
Abstract
Background
Research involving expressed sequence tags (ESTs) is intricately coupled to the existence of large, well-annotated sequence repositories. Comparatively complete and satisfactory annotated public sequence libraries are, however, available only for a limited range of organisms, rendering the absence of sequences and gene structure information a tangible problem for those working with taxa lacking an EST or genome sequencing project. Paralogous genes belonging to the same gene family but distinguished by derived characteristics are particularly prone to misidentification and erroneous annotation; high but incomplete levels of sequence similarity are typically difficult to interpret and have formed the basis of many unsubstantiated assumptions of orthology.
In these cases, a phylogenetic study of the query sequence together with the most similar sequences in the database may be of great value to the identification process. In order to facilitate this laborious procedure, a project to employ automated phylogenetic analysis in the identification of ESTs was initiated.
Results
galaxieEST is an open source Perl-CGI script package designed to complement traditional similarity-based identification of EST sequences through employment of automated phylogenetic analysis. It uses a series of BLAST runs as a sieve to retrieve nucleotide and protein sequences for inclusion in neighbour joining and parsimony analyses; the output includes the BLAST output, the results of the phylogenetic analyses, and the corresponding multiple alignments. galaxieEST is available as an on-line web service for identification of fungal ESTs and for download / local installation for use with any organism group at http://galaxie.cgb.ki.se/galaxieEST.html.
Conclusions
By addressing sequence relatedness in addition to similarity, galaxieEST provides an integrative view on EST origin and identity, which may prove particularly useful in cases where similarity searches return one or more pertinent, but not full, matches and additional information on the query EST is needed.
Similar content being viewed by others

Background
The capture of mRNA from living cells represents an intriguing opportunity to study gene expression at various stages and conditions. Using the mRNA as template, DNA is synthesized and subsequently sequenced as expressed sequence tags (ESTs), numerous small sequence fragments, which singly or when assembled form the initial sequences in question. The genes can then be identified through similarity searches like BLAST [1] on Genbank or other dedicated sequence repositories.
The identification process is, however, often hampered by the lack of fully matching sequences in the reference databases; comparatively complete and satisfactory annotated public libraries exist only for a limited number of organisms. Ideally, even in the absence of full matches, the reference database holds sequences that are related to the query sequence through common ancestry. Such homologues are likely to constitute an invaluable information source for the elucidation of identity and function of the gene which the query EST represents, and unless they are very divergent, these sequences will typically be retrieved by a standard BLAST search. A sequence similarity search does not per se reveal information about the sequence relatedness, but it does provide a qualified platform for further analyses such as phylogenetic inference. The use of phylogenetic inference in EST annotation may prove to be of particular relevance when dealing with large gene families where the individual genes show secondarily derived characteristics / high mutation rates.
Carrying out phylogenetic analysis on Genbank EST sequences is, however, an onerous undertaking, generally calling for application of several external computer programs and numerous manual steps. The authors present a Perl-CGI script package, galaxieEST, designed to facilitate the identification of EST sequences through automated BLAST searches and phylogenetic analysis of the results. The package collects an EST sequence from the user through a web interface and uses the EST as a query in a series of BLAST runs on nucleotide, EST, and protein databases. Each positive BLAST outcome is subjected to a joint phylogenetic analysis with the query EST and the best BLAST matches from each stage. The output includes the BLAST results, the outcome of the phylogenetic analysis and the corresponding multiple alignment. galaxieEST is available as a web service for identification of fungal EST sequences and for download / local installation, in which case the organism group and EST coverage can be set as seen fit.
Implementation
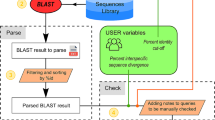
galaxieEST is based on the galaxie package [2]. It is written in Perl [3] and runs as a CGI script under the Apache httpd web server [4] on a Red Hat Linux server [5]. All sequences are stored in local MySQL databases [6] from which data is extracted using the DBI Perl-MySQL package [7]. BLAST, Clustal W [8], and PHYLIP [9] are used for sequence similarity searches, multiple alignment, and phylogenetic analysis, respectively.
For the web service installation, three reference sets of fungal sequences were collected from Genbank and stored in separate MySQL databases (May 25; 2004): all Genbank / EMBL / DDBJ nucleotide sequences (excluding ESTs) between 110 and 1100 base paires (bp) in length (76,064 sequences), all EST sequences between 60 and 1100 bp in length (306,307 sequences), and all protein sequences between 10 and 400 amino acids in length (66,873 sequences). No patented sequences or working drafts were included. A parser script to format Genbank sequence files for easy addition into MySQL tables was written and is available for download.
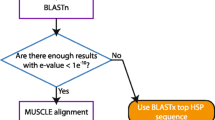
galaxieEST has a six-step analysis procedure. It collects the query EST and analysis parameters from the web interface and runs BLASTn locally against the nucleotide database. Significant matches – as judged by the BLAST e-value – are displayed and aligned together with the query sequence for joint phylogenetic analysis using either neighbour joining (NJ) or the maximum parsimony (MP) optimality criterion. The phylogenetic analyses feature random sequence addition, outgroup rooting, bootstrapping, and branch swapping (MP). The user specifies whether to include all significantly matching sequences or only matches of unique e-values from the BLAST run; the latter option can be used to reduce the impact of identical sequences on the analysis (the inclusion of too many identical sequences in a phylogenetic analysis is likely to yield a tree with little, if any, resolution, such trees being unsound bases for inferences on sequence relatedness). The results from BLAST and the phylogenetic tree are displayed and the multiple alignment is accessed through a hyperlink. The output of galaxieEST is hyperlinked into Genbank for rapid retrieval of complementary information, and several of the information items of the output can be copied into external tools for further processing.
The second analysis step is similar to the first one but uses the EST database as reference. For the third step, all three forward reading frames of the query EST are translated into amino acid sequences and compared with the protein database records using BLASTx. Significant matches are collected and analysed as above. Finally, the query sequence is reversed and its complementary DNA string is computed. Steps one through three are repeated for the reverse complementary strand of the query EST; this accounts for cases where, unintentionally or otherwise, the direction of either the query EST or (some of) the reference sequences is reverse and complementary.
Results and discussion
Research involving EST sequences relies heavily upon the existence of wide-scoped and well-annotated sequence repositories. Many tools have been developed to annotate EST sequences; the examples include Gene2EST [10], cDNA2Genome [11], and EST_Genome [12]. Despite recent progress, many newly sequenced ESTs are left poorly matched when performing traditional similarity searches on available databases. It might in these cases be a good idea to set up separate BLAST runs against different databases, such as EST-nucleotide, EST-EST, and EST-protein. galaxieEST automates these searches for any given query EST. Moreover, for each positive outcome, galaxieEST collects the best matches and performs a phylogenetic analysis of these sequences together with the query EST.
Although the parameters of the phylogenetic analysis can be set as extensive as seen fit upon script installation, the phylogenetic analyses of galaxieEST should not be seen as a rapid way to generate phylogenetic trees for publication or to establish the true branching order amongst a set of sequences. Such tasks call for more extensive analyses (e.g., multiple outgroups, more thorough computer runs / analysis schemes, and manually adjusted alignments); galaxieEST should rather be looked upon as an attempt to utilise the powerful properties of phylogenetic inference to get an alternative estimation of sequence relatedness. Sequence coverage is another important issue; the fact that BLAST is used as a sieve to find sequences for further processing means that galaxieEST will not proceed with the step in question unless there are significant BLAST matches in the first place. The authors intend to update the sequences of the fungal EST web service on a regular basis.
The galaxieEST output is composite, consisting of the results from each of the six BLAST runs and up to six phylogenetic analyses. While the information from each analysis is easily interpreted, the penetration of the combined results may be less straightforward. galaxieEST is a common roof for a series of analyses, each of which may give a small contribution to the true nature of the EST at hand and the totalling of which is likely to give more information on the attributes and ancestry of the query EST than any single similarity search. galaxieEST gives a quick overview of what is present in the public databases and uses several different tools to try to retrieve as much information as possible pertaining to the query EST. The price that one pays – some 30 seconds for the analyses to complete – may well be a negligible cost for examining information otherwise partially overlooked.
Conclusions
The elucidation of sequence identity, origin, and properties is best done in an evolutionary context. Sequences but partly matched in publicly available sequence repositories are typically difficult to process, but much information may be gained from the investigation of related sequences and the information they are likely to convey. The Perl-CGI package galaxieEST was written for that purpose in that it represents an attempt to employ automated phylogenetic analysis in the EST identification process; it is freely available as an on-line web service for fungal ESTs and for download / local installation for use with any organism group.
Availability and requirements
Project name: galaxieEST – addressing EST identity through automated phylogenetic analysis
Project home page: http://galaxie.cgb.ki.se/galaxieEST.html
Operating system(s): primarily Linux and UNIX / BSD
Programming language: Perl, SQL
Other requirements: Apache, MySQL, PHYLIP, DBI / DBD, and Clustal W
License: GNU type
Any restrictions to use by non-academics: none
Abbreviations
- CGI:
-
Common Gateway Interface
- DBD:
-
Database Driver
- DBI:
-
Database independent Interface [for Perl]
- EST:
-
Expressed Sequence Tag
- MP:
-
Maximum Parsimony
- NJ:
-
Neighbour-Joining
- SQL:
-
Structured Query Language
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25: 3389–3402. 10.1093/nar/25.17.3389
Nilsson RH, Larsson K-H, Ursing BM: galaxie – CGI scripts for sequence identification through automated phylogenetic analysis. Bioinformatics 2004, 20: 1447–1452. 10.1093/bioinformatics/bth119
The Perl Directory[http://www.perl.org]
The Apache http Server Project[http://httpd.apache.org]
Red Hat, Inc[http://www.redhat.com]
MySQL AB[http://www.mysql.com]
DBI Database Interface Module[http://dbi.perl.org]
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22: 4673–4680.
Felsenstein J: PHYLIP – Phylogeny Inference Package (Version 3.2). Cladistics 1989, 5: 164–166.
Gemünd C, Ramu C, Altenberg-Greulich B, Gibson TJ: Gene2EST: a BLAST2 server for searching expressed sequence tag (EST) databases with eukaryotic gene-sized queries. Nucleic Acids Res 2001, 29: 1272–1277. 10.1093/nar/29.6.1272
del Val C, Glatting K-H, Suhai S: CDNA2Genome: A tool for mapping and annotation cDNAs. BMC Bioinformatics 2003, 4: 39. 10.1186/1471-2105-4-39
Mott R: EST_GENOME: A program to align spliced DNA sequences to unspliced genomic DNA. Comput Appl Biosci 1997, 13: 477–478.
Acknowledgements
This work was supported by the Helge Ax:son Johnson Foundation and the Wilhelm and Martina Lundgren Science Foundation with grants to RHN; by FORMAS with grant no. 23.0697/00 to K-HL; by the Knut and Alice Wallenberg Foundation with a fellowship and Medivir AB with a grant to BMU; and by NorFA with grant no. 020725 to the UNITE group. The Göteborg University / Chalmers Research School in Genomics and Bioinformatics is gratefully acknowledged for bringing the authors together for this project and for valuable support on related matters. Valuable comments from two anonymous reviewers are also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
RHN wrote large parts of the Perl source and the MySQL interface. BR initiated the project and contributed with advice on fungal ESTs and their processing. K-HL contributed with ideas on sequence sampling and phylogenetic analysis. BMU was responsible for the web-interface and the scientific aspects of the scripts. All authors drafted the manuscript and approved the final version.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Nilsson, R.H., Rajashekar, B., Larsson, KH. et al. galaxieEST: addressing EST identity through automated phylogenetic analysis. BMC Bioinformatics 5, 87 (2004). https://doi.org/10.1186/1471-2105-5-87
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-5-87