
Abstract
Background
Rates of substitution in protein-coding sequences can provide important insights into evolutionary processes that are of biomedical and theoretical interest. Increased availability of coding sequence data has enabled researchers to estimate more accurately the coding sequence divergence of pairs of organisms. However the use of different data sources, alignment protocols and methods to estimate substitution rates leads to widely varying estimates of key parameters that define the coding sequence divergence of orthologous genes. Although complete genome sequence data are not available for all organisms, fragmentary sequence data can provide accurate estimates of substitution rates provided that an appropriate and consistent methodology is used and that differences in the estimates obtainable from different data sources are taken into account.
Results
We have developed FRAGS, an application framework that uses existing, freely available software components to construct in-frame alignments and estimate coding substitution rates from fragmentary sequence data. Coding sequence substitution estimates for human and chimpanzee sequences, generated by FRAGS, reveal that methodological differences can give rise to significantly different estimates of important substitution parameters. The estimated substitution rates were also used to infer upper-bounds on the amount of sequencing error in the datasets that we have analysed.
Conclusion
We have developed a system that performs robust estimation of substitution rates for orthologous sequences from a pair of organisms. Our system can be used when fragmentary genomic or transcript data is available from one of the organisms and the other is a completely sequenced genome within the Ensembl database. As well as estimating substitution statistics our system enables the user to manage and query alignment and substitution data.
Similar content being viewed by others


Background
Substitution rates of coding sequences provide a valuable means of characterising the evolutionary divergence of homologues. A significant excess in the rate of non-synonymous substitution (K a ) compared to the rate of nearly neutral synonymous substitution (K s ) is widely used as evidence that a sequence has evolved under positive selective pressure [1]. The identification of individual genes under positive selection or strong negative selection has important implications for the understanding of processes such as the evolution of drug resistance and evasion of the immune system by pathogens [2, 3]. In the case of genes not evolving under positive selection the relative rates of non-synonymous to synonymous substitutions (K a /K s ) provide an indication of selective constraint and a means of estimating the strength of purifying selection acting on the sequences.
The average value of K a /K s from pairwise alignments of coding sequences has been used to compare the evolutionary rates of duplicated and unduplicated genes in order to investigate the affect of gene duplication on the rate of sequence evolution [4–6]. More recently, coding sequence substitution rates of collinear and rearranged chromosomes from closely related organisms have been proposed to contain clearly detectable traces of the speciation process [7]. Coding sequence substitution rate estimates have also permitted investigation of the relationship between level and breadth of gene expression and the rate of coding sequence evolution [8, 9]. All of these comparisons have required the construction of large datasets of pair-wise in-frame alignments of orthologous sequences. FRAGS automates the process of generating these kinds of datasets and provides a framework for the management of the results generated.
Databases designed specifically to contain molecular evolutionary information have recently been established, prompted by the necessity to manage large quantities of data generated by genomic studies. The Adaptive Evolution Database, TAED [10], which contains coding sequence substitution rates estimated using the approximate method of Li et al. (Li, Pamillo and Bianchi [11–13]), is one example of these developments. Software for genomic scale molecular evolutionary studies has also been released, e.g. GenomeHistory [14], which uses a Maximum Likelihood estimation method following Muse and Gaut [15], and Goldman and Yang's method [16]. Unfortunately, it is often difficult to obtain both the software and databases used in these kinds of studies, and consequently the reproduction of results can be both laborious and inexact. Database resources such as Ensembl [17–19], Hovergen [20] and TOGA [21] are being constructed with eukaryotic comparative genomics in mind, but still need further development to provide adequate evolutionary information.
Implementation
FRAGS (the FRAGment Substitution rate estimation system) is a Python [22] based system, designed for obtaining and managing coding sequence substitution data from in-frame alignments of sequence fragments, and coding sequences from a related completely sequenced genome within the Ensembl database [23]. FRAGS uses existing Open Source or academically available software, including modules and code from the Biopython project [24]. Embedded SQL statements are used for database manipulation.
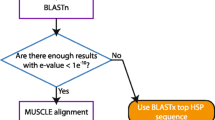
In order to obtain coding sequence substitution distances it was necessary to develop a system that was able to produce in-frame alignments from any available fragmentary sequences (such as shotgun sequences, BAC end sequences and ESTs) and orthologous sequences from a completely sequenced organism. The requirement of a completely sequenced reference organism comes from our approach to identifying orthologues. Sequence fragments with similarity to just one coding sequence in the reference organism and that do not have significant similarity to non-coding regions are considered orthologous (see below). The complete strategy used to generate in-frame alignments from fragmentary data is shown in Figure 1.

Flow diagram illustrating the strategy used to generate in-frame alignments from fragmentary sequences.
The input data to FRAGS consists of sequence fragments in fasta format. The sequence fragments are filtered for repeats and vector contamination using RepeatMasker [25] and BLASTN [26] against the Univec_core database [27]. The system is also capable of filtering for low-quality sequence regions, using an adjustable cut-off, if Phred [28] quality scores are provided. The sequence fragments are searched against all known and predicted proteins in the Ensembl database using BLASTX [26] with the default parameters. In order to eliminate paralogous matches that would distort estimates of substitution rates, nucmer, from the MUMmer package [29, 30], is used to search the fragments against the complete genome sequence underlying the Ensembl database. Sequence fragments that do not match to the genome sequence with a similarity and match-length above pre-defined cut-off values, or sequences that have more than one match to the genome, are excluded from further analysis. The parameters that determine whether a fragment has matched unambiguously are decided by the user. The co-ordinates of unambiguous matches of fragments to the genome are compared to the genomic co-ordinates of the Ensembl protein matching the fragment. If these co-ordinates are within a predetermined distance, the fragment and protein are retained for further analysis (Figure 1.).
In-frame alignments of protein coding sequences are produced through a two-step procedure using GeneWise [31, 32]. First the nucleotide sequence fragment is aligned against the Ensembl protein and then the corresponding Ensembl transcript is aligned against the putative translation of the nucleotide fragment. Maximum likelihood estimates of coding sequences substitution distances are then derived from the in-frame alignments using the codeml program from the Paml [33] package. Substitution distances are estimated from individual sequence alignments as well as from a concatenated alignment of all of the sequences.
Data sets and program parameters
We used FRAGS to estimate coding sequence substitution distances for human and chimpanzee orthologue-pairs using genomic, EST and complete coding sequence datasets from chimpanzee and the Ensembl Human database (version 8.3 based on the June 2002 Golden Path build – NCBI30). In total 148,102 genomic sequence fragments were obtained from the Riken Chimpanzee Sequencing Initiative [34, 35]. A separate set of 6930 chimpanzee cDNA sequences was obtained from the Genbank EST division. A smaller set of complete chimpanzee coding sequences from Genbank was also obtained [4].
BLASTX matches between chimpanzee sequence fragments and human proteins from the Ensembl database were required to be more than 60 amino acids in length and to have at least 90% identity. The chimpanzee sequence fragments were searched against the human genome (June 2002 Golden Path genome assembly [36]) using nucmer [29, 30]. Chimpanzee sequence fragments that did not match the genome in the same location as the homologous human protein were omitted from further analysis. Sequence fragments that had a second match to the genome, with at least 90% of the number of identical residues as the best match, were also removed from the analysis.
The pairwise comparison mode of codeml (runmode = -2) was used to provide maximum likelihood estimates of substitution distances from the in-frame alignments generated by GeneWise (see Implementation). Estimates were carried out separately for alignments of individual fragments as well as for a concatenated alignment of all fragments. Ambiguity characters were removed from the data by codeml before the maximum likelihood estimates were performed (clean_data = 1). Default values were used for the remaining parameters of codeml.
Estimates of sampling error for the concatenated alignments were produced using a non-parametric bootstrap resampling procedure, rather than from the output of codeml, as error estimates from codeml may not be reliable [37]. A sampling error sufficient to include 90% of the resampled data was estimated from 100 bootstrap replicates of the concatenated alignments.
Database and graphical interface
MySQL® [38] was used as the management system for the relational database. FRAGS was developed for Linux and FreeBSD systems. In addition to the MySQL® SQL interpreter, a number of graphical interfaces for data manipulation are available under Open Source licenses. The MySQL® Control centre and the database component of OpenOffice.org [39] are cross-platform GUI's that can be used for data manipulation. The databases can also be accessed via the phpMyAdmin [40] interface ([41]; Figure 2.) which can be used for querying, sorting and export of data from a web browser.

Screenshot of the user interface to the MySQL database. The screenshot shows a subset of the data available from the substitution statistics table and also illustrates the query functionality available from the web-based phpMyAdmin interface [41].
Results and Discussion
We used FRAGS to estimate substitution rates from alignments of human and chimpanzee coding sequences. Three different sources of chimpanzee sequence data were analysed separately, i.e. genomic sequence fragments, ESTs and annotated coding sequences from Genbank. We applied a maximum likelihood [16] as well as an approximate method [42] to infer substitution rates from individual alignments as well as from concatenated alignments consisting of all sequences of a specific type. Estimates derived from the approximate method were similar to the estimates derived from the maximum likelihood method and can be obtained from our web site [43]. Sequence quality scores were available for the genomic sequences and for these we report substitution rates derived from a quality filtered dataset and for the complete dataset.
Sequence pairs with zero substitutions at synonymous sites were omitted from substitution rate calculations for individual sequences, because K a /K s is undefined (Table 1). Estimates of K a /K s from concatenated sequences are not affected by individual sequences with K s = 0. Furthermore concatenated alignments have been shown to give more accurate estimates of genetic distances than estimates derived by averaging over individual genes or proteins [44]. In spite of this substitution rates derived from concatenated and individual sequences were very similar. The results presented in Table 2. are for concatenated alignments and represent the most accurate estimates of substitution rates that we have produced. Intermediate data generated prior to the final estimates of substitution rates can be accessed from the MySQL databases [43].
We have found that, at least in the case of human and chimpanzee, the source of the sequence data used has a larger impact on the estimate of substitution rates than the methodology employed. Our estimate of the non-synonymous substitution distance derived from concatenated alignments of genomic sequences was approximately twice the estimate derived from the EST sequences, even when the genomic sequences were filtered for sequence quality (K a = 0.012 and 0.006 for genomic and EST sequences respectively; Table 2.). The estimate of the synonymous substitution distance was also slightly elevated in the genomic sequences (K s = 0.033 compared to 0.026 for EST sequences), but, in spite of the increase in K s the estimate of K a /K s remained far higher in the genomic sequences compared to the ESTs (K a /K s = 0.37 and 0.24 respectively). The estimate of K a /K s that we derived from the Genbank coding sequences was far higher than the one obtained from either the ESTs or the quality filtered genomic sequences (K a /K s = 0.47, 0.37, 0.24 for the concatenated Genbank, genomic and EST sequences respectively; Table 2). These higher estimates could be a consequence of the over-representation of immune system proteins, with relatively high K a values, in this data set [45].
The average value of K a /K s that we obtained from the Genbank EST sequences using FRAGS (K a /K s = 0.24) was in good agreement with two previous estimates. The estimate of Hellmann and colleagues (K a /K s = 0.20; [45]) was based on 1226 coding sequences obtained from an EST dataset. A similar figure of 0.20 was obtained by Sakate et al. [46] from 226 consensus sequences derived from a slightly larger set of 5' cDNA sequence ends from brain, skin, and liver sequence libraries. However, in a study that focussed on differences in the rates of evolution between rearranged and unrearranged chromosomes, Navarro and Barton [7] have recently published an estimate of K a /K s derived from human and chimpanzee coding sequence orthologues from Genbank. Their estimate of 61% is considerably higher than any previous estimate and is also higher than the one derived here from a similar Genbank dataset.
The values of K a /K s derived from EST sequences, both here and previously [45], are significantly lower than the values derived from the other sources. Highly expressed genes are known to experience higher levels of purifying selection and to have lower average values of K a /K s [8, 9, 47, 48]. Hellmann et al. [45] have suggested that low values of K a /K s estimated from EST data may result from over-representation of highly expressed genes in cDNA libraries. Because highly expressed genes are over-represented in EST data, estimates of K a /K s derived from EST data should not be compared to estimates derived from genomic data or from a random set of complete coding sequences.
Methods used to align sequences have also been shown to contribute to the variability in estimates of substitution rates. In one comparison of mouse and human orthologues [49] sequence identity was found to be 5–6% higher than in an earlier estimate [50]. The higher estimate was proposed to result from the use of local rather than global alignments [49]. Our study uses identical alignment strategies for the different kinds of data studied and therefore results derived from the different data sources are more easily compared.
Errors in estimation of substitution can be introduced by inclusion of low-quality sequence (sequence that is more likely to be incorrectly identified by base calling software). Because sequencing errors are independent of translation, they tend to result in values of K a /K s that are closer to unity. The average value of K a /K s across all genes in a pair of organisms is normally far less than one and, as a result, sequence error normally increases the estimate of the average of K a /K s . The substantial decrease in the average value of K a /K s derived from quality-filtered BES sequences compared to the unfiltered sequences (Table 2) indicates that a significant amount of sequence error was removed through filtering. Interestingly, in spite of being error-prone, EST sequences provide the lowest values of K a /K s .
No publicly accessible quality data was available for the EST sequences, however, an upper-bound on the rate of sequencing error in the ESTs was estimated from the value of K a /K s . Because sequencing error is random, the number of synonymous sequence errors per synonymous site is expected to be equal to the number of non-synonymous sequence errors per non-synonymous site. In the worst-case all non-synonymous substitutions in the EST data are sequencing errors. For the EST data studied here this implied a maximum error-rate of 0.0044, or one sequencing error per 225 base-pairs. Given that non-synonymous substitutions do occur, this estimate is very conservative and the true error-rate is likely to be much lower than the upper-bound estimated here. A similar calculation, based on a comparison of the number of non-synonymous substitutions per non-synonymous site in the filtered and unfiltered genomic data, yielded a high estimate of one error per 100 base-pairs in the BES data.
It is important to emphasize that FRAGS is not intended to replace existing tree-based methods of inferring positive selection. When multiple homologous coding sequences are available for a gene of interest, tree-based methods are expected to be the most sensitive way to infer positive selection. Swanson et al. [51] have shown, however, that pairwise comparisons of fragmentary coding sequences can provide an initial screen for genes evolving under positive selection and can reveal differing selective pressures acting on different gene classes.
Future Prospects
In order to stimulate further modular development we will continue to integrate this software more tightly within the frameworks of existing projects (Biopython [24], BioSQL [52] and Ensembl [23]. This will assist in the accommodation of software components (plugins), that are 'wrapped' by modules that enforce adherence to the underlying database structure, making it more easily adaptable for use with different alignment strategies and methods of estimating substitution rates. Currently FRAGS uses codeml [33] to estimate substitution rates from the in-frame alignments, however we intend to extend the software to incorporate additional methods for estimating substitution rates that are released under Academic or Open Source licenses [53].
Module tests are being written to enable repeatability of 'experiments', and will be included in subsequent releases of the package. We hope that other researchers using this software will contribute to the development of these module tests, and that they may, in future, serve as the basis for standardizing these types of analyses.
In order to deal with larger quantities of sequence data, especially data derived from shotgun sequencing projects, time-consuming data processing stages of the software will be fully parallelized. Analysis of larger quantities of data will also benefit from clustering and assembly of sequence data in order to reduce redundancy of the alignments and improve speed.
Conclusions
Different methodologies and data sources have yielded significantly different estimates of the important evolutionary parameter K a /K s for pairs of organisms. The difficulty in obtaining an accurate estimate of K a /K s for orthologous sequences is particularly acute for organisms for which limited amount of fragmentary data is available, as different data types (genomic or transcript) and genes from different functional classes can yield very different estimates. For such organisms, the limited set of genes for which complete coding sequences are available are often highly functionally biased.
Our software facilitates significantly the task of estimating substitution rates of coding sequences from fragmentary data and enables the researcher to repeat the analysis for different datasets, or with different parameter values. This software can be of considerable utility to researchers who are performing initial characterisation of sequenced fragments from organisms for which a related completely sequenced organism is available. As the number of completely sequenced and well-annotated organisms in databases such as Ensembl increases, we anticipate that our software will become even more widely applicable.
Availability and requirements
Project name: FRAGS – A FRAGment Substitution rate estimation system
Project home page: http://sourceforge.net/projects/frags
Operating system(s): Linux, FreeBSD
Programming language: Python, SQL
Other requirements: Python 2.3 or higher, RepeatMasker, MUMmer 2.12 or higher, NCBI BLAST, GeneWise, PAML, MySQL, phpMyAdmin (optional), BioPython, MySQLdb, TextTools, Numeric Python. Data requirements: relevant EnsemblMart tables and 'Golden' path genomes, the UniVec_Core [27] database which is part of NCBI's VecScreen [54]
License: BSD
Any restrictions to use by non-academics: Consult the BSD License.
Abbreviations
- BAC:
-
Bacterial Artificial Chromosome
- BES:
-
BAC end sequence
- EST:
-
Expressed Sequence Tag
- cDNA:
-
Complementary DNA
- SQL:
-
Structured Query Language
- CDS:
-
Coding Sequence
References
Yang Z, Bielawski JP: Statistical methods for detecting molecular adaptation. Trends Ecol Evol 2000, 15: 496–503. 10.1016/S0169-5347(00)01994-7
Varki A: A chimpanzee genome project is a biomedical imperative. Genome Res 2000, 10: 1065–70. 10.1101/gr.10.8.1065
Gagneux P, Varki A: Genetic differences between humans and great apes. Mol Phylogenet Evol 2001, 18: 2–13. 10.1006/mpev.2000.0799
Nembaware V, Crum K, Kelso J, Seoighe C: Impact of the presence of paralogs on sequence divergence in a set of mouse-human orthologs. Genome Res 2002, 12: 1370–6. 10.1101/gr.270902
Kondrashov FA, Rogozin IB, Wolf YI, Koonin EV: Selection in the evolution of gene duplications. Genome Biol 2002, 3: RESEARCH0008. 10.1186/gb-2002-3-2-research0008
Lynch M, Conery JS: The evolutionary fate and conseuqences of duplicate genes. Science 2000, 290: 1151–1155. 10.1126/science.290.5494.1151
Navarro A, Barton NH: Chromosomal speciation and molecular divergence – accelerated evolution in rearranged chromosomes. Science 2003, 300: 321–4. 10.1126/science.1080600
Duret L, Mouchiroud D: Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci U S A 1999, 96: 4482–7. 10.1073/pnas.96.8.4482
Duret L, Mouchiroud D: Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol 2000, 17: 68–74.
Liberles DA, Schreiber DR, Govindarajan S, Chamberlin SG, Benner SA: The adaptive evolution database (TAED). Genome Biol 2001, 2: RESEARCH0028.
Li WH, Wu CI, Luo CC: A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol Biol Evol 1985, 2: 150–174.
Pamillo P, Bianchi NO: Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mol Biol Evol 1993, 10: 271–281.
Li WH: Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol 1993, 36: 96–9.
Conant GC, Wagner A: GenomeHistory: a software tool and its application to fully sequenced genomes. Nucleic Acids Res 2002, 30: 3378–86. 10.1093/nar/gkf449
Muse SV, Gaut BS: A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol 1994, 11: 715–24.
Goldman N, Yang Z: A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 1994, 11: 725–36.
Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, Gilbert J, Hammond M, Huminiecki L, Kasprzyk A, Lehvaslaiho H, Lijnzaad P, Melsopp C, Mongin E, Pettett R, Pocock M, Potter S, Rust A, Schmidt E, Searle S, Slater G, Smith J, Spooner W, Stabenau A, Stalker J, Stupka E, Ureta-Vidal A, Vastrik I, Clamp M: The Ensembl genome database project. Nucleic Acids Res 2002, 30: 38–41. 10.1093/nar/30.1.38
Clamp M, Andrews D, Barker D, Bevan P, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, Gilbert J, Hammond M, Hubbard T, Kasprzyk A, Keefe D, Lehvaslaiho H, Iyer V, Melsopp C, Mongin E, Pettett R, Potter S, Rust A, Schmidt E, Searle S, Slater G, Smith J, Spooner W, Stabenau A, Stalker J, Stupka E, Ureta-Vidal A, Vastrik I, Birney E: Ensembl 2002: accommodating comparative genomics. Nucleic Acids Res 2003, 31: 38–42. 10.1093/nar/gkg083
Ensembl[http://www.ensembl.org]
Duret L, Mouchiroud D, Gouy M: HOVERGEN: a database of homologous vertebrate genes. Nucleic Acids Res 1994, 22: 2360–5.
Lee Y, Sultana R, Pertea G, Cho J, Karamycheva S, Tsai J, Parvizi B, Cheung F, Antonescu V, White J, Holt I, Liang F, Quackenbush J: Cross-referencing eukaryotic genomes: TIGR Orthologous Gene Alignments (TOGA). Genome Res 2002, 12: 493–502. 10.1101/gr.212002
Python[http://www.python.org]
Ensembl[http://www.ensembl.org]
Biopython[http://www.biopython.org]
RepeatMasker[http://repeatmasker.genome.washington.edu/RM/webrepeatmaskerhelp.html]
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215: 403–10. 10.1006/jmbi.1990.9999
UniVec Core[ftp://ftp.ncbi.nih.gov/pub/UniVec/UniVec_Core]
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 1998, 8: 186–94.
Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL: Alignment of Whole Genomes. Nucleic Acids Res 1999, 27: 2369–76. 10.1093/nar/27.11.2369
Delcher AL, Phillippy A, Carlton J, Salzberg SL: Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 2002, 30: 2478–83. 10.1093/nar/30.11.2478
Birney E, Thompson JD, Gibson TJ: PairWise and SearchWise: finding the optimal alignment in a simultaneous comparison of a protein profile against all DNA translation frames. Nucleic Acids Res 1996, 24: 2730–9. 10.1093/nar/24.14.2730
Birney E, Durbin R: Dynamite: a flexible code generating language for dynamic programming methods used in sequence comparison. Proc Int Conf Intell Syst Mol Biol 1997, 5: 56–64.
Yang Z: PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 1997, 13: 555–6.
Riken Chimpanzee Sequencing Initiative[http://hgp.gsc.riken.go.jp]
Fujiyama A, Watanabe H, Toyoda A, Taylor TD, Itoh T, Tsai SF, Park HS, Yaspo ML, Lehrach H, Chen Z, Fu G, Saitou N, Osoegawa K, de Jong PJ, Suto Y, Hattori M, Sakaki Y: Construction and analysis of a human-chimpanzee comparative clone map. Science 2002, 295: 131–4. 10.1126/science.1065199
UCSC Golden Path ftp site[ftp://genome.cse.ucsc.edu/goldenPath/]
Yang Z: Phylogenetic Analysis by Maximum Likelihood (PAML). 2002.
MySQL[http://www.mysql.com]
OpenOffice.org[http://www.openoffice.org]
phpMyAdmin[http://www.phpmyadmin.net]
phpMyAdmin interface to Frags databases[http://www.sanbi.ac.za/~estienne/phpMyAdmin/index.php]
Yang Z, Nielsen R: Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol 2000, 17: 32–43.
Additional tables and data[http://www.sanbi.ac.za/~estienne/data_tables.html]
Nei M, Xu P, Glazko G: Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. Proc Natl Acad Sci U S A 2001, 98: 2497–502. 10.1073/pnas.051611498
Hellmann I, Zollner S, Enard W, Ebersberger I, Nickel B, Paabo S: Selection on human genes as revealed by comparisons to chimpanzee cDNA. Genome Res 2003, 13: 831–7. 10.1101/gr.944903
Sakate R, Osada N, Hida M, Sugano S, Hayasaka I, Shimohira N, Yanagi S, Suto Y, Hashimoto K, Hirai M: Analysis of 5'-end sequences of chimpanzee cDNAs. Genome Res 2003, 13: 1022–6. 10.1101/gr.783103
Gouy M, Gautier C: Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res 1982, 10: 7055–74.
Sharp PM, Li WH: Codon usage in regulatory genes in Escherichia coli does not reflect selection for 'rare' codons. Nucleic Acids Res 1986, 14: 7737–49.
Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES, Mouse Genome Sequencing Consortium: Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420: 520–62. 10.1038/nature01262
Makalowski W, Boguski MS: Synonymous and nonsynonymous substitution distances are correlated in mouse and rat genes. J Mol Evol 1998, 47: 119–21.
Swanson WJ, Clark AG, Waldrip-Dail HM, Wolfner MF, Aquadro CF: Evolutionary EST analysis identifies rapidly evolving male reproductive proteins in Drosophila. Proc Natl Acad Sci 2001, 98: 7375–9. 10.1073/pnas.131568198
BioSQL[http://www.obda.open-bio.org]
Open Source Licenses[http://www.opensource.org/licenses/index.php]
NCBI VecScreen[http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html]
Acknowledgements
ES would like to acknowledge the South African NRF funding (GUN 2050323) received during the course of this research. CS is grateful to the South African National Bioinformatics Network. Thanks to Victoria Nembaware, Faisel Mosoval and Rüdiger Bräuning for constructive comments. This research would not have been possible without the Open Source community.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ES conceived and coded the software and prepared the manuscript draft. CS supervised the research and the development of the software, and assisted in preparation of the manuscript draft. WH provided user feedback, reviewed the development of the system, edited the draft and provided guidance as to the applicability and design of the system. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Swart, E.C., Hide, W.A. & Seoighe, C. FRAGS: estimation of coding sequence substitution rates from fragmentary data. BMC Bioinformatics 5, 8 (2004). https://doi.org/10.1186/1471-2105-5-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-5-8