
Abstract
Background
Once specific genes are identified through high throughput genomics technologies there is a need to sort the final gene list to a manageable size for validation studies. The triaging and sorting of genes often relies on the use of supplemental information related to gene structure, metabolic pathways, and chromosomal location. Yet in disease states where the genes may not have identifiable structural elements, poorly defined metabolic pathways, or limited chromosomal data, flexible systems for obtaining additional data are necessary. In these situations having a tool for searching the biomedical literature using the list of identified genes while simultaneously defining additional search terms would be useful.
Results
We have built a tool, BEAR GeneInfo, that allows flexible searches based on the investigators knowledge of the biological process, thus allowing for data mining that is specific to the scientist's strengths and interests. This tool allows a user to upload a series of GenBank accession numbers, Unigene Ids, Locuslink Ids, or gene names. BEAR GeneInfo takes these IDs and identifies the associated gene names, and uses the lists of gene names to query PubMed. The investigator can add additional modifying search terms to the query. The subsequent output provides a list of publications, along with the associated reference hyperlinks, for reviewing the identified articles for relevance and interest. An example of the use of this tool in the study of human prostate cancer cells treated with Selenium is presented.
Conclusions
This tool can be used to further define a list of genes that have been identified through genomic or genetic studies. Through the use of targeted searches with additional search terms the investigator can limit the list to genes that match their specific research interests or needs. The tool is freely available on the web at http://prostategenomics.org[1], and the authors will provide scripts and database components if requested mdatta@mcw.edu
Similar content being viewed by others


Background
The use of high throughput genomic and proteomic technologies has resulted in the creation of large datasets of differentially expressed genes and proteins. Even after further statistical analysis these datasets may be sufficiently large such that the validation of all possibilities are outside the resources of the investigators. In these situations there is a need to efficiently triage and sort the dataset to identify the genes of highest interest to the scientists. In many situations the experimental design takes advantage of specific biological samples available to the investigator. Thus the investigator often has additional scientific data and personal insight that may be helpful in guiding the examination of the genomic output. Yet many tools developed to sort and add supplemental information to the genomic data use global processes such as metabolic pathway mapping [2–4], promoter binding [5], chromosomal location, or Gene function/GO terminology [6, 7], and thus may not leverage the additional knowledge of the investigator. This leaves the scientist with the time consuming task of manually sorting through the dataset with the appended data to identify genes that may provide useful information. Here we present an automated tool, BEAR GeneInfo, which allows a user to simultaneously query the biomedical literature with lists composed of multiple gene names while using additional tailored search terms. The associated output of biomedical references is provided for further review and subsequent query modification, allowing the user to follow-up on interesting trends in the data, thus maximizing the potential of the genomic data. This tool joins the list of additional tools including PubMatrix [8], MatchMiner [9], and XplorMed [10] that are enhancing the ability of scientists to perform integrated searches of large complex datasets, and by doing so identify new trends and associations within the scientific data.
Implementation
Interface and database design
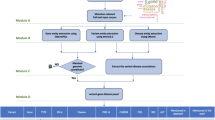
BEAR GeneInfo consists of five components (figure 1); A web based interface for user data input and results display, a CGI script for user data processing and results display, an underlying database to store gene related information, Perl scripts for database maintenance and data updates, and link-outs to NCBI. The database architecture was created in Rational Rose using an object oriented design and implemented in Oracle 9 (figure 2). The database was populated through downloads and updates derived from Unigene [11], Locuslink [12] and the UCSC Genome Browser [13]. Additional gene names were identified through MatchMiner [14] based on queries of individual Unigene IDs. These MatchMiner queried gene names were designated as gene name aliases, and used to populate the Unigene (gene name) alias table (figure 2). In order to limit the number of GenBank accession numbers displayed in association with a given Unigene or Locuslink ID, SOURCE [15] was queried with the respective IDs and their defined "representative GenBank accession number" was downloaded [16]. The associated GenBank accession numbers, Unigene IDs, and Locuslink IDs were preserved within the tables for use in the querying of user uploaded gene lists.

GeneInfo user interface

GeneInfo database in Rational Rose
Pathways for database queries to derive gene name lists
BEAR GeneInfo allows for the querying of PubMed with gene names and gene symbols based on the entry of either Unigene ID, GenBank accession number, or LocusLink ID, and as such pathways for the identification of gene names based on each type of data entry were created. If the user enters in Unigene IDs which have been retired BEAR GeneInfo uses a CGI subroutine to query Unigene and updates the Ungiene ID, using the new Unigene ID to continue the query. When the list of genes is provided as either GenBank accession numbers or Unigene IDs the queries utilize gene names associated with the Unigene IDs, and also identify gene symbols associated with the corresponding Unigene and Locuslink IDs. After these have been identified BEAR GeneInfo adds the gene names derived from MatchMiner based on a query with the Unigene ID. All these gene names and symbols are used to search PubMed. When the list of genes is provided as LocusLink IDs the same process is used, only one uses the official LocusLink gene name instead of the official Unigene gene name. Due to the complexity of the gene naming conventions, we have sought to offer different methods for searching with different gene name options. The lists of gene names are filtered for general "user unfriendly" elements such as parentheses or descriptive words such as "ESTs weakly similar to" or "homo sapiens".
PubMed queries based on the gene name lists
The textual complexity of the gene names can lead to very different search results. For this reason we have incorporated a series of filters into BEAR GeneInfo for use in choosing the format of the gene name searches. There are two choices; a default filter that uses processes we have developed in the construction of the BEAR GeneSifter tool (X. Wen, G. Zhou, H. Liu and M. Datta, unpublished), and a gene search term splitter. The default filter takes a given gene name, and removes any parenthesis i.e. "(", "[", ")", or "]" or semicolons ";" and colons ":" from the text, replaces commas (,) with a Boolean AND term, and places quotation marks (") around the text groups for searching. The gene search term splitter takes the gene name and also removes any parenthesis i.e. "(", "[", ")", or "]" or semicolons ";" and colons ":" from the text, and uses comma replacement with the Boolean AND term, but then takes each search word in the gene name and separates them, and searches each individual term against the user added search terms. The results are then combined and reported for the gene name. In each case an additional general "nonsense" filter is applied, such that if a single search string returns more than 2,500 references, the term is deemed to be "nonsense" and its references are not included in the query results. Figure 3 demonstrates an example of the text filters for a given gene. Of note, different numbers of references can be retrieved based on the choice of search text filters. In addition to the gene names, the user may enter additional search terms based on their specific knowledge of the experimental system. These terms can be entered using Boolean logic, and will be searched with respect to each individual gene name that has been uploaded in an "AND" or "NOT" format. Queries of the NCBI PubMed database are restricted to nights (9 pm – 5 am EST) and parsed over time to limit the user time on the PubMed database. More effective queries for high throughput users would use a locally downloaded copy of PubMed. Results are then sent to the user by email, which directs the user to a web site for receipt of their output data. The output data is stored for ten days on the site before it is deleted. The resultant output is provided to the user in two formats, http and txt. In each case the user receives a list of the genes and the user's additional query specifications along with the number of associated biomedical references. In the http file the user receives hyperlinks to the references while in the txt file the user receives a list of the PubMed IDs for further analysis. In the http file additional hyperlinks are supplied for each individual search term within a gene ID query, allowing a user to examine the effect each search term has on the final compiled list of references for that gene ID (figure 4). The resultant output data can be examined by the user and used to refine subsequent queries that are performed.

Example output based on the two different word filtering options. The default filter searches the gene names with the users search terms. The gene search term splitter takes each gene name and separates the terms, combining each with the user search terms, and then combining the results. The default filter results in a higher number of references than the gene search term filter.

Example output evaluated by individual search queries. In the example the gene ID generated six gene name queries linked to the user generated search term "antioxidant". The individual queries generated different numbers of references, the results of which were combined in the final result. Hyperlinks to each query link to PubMed, allowing the user to determine which queries contribute specific references to the final result.
Results
Example use: Selenium treatment of prostate cancer cells
The experimental data presented here is a component of a published study described in the companion article by Schlicht et al. (ref-pending). The methods for the derivation of the data are present in that study. Portions of the data are reanalyzed here and presented not as new data, but as an example of how the BEAR GeneInfo tool can be used. A more detailed study of the data can be obtained from the Schlicht et al. reference (ref-pending). Selenium is currently being investigated as a potential chemopreventive in prostate cancer. While clinical trials have suggested some potential benefit, the mechanism of action of Selenium is unclear, and is an area of active research. Using gene expression microarray technology a dataset reflecting the differential expression of the human metastatic prostate cancer cell line PC3 was developed after treatment with Selenium for either 6 hours (1123 genes) or 5 days (1053 genes). Twenty-four of the genes demonstrated differential gene expression with respect to Selenium at both 6 hours and 5 days. Using this set of 24 genes, additional prioritization and sorting was attempted using the BEAR GeneInfo tool (table 1). A list of the 24 genes was uploaded into BEAR GeneInfo as either GenBank accession numbers, Unigene IDs, or Locuslink IDs, and run in comparison with additional user provided search terms including "prostate cancer", "Selenium", "metastasis", "apoptosis" and "antioxidant". The majority of the genes (average 13.8/24 genes, median 15/24 genes, range 10/24 to 16/24 genes) returned no references for the associated search terms. The average number of references was 5583 references per gene, although the median was 0 references per gene, with a range of 0 to 57,693. Of note, the ability to generate lists of gene names was similar for different gene IDs. The Unigene ID and GenBank accession number yielded similar results. Five genes returned more than 1,000 references for each search. Upon review these gene names identified common phrases or word groups, and as such were disregarded. The evaluation of the returned references was made possible through a search term query, in which each search term used for a given gene ID is hyperlinked to the related references (figure 4). Examining the associated references with the search terms allows the user to identify inappropriately identified references, and thus disregard specific search results. BEAR GeneInfo searches were repeated for the list of 24 genes using the two types of search filters (table 2). In addition, the genes names were individually searched with the associated query terms in PubMed using the EndNote reference program and then number of returned references counted. All the associated references were read to evaluate the relevance of the publications with regard to the search terms. This review confirmed that the return of large (greater than 1,000) references was due to misrepresented words or phrases in genes with complex naming features. A comparison of the search filters revealed different results for the default and gene search term splitter filters, with the latter returning larger numbers of references, but not affecting the most significant numbers of references. These differences did not affect the relevance of the references identified. The results were similar to that obtained by manually searching PubMed. In all three cases where BEAR GeneInfo returned over 1,000 references, no references were identified through manual searching, confirming the non-specific nature of the automated results. Thus it appears that the most interesting genes would be characterized as having limited, but not too much, associated data in PubMed. This would reflect genes where early studies had been attempted, but for which extensive studies have not already been completed (thus leaving more to examine). Alternatively they may represent genes where connections to the user's study have yet to be identified. There was no situation where BEAR GeneInfo failed to identify references while a manual search using Endnote was able to return references. In table 2 using BEAR GeneInfo three genes met the arbitrary criteria of having references for both prostate cancer and the majority of the additional search terms; peroxiredoxin-1, surfeit-5, and lysozyme homolog. Manual searching only identified two of these three genes. The third, the lysozyme homolog, was identified through the use of multiple gene name queries generated by BEAR GeneInfo, and as such identified the significance of this type of protein with respect to prostate cancer and selenium. Thus it appears that the use of an automated system for data mining of the biomedical literature was at least as accurate as manual data mining, and offers significant advantages in time savings. The identification of peroxiredoxin-1 was interesting as there are references studying this gene in prostate cancer in both the rat and the human, a design in the initial study [17, 18]. Peroxiredoxin-1 was also studied in Selenium, but had not been examined with respect to Selenium in prostate cancer. Lysozyme has been used to study cellular differentiation in tumor diagnosis for years, and has been described in variant forms of prostate cancer [19–21]. Its identification here may imply a role not just in tumor identification, but also in function. In a search across multiple terms in (table 1) the identification of an F-Box protein is intriguing as other F-Box proteins have already been implicated in both prostate and prostate cancer cellular functions [22–24]. F-Box proteins have yet to be implicated in Selenium action, but have been identified based on their roles in antioxidant protection [25]. Thus the resulting identification of 274 query-specific references associated with three genes allowed for the focused examination of these genes, and reduced the list of genes to be used in subsequent validation studies by one eighth (87.5%) and provided interesting genes for further validation studies.
Discussion
The use of high throughput techniques in biology often results in large dataset outputs where data needs to be triaged and further analyzed. In the process of data triage one hopes to leverage as much information as possible to allow for the correct sorting of genes. This process needs access to comprehensive databases of information on genes so that opportunities, in particular poorly defined or ignored gene targets, are not missed. A rich source of gene data is the corpus of biomedical literature present in PubMed [26]. This corpus provides a large dataset of gene information that can be mined for supplementary information related to genomic data analysis. Approaches have sought to create comprehensive datasets that identify all the relationships present in the biomedical literature between genes, genes and drug targets, and along metabolic pathways [9, 27, 28]. These approaches have used either curated or automated data compilation. The curated databases have been of great value, but are limited by their level of coverage necessitated by the labor-intensive nature of the process. Data mining techniques have been used to identify associated information within the biomedical literature (XplorMed [10]) or between the biomedical literature and lists of gene and drug names (MedMiner [5] and PubGene [29]). The former tool does not allow for the use of large gene names lists while the latter two tools are limited in their inability to modify search terms based on user-specific knowledge, unpublished results, or the unique characteristics of the biological experiment. BEAR GeneInfo is a web based tool designed to allow a more flexible user-driven data mining process. A recent tool that does offer the ability to combine a list of gene names with additional search terms is the PubMatrix system for multiplex literature mining [8]. PubMatrix offers unique value in its integration of genes with user derived search terms, but is different from BEAR GeneInfo in its use of gene names. Both PubMatrix and BEAR GeneInfo can be used to identify references from NCBI PubMed, when queried by gene namelists, while BEAR GeneInfo can derive gene namelists from GenBank Accession numbers, Unigene or LocusLink ID lists. The use of compound search terms, as one uses in the PubMed query interface is common to PubMatrix and BEAR GeneInfo, although the integration of search terms is based on the Boolean "AND" operator in PubMatrix, while BEAR GeneInfo offers both "AND" or "OR" Boolean operators. Both tools are limited by the quality of reference curation and do not use statistical filtering of query results. BEAR GeneInfo will return the search result in a web page display of the PubMed records for review by investigators, while PubMatrix utilizes more graphical based data presentation. The information it provides include two link buttons, one for direct link to the PubMed references, and one for a separate page displaying the URLs for each matched PubMed reference. The search results also display the gene name, and hyperlinked GenBank accession number, Unigene ID and LocusLink ID. BEAR GeneInfo provides an interface for the user to modify the searches with user-driven Boolean search terms along with the gene so the result will yield more focused information for researchers to review. BEAR GeneInfo is available at: http://www.prostategenomics.org
Use of the BEAR GeneInfo system: Selenium treatment in prostate cancer
The true value in the use of a tool such as BEAR GeneInfo is the ability for the user to prioritize and sort genes identified through high throughput methods such as microarray studies. By allowing the user to define additional search terms based on the biological experiment focused searches can be undertaken that leverage the knowledge of the user and allow for the tracing of specific links not always identified through metabolic pathways, chromosomal location, or simple differential expression. This is demonstrated by our identification of differentially expressed genes of interest with respect to Selenium treatment of prostate cancer cells. A weakness of this process is the limitation of the use of known biological links within a system based on the scientific data available to the user. This does limit the possibility of unbiased gene discovery, but this process can still be exercised through the examination of differentially expressed genes by purely statistical methods, without associated gene data present in the biomedical literature and based on previous scientific data. One of the main values of this process may be the ability to link diverse scientific fields and disciplines, with the only limitation being the ability of the user to imagine a possible link when querying the tool.
Conclusions
Here we have presented a tool, BEAR GeneInfo, that can be used to further define a list of genes that have been identified through genomic or genetic studies. Through the use of targeted searches with additional search terms the investigator can filter a list of genes, in the process prioritizing the ones that match their specific research interests. In addition, BEAR GeneInfo provides an initial point for launching further refinements of text based query tools that examine the biomedical literature. The tool is freely available on the web at http://prostategenomics.org[1], and the authors will provide scripts and database components if requested
Availability and requirements
The BEAR GeneInfo database is currently available as a web based tool at http://prostategenomics.org[1] but the components can be obtained after contacting the author at mdatta@mcw.edu. The database is built in Oracle 9 and the scripts are all Perl based.
References
Prostategenomics.org: . http://www.prostategenomics.org
Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR: GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet 2002, 31: 19–20. 10.1038/ng0502-19
Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR: MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol 2003, 4: R7. 10.1186/gb-2003-4-1-r7
Voit EO, Radivoyevitch T: Biochemical systems analysis of genome-wide expression data. Bioinformatics 2000, 16: 1023–1037. 10.1093/bioinformatics/16.11.1023
Vadigepalli R, Chakravarthula P, Zak DE, Schwaber JS, Gonye GE: PAINT: a promoter analysis and interaction network generation tool for gene regulatory network identification. Omics 2003, 7: 235–252. 10.1089/153623103322452378
Draghici S, Khatri P, Bhavsar P, Shah A, Krawetz SA, Tainsky MA: Onto-Tools, the toolkit of the modern biologist: Onto-Express, Onto-Compare, Onto-Design and Onto-Translate. Nucleic Acids Res 2003, 31: 3775–3781. 10.1093/nar/gkg624
Khatri P, Draghici S, Ostermeier GC, Krawetz SA: Profiling gene expression using onto-express. Genomics 2002, 79: 266–270. 10.1006/geno.2002.6698
Becker KG, Hosack DA, Dennis G., Jr., Lempicki RA, Bright TJ, Cheadle C, Engel J: PubMatrix: a tool for multiplex literature mining. BMC Bioinformatics 2003, 4: 61. 10.1186/1471-2105-4-61
Bussey KJ, Kane D, Sunshine M, Narasimhan S, Nishizuka S, Reinhold WC, Zeeberg B, Ajay W, Weinstein JN: MatchMiner: a tool for batch navigation among gene and gene product identifiers. Genome Biol 2003, 4: R27. 10.1186/gb-2003-4-4-r27
Perez-Iratxeta C, Perez AJ, Bork P, Andrade MA: Update on XplorMed: A web server for exploring scientific literature. Nucleic Acids Res 2003, 31: 3866–3868. 10.1093/nar/gkg538
Unigene: . http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene
Locuslink: . http://www.ncbi.nlm.nih.gov/LocusLink/
Browser UCSC Genome: . http://genome.ucsc.edu/
MatchMiner: . http://discover.nci.nih.gov/matchminer/html/index.jsp
SOURCE: . http://source.stanford.edu/cgi-bin/source/sourceSearch
Diehn M, Sherlock G, Binkley G, Jin H, Matese JC, Hernandez-Boussard T, Rees CA, Cherry JM, Botstein D, Brown PO, Alizadeh AA: SOURCE: a unified genomic resource of functional annotations, ontologies, and gene expression data. Nucleic Acids Res 2003, 31: 219–223. 10.1093/nar/gkg014
Shen C, Nathan C: Nonredundant antioxidant defense by multiple two-cysteine peroxiredoxins in human prostate cancer cells. Mol Med 2002, 8: 95–102. 10.1007/s00894-002-0075-z
Tam NN, Gao Y, Leung YK, Ho SM: Androgenic Regulation of Oxidative Stress in the Rat Prostate: Involvement of NAD(P)H Oxidases and Antioxidant Defense Machinery during Prostatic Involution and Regrowth. Am J Pathol 2003, 163: 2513–2522.
Adlakha H, Bostwick DG: Paneth cell-like change in prostatic adenocarcinoma represents neuroendocrine differentiation: report of 30 cases. Hum Pathol 1994, 25: 135–139. 10.1016/0046-8177(94)90268-2
Frydman CP, Bleiweiss IJ, Unger PD, Gordon RE, Brazenas NV: Paneth cell-like metaplasia of the prostate gland. Arch Pathol Lab Med 1992, 116: 274–276.
Weaver MG, Abdul-Karim FW, Srigley J, Bostwick DG, Ro JY, Ayala AG: Paneth cell-like change of the prostate gland. A histological, immunohistochemical, and electron microscopic study. Am J Surg Pathol 1992, 16: 62–68.
Lu L, Schulz H, Wolf DA: The F-box protein SKP2 mediates androgen control of p27 stability in LNCaP human prostate cancer cells. BMC Cell Biol 2002, 3: 22. 10.1186/1471-2121-3-22
Yang G, Ayala G, Marzo AD, Tian W, Frolov A, Wheeler TM, Thompson TC, Harper JW: Elevated Skp2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p27 and PTEN and with reduced recurrence-free survival. Clin Cancer Res 2002, 8: 3419–3426.
Shim EH, Johnson L, Noh HL, Kim YJ, Sun H, Zeiss C, Zhang H: Expression of the F-box protein SKP2 induces hyperplasia, dysplasia, and low-grade carcinoma in the mouse prostate. Cancer Res 2003, 63: 1583–1588.
Swaroop M, Gosink M, Sun Y: SAG/ROC2/Rbx2/Hrt2, a component of SCF E3 ubiquitin ligase: genomic structure, a splicing variant, and two family pseudogenes. DNA Cell Biol 2001, 20: 425–434. 10.1089/104454901750361488
McEntyre J, Lipman D: PubMed: bridging the information gap. Cmaj 2001, 164: 1317–1319.
Weinstein JN: Searching for pharmacogenomic markers: the synergy between omic and hypothesis-driven research. Dis Markers 2001, 17: 77–88.
Sun LZ, Ji ZL, Chen X, Wang JF, Chen YZ: ADME-AP: a database of ADME associated proteins. Bioinformatics 2002, 18: 1699–1700. 10.1093/bioinformatics/18.12.1699
Jenssen TK, Laegreid A, Komorowski J, Hovig E: A literature network of human genes for high-throughput analysis of gene expression. Nat Genet 2001, 28: 21–28. 10.1038/88213
Prostategenomics.org: . http://www.prostategenomics.org
Prostategenomics.org: . http://www.prostategenomics.org
Acknowledgements
The authors would like to acknowledge the support of NCI grant R21CA098032 to MWD and the Milwaukee Breast Cancer Showhouse Foundation Award to MWD in support of this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
M.W.D. was responsible for the conception and implementation of this project in association with P.J.T. H.L., X.W., and G.Z. were actively involved in the programming, database construction, and testing of the software. M.S. and M.H. were responsible for spotted cDNA construction, hybridization, and experimental analysis along with M.W.D. All the authors reviewed and accepted the final version of the paper.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zhou, G., Wen, X., Liu, H. et al. B.E.A.R. GeneInfo: A tool for identifying gene-related biomedical publications through user modifiable queries. BMC Bioinformatics 5, 46 (2004). https://doi.org/10.1186/1471-2105-5-46
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-5-46