information, e.g., the secondary structure inspector. All current standard alignment editors can handle secondary structures as character sequence only.
By using standard greedy protein alignment algorithms we inherit their time efficiency. In contrast to, e.g., MARNA [14] or RNAforester [15], the time complexity of calculation grows not rapidly with large files. We present a completely new approach using nucleotides and every single secondary structure for building and improving RNA sequence alignments in comparison to others, which just take the consensus structure information.
The structure output converted to Vienna style DotBracket can be created from any desired RNA folding program, e.g., RNAfold, Mfold [33] or RNAStructure [34]. It is then aligned by using a suitable substitution matrix, which in our case is based on information of the ITS2 Database.
Due to the natural limitation that two structures can be hidden in one sequence, in general only one will be considered by our approach.
A future version of 4SALE will integrate in addition to RNAforester [15] more real structural alignment methods as WebServices via the SOAP interface. Also secondary structure prediction algorithms as an alternative to RNAfold will be included. Furthermore, more visualizations like secondary structure drawings can be implemented.
Conclusion
4SALE is easy to use and has a fast (<O(N3)) and good heuristic to globally align multiple RNA sequences and their associated secondary structures simultaneously.
Availability and requirements
4SALE is freely available at http://4sale.bioapps.biozentrum.uni-wuerzburg.de. A JAVA virtual machine 5.0 is needed to run the application. Furthermore, for automatic sequence and structure based alignments a local installation of CLUSTAL W and/or internet connection for WebService based alignments is required.
Abbreviations
- CBC:
-
compensatory base change
- ITS2:
-
internal transcribed spacer 2
References
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22(22):4673–4680.
Edgar RC: MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004, 5: 113. 10.1186/1471-2105-5-113
Morgenstern B: DIALIGN: multiple DNA and protein sequence alignment at BiBiServ. Nucleic Acids Res 2004, (32 Web Server):W33-W36.
Notredame C, Higgins DG, Heringa J: T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 2000, 302: 205–217. 10.1006/jmbi.2000.4042
Stoye J, Moulton V, Dress AW: DCA: an efficient implementation of the divide-and-conquer approach to simultaneous multiple sequence alignment. Comput Appl Biosci 1997, 13(6):625–626.
Clamp M, Cuff J, Searle SM, Barton GJ: The Jalview Java alignment editor. Bioinformatics 2004, 20(3):426–427. 10.1093/bioinformatics/btg430
Galtier N, Gouy M, Gautier C: SEAVIEW and PHY LO WIN : two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci 1996, 12(6):543–548.
Parry-Smith DJ, Payne AW, Michie AD, Attwood TK: CINEMA-a novel colour INteractive editor for multiple alignments. Gene 1998, 221: GC57-GC63. 10.1016/S0378-1119(97)00650-1
Hepperle D: Align Ver.07/04©. Multisequence alignment-editor and preparation/manipulation of phylogenetic datasets. Win32-Version. 2004.http://www.sequentix.de [Distributed by the author via: ].
Schultz J, Müller T, Achtziger M, Seibel PN, Dandekar T, Wolf M: The internal transcribed spacer 2 database-a web server for (not only) low level phylogenetic analyses. Nucleic Acids Res 2006, (34 Web Server):W704-W707.
Schultz J, Maisel S, Gerlach D, Müller T, Wolf M: A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA 2005, 11(4):361–364. 10.1261/rna.7204505
Wolf M, Achtziger M, Schultz J, Dandekar T, Müller T: Homology modeling revealed more than 20,000 rRNA internal transcribed spacer 2 (ITS2) secondary structures. RNA 2005, 11(11):1616–1623. 10.1261/rna.2144205
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR: Rfam: an RNA family database. Nucleic Acids Res 2003, 31: 439–441. 10.1093/nar/gkg006
Siebert S, Backofen R: MARNA: multiple alignment and consensus structure prediction of RNAs based on sequence structure comparisons. Bioinformatics 2005, 21(16):3352–3359. 10.1093/bioinformatics/bti550
Höchsmann M, Voss B, Giegerich R: Pure Multiple RNA Secondary Structure Alignments: A Progressive Profile Approach. IEEE/ACM Transactions on Computational Biology and Bioinformatics 2004, 1: 53–62. 10.1109/TCBB.2004.11
Griffiths-Jones S: RALEE-RNA ALignment editor in Emacs. Bioinformatics 2005, 21(2):257–259. 10.1093/bioinformatics/bth489
Rijk PD, Wachter RD: DCSE, an interactive tool for sequence alignment and secondary structure research. Comput Appl Biosci 1993, 9(6):735–740.
Jeon YS, Chung H, Park S, Hur I, Lee JH, Chun J: jPHYDIT: a JAVA-based integrated environment for molecular phylogeny of ribosomal RNA sequences. Bioinformatics 2005, 21(14):3171–3173. 10.1093/bioinformatics/bti463
Andersen ES, Lind-Thomsen A, Knudsen B, Kristensen SE, Havgaard JH, Larsen N, Sestoft P, Kjems J, Gorodkin J: Detection and editing of structural groups in RNA families. submitted 2006.
Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P: Fast Folding and Comparison of RNA Secondary Structures. Monatsh Chem 1994, 125: 167–188. 10.1007/BF00818163
Seibel PN, Kruger J, Hartmeier S, Schwarzer K, Lowenthal K, Mersch H, Dandekar T, Giegerich R: XML schemas for common bioinformatic data types and their application in workflow systems. BMC Bioinformatics 2006, 7: 490. 10.1186/1471-2105-7-490
Schneider TD, Stephens RM: Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 1990, 18(20):6097–6100.
Hudelot C, Gowri-Shankar V, Jow H, Rattray M, Higgs PG: RNA-based phylogenetic methods: application to mammalian mitochondrial RNA sequences. Mol Phylogenet Evol 2003, 28(2):241–252. 10.1016/S1055-7903(03)00061-7
Smith AD, Lui TWH, Tillier ERM: Empirical models for substitution in ribosomal RNA. Mol Biol Evol 2004, 21(3):419–427. 10.1093/molbev/msh029
Müller T, Vingron M: Modeling amino acid replacement. J Comput Biol 2000, 7(6):761–776. 10.1089/10665270050514918
Müller T, Spang R, Vingron M: Estimating amino acid substitution models: a comparison of Dayhoff's estimator, the resolvent approach and a maximum likelihood method. Mol Biol Evol 2002, 19: 8–13.
Wolf M, Friedrich J, Dandekar T, Müller T: CBCAnalyzer: inferring phylogenies based on compensatory base changes in RNA secondary structures. In Silico Biol 2005, 5(3):291–294.
Coleman AW: The significance of a coincidence between evolutionary landmarks found in mating affinity and a DNA sequence. Protist 2000, 151: 1–9. 10.1078/1434-4610-00002
Coleman AW, Vacquier VD: Exploring the phylogenetic utility of its sequences for animals: a test case for abalone (haliotis). J Mol Evol 2002, 54(2):246–257. 10.1007/s00239-001-0006-0
Coleman AW: ITS2 is a double-edged tool for eukaryote evolutionary comparisons. Trends Genet 2003, 19(7):370–375. 10.1016/S0168-9525(03)00118-5
Felsenstein J: PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle 2005.
Felsenstein J: PHYLIP – Phylogeny Inference Package (Version 3.2). Cladistics 1989, 5: 164–166.
Zuker M: Computer prediction of RNA structure. Methods Enzymol 1989, 180: 262–288.
Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH: Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci USA 2004, 101(19):7287–7292. 10.1073/pnas.0401799101
Acknowledgements
Special thanks go to Jan Krüger and Sven Hartmeier (both University of Bielefeld, Germany) for their great help with integrating the WebServices into 4SALE. Furthermore, we thank Andreas Pokorny and Joachim Friedrich (both University of Würzburg, Germany) for fruitful discussions and gratefully acknowledge the funding from the "Impuls- und Vernetzungsfonds der Helmholtz-Gemeinschaft Deutscher Forschungszentren e.V." (HOBIT VH-VI-023).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MW conceived the study. TM and MW provided the alignment algorithm. TM estimated the sequence and secondary structure substitution model and its associated score matrix. Architecture, implementation and graphical design by PS. MW, PS and TM drafted the manuscript. MW, TM, JS and TD participated in study design and coordination. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Seibel, P.N., Müller, T., Dandekar, T. et al. 4SALE – A tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinformatics 7, 498 (2006). https://doi.org/10.1186/1471-2105-7-498
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-7-498