
Abstract
Background
Spectral counting methods provide an easy means of identifying proteins with differing abundances between complex mixtures using shotgun proteomics data. The crux spectral-counts command, implemented as part of the Crux software toolkit, implements four previously reported spectral counting methods, the spectral index (SI N ), the exponentially modified protein abundance index (emPAI), the normalized spectral abundance factor (NSAF), and the distributed normalized spectral abundance factor (dNSAF).
Results
We compared the reproducibility and the linearity relative to each protein’s abundance of the four spectral counting metrics. Our analysis suggests that NSAF yields the most reproducible counts across technical and biological replicates, and both SI N and NSAF achieve the best linearity.
Conclusions
With the crux spectral-counts command, Crux provides open-source modular methods to analyze mass spectrometry data for identifying and now quantifying peptides and proteins. The C++ source code, compiled binaries, spectra and sequence databases are available athttp://noble.gs.washington.edu/proj/crux-spectral-counts.
Similar content being viewed by others


Background
Existing methods for differential proteomics (reviewed by[1]) fall into two categories: spectral counting methods that rely on counting the number of spectra that map to a given protein across multiple experiments, and peptide chromatographic peak intensity methods that use the area under the peptide precursor ion peak as a measure of peptide abundance. In principle, methods based on mass spectrometry peak areas are potentially much more accurate, but these methods require highly reproducible liquid chromatography as well as accurate methods for chromatographic alignment and identification of peaks within the profile spectra. In contrast, spectral counting methods are straightforward to employ and have been shown to correctly detect known differences between samples[2], which contributes to their wide use.
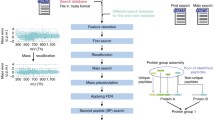
The command line tool crux spectral-counts implements four popular spectral counting methods: the spectral index (SI N )[3], the exponentially modified protein abundance index (emPAI)[4], the normalized spectral abundance factor (NSAF)[5], and the distributed normalized spectral abundance factor (dNSAF)[6]. The crux spectral-counts command is integrated within the Crux software toolkit, which provides actively maintained open-source methods to identify and now quantify peptides and proteins from shotgun mass spectrometry datasets. Crux supports a variety of input spectra formats, and the tools can easily be incorporated into proteomic analysis pipelines, such as the Trans-Proteomic Pipeline (TPP)[7]. Finally, the modular design of Crux allows improvements to one part of the toolkit to be propagated through downstream analyses.
Currently, several software packages offer spectral counting protein quantification methods[8]. ProteoIQ (http://www.bioinquire.com) and Scaffold[9] are commercial software products that post-process results from a variety of database search programs. Freely available tools such as APEX[10], emPAI calc[11], and PepC[12] each offer a single spectral counting method. Table1 compares the features of six software spectral counting tools. Crux offers more spectral counting methods than other tools and is the only method to provide peptide-level in addition to protein-level counts.
Using crux spectral-counts, we compared and contrasted the reproducibility and linearity of the four spectral counting methods. Our experiments suggest that the NSAF metric provides the most reproducible protein quantification. In contrast, our linearity experiments show that SI N and NSAF provide the best performance, with dNSAF providing intermediate performance and emPAI yielding the worst linearity.
The contributions of this paper are thus two-fold: we describe a performance comparison of the reproducibility and linearity of the SI N , emPAI, NSAF, and dNSAF protein quantification methods, and we provide to the proteomics community a flexible, open source spectral counting software tool.
Implementation
Software
The crux spectral-counts command is implemented as part of the Crux proteomics software toolkit[13]. The toolkit is implemented in C++ as a single binary that supports commands for database searching and a variety of downstream analyses[14–18].
The crux spectral-counts command takes as input a protein database in FASTA format and a collection of peptide-spectrum matches (PSMs) produced by a database search procedure. The PSMs may be in Crux’s tab-delimited text format, PeptideProphet’s PepXML or mzIdentML[19]. To compute the SI N score, a set of spectra must also be provided as input in MS2, mzXML, or mgf format. By default, crux spectral-counts will select the PSMs in the input by a user modifiable threshold of q-value ≤ 0.01.
For each protein with at least one spectral count, the program then computes the NSAF, dNSAF, emPAI, or the SI N score. The NSAF metric is defined as
where N is the protein index, s N is the number of spectra matched to protein N, L N is the length of protein N, and n is the total number of proteins in the input database.
The dNSAF metric is given by
where is the spectral count for the peptides uniquely mapping to protein N, is the spectral count of degenerate peptide j (out of the protein’s k degenerate peptides) mapped to protein N, and dj,N is the distribution factor of peptide shared counts, defined by the equation
The metric emPAI is defined as
where is the number of unique peptides observable for protein N and is the number of unique peptides observed for protein N.
Finally, the SI N score is calculated using
where p N is number of unique peptides in protein N, s j is the number of spectra assigned to peptide j, and i k is the total fragment ion intensity of spectrum k. Analogous scores can also be computed for each peptide, rather than for each protein. A detailed description of the peptide-level scoring metrics is available in the on-line documentation. As output, crux spectral-counts produces a tab-delimited file listing proteins and their corresponding counts, in reverse sorted order.
The crux spectral-counts command also computes a parsimonious set of proteins, using the greedy set cover approach used by IDPicker[20]. Users thus have the option of considering spectral counts only for proteins within the parsimonious set.
Data Collection
For the reproducibility experiments, proteins were extracted from the cochlear nucleus of the developing mouse brain at postnatal day 7 and postnatal day 21. Two biological replicates were generated for each age by dissecting out the cochlear nuclei from two separate mice at each age. One of the 21-day mice was used to generate two samples, thereby providing a technical replicate in addition to a biological replicate. The samples prepared from the chicken brain were derived from nucleus laminaris, an auditory region in the brain stem. Samples were taken from the dorsal (D) and ventral (V) regions of this area. For each region, two biological replicates were generated, and one of those replicates was also subjected to technical replication. Each sample was digested with trypsin and subjected to liquid chromatography followed by tandem mass spectrometry.
For the linearity experiments, we used eight samples that represent a dilution curve of 48 known proteins synthesized by Sigma (UPS1,http://www.sigmaaldrich.com). These data sets are mixtures (Std1–Std8) of the C. elegans lysate at equal concentrations and the 48 proteins, diluted by a two-fold in each successive standard. Std 8 has the lowest concentration of the known proteins (6 fmol) and Std 1 has the highest concentration (870 fmol).
All three data sets are publicly available athttp://noble.gs.washington.edu/proj/crux-spectral-counts.
Data analysis
The fragmentation spectra from the experiments were searched against their respective mouse, chicken, or the C. elegans+UPS1 protein database using crux sequest-search followed by crux q-ranker, with the default parameters. crux spectral-counts was applied to the peptide-spectrum matches (PSMs) that received q-values ≤ 0.01. The resulting data sets for the mouse and chicken replicates are summarized in Additional file1: Table S1, and the UPS1 dilution curve data sets are summarized in Additional file1: Table S2.
Results
Testing reproducibility between replicates
To investigate the reproducibility of the four spectral count methods, we analyzed mass spectrometry data from technical and biological replicates from chicken and mouse samples. We then produced a scatter plot for each pair of biological or technical replicates and computed the corresponding Spearman correlation. For these comparisons, proteins identified in only one of the two datasets were ignored. Figure1 shows sixteen such plots, corresponding to one biological and one technical replicate for chicken and mouse, respectively. The complete collection of 76 plots is provided as Additional file1: Figures S1–S2. From these analyses, as summarized in Table2, we draw two primary conclusions. First, the spectral counts are generally reproducible: the mean correlation value across all 76 pairs is 0.867, and the minimum correlation is 0.719. Second, reassuringly, the technical replicates produce higher correlations than the biological replicates: the mean correlation among 24 pairs of technical replicates is 0.885, whereas the corresponding value for the 52 pairs of biological replicates is 0.859 (two-tailed Wilcoxon rank-sum test p-value=0.026).

Reproducibility of spectral counts across biological and technical replicate experiments. Each plot compares either the SI N , emPAI, NSAF or dNSAF measure for proteins that were reproducibly identified across two replicate experiments. For visualization purposes, the counts are plotted on a logarithmic scale. The number in the lower right corner of each panel is the corresponding Spearman correlation and the number in the upper left is the number of datapoints compared.
To test whether the observed differences in correlations among the four metrics are significant, we applied a Wilcoxon signed-rank test to paired sets of correlations. With four metrics, there are six possible paired comparisons. Figure2 shows the results of this analysis, where one metric attaining a significant increase (using a Bonferroni p-value of 0.05/6=0.008333) over another is indicated by a directed edge. From this graph we conclude that, for the biological and technical replicates, NSAF yields significantly more reproducible quantification values than SI N , dNSAF and emPAI. Our reproducibility results agree with Colaert et al., who claim that NSAF is more reproducible than SI N and emPAI[21]. However, in contrast to our results, Griffen et al. report better reproducibility across replicates for SI N compared to NSAF[3].

Comparison of spectral counts across replicates. This graph summarizes the statistical analysis of the reproducibility measurements. An edge leading out from node A to node B indicates a statistically significant improvement in reproducibility for method A relative to method B.
Testing linear response for protein abundance across samples
To determine the linear response of each of the spectral count metrics, we analyzed mass spectra from a dataset of samples that form a dilution curve of forty-eight proteins with known amounts spiked into a C. elegans lysate. We performed linear regression between each protein spectral count and the associated amounts across the dilution curve samples. For this analysis, we only included proteins that obtain a positive spectral count in three or more of the data sets, which results in a comparison of forty-two proteins across the four metrics. We then carried out a Wilcoxon signed rank test analysis separately on the average correlation, R2, and the mean percent error (MPE). The results of these tests (Figure3) are fairly consistent with one another: NSAF significantly outperforms dNSAF, and dNSAF and SI N significantly outperform emPAI.

Comparison of spectral counts across UPS1 dilution curve. This graph summarizes the statistical analysis of the linearity measurements. Two types of analysis were performed, using the linear regression correlation, R2 and mean percent error (MPE) for the C. elegans + UPS1 dilution curve dataset. An edge leading out from node A to node B indicates a statistically significant improvement in linearity for method A relative to method B.
Colaert et al. (2011) claim that SI N is more accurate than both NSAF and emPAI[21], but we find evidence only to support the former claim, even though our experiments employ a wider dynamic range of protein abundance (6.7–20 fmol versus 6–870 fmol) and more data sets (two versus eight). Based on our experiments, we conclude that NSAF or SI N are the methods of choice for ensuring an accurate linear response between a protein’s change in abundance across different samples.
It is worth noting that Griffin et al. (2010) observe a good linear fit between SI N and protein quantification. However, their evaluation methodology fits a single line to all of the SI N values from many proteins, whereas we have fit a separate line for each protein. This difference reflects our belief that spectral counting methods are most useful as measures of the relative abundance of a single protein between two experiments. We did not test the claim that SI N provides an accurate absolute protein abundance metric.
Discussion
Overall, our experiments suggest a relative ordering of spectral counting methods according to their reproducibility and the linearity of their response, but we can only speculate as to the reasons for the ranking that we observe. For example, we note that NSAF outperforms the emPAI metric in both of our experiments. The emPAI measure takes into account the least information—not only does it ignore fragment ion intensities, but emPAI also fails to account for the length of the protein. Apparently, this relatively simple approach is insufficient to accurately estimate protein abundance. The relative performance of NSAF and SI N , on the other hand, is less clear: NSAF yields more reproducible results than SI N but the two methods are statistically indistinguishable with respect to linearity. The main difference between SI N and the other three metrics is that SI N is the only metric that takes into account the intensities of the fragment ion peaks. In this sense, SI N goes a bit beyond the strict definition of “spectral counting.” Our experiments do not support the claim that such intensity information is valuable for quantification. However, the conflicting results of our study and Collaert et al., on the one hand, versus Griffin et al. on the other hand, suggests perhaps that further comparison of these methods is warranted.
An additional direction for future work involves quantifying the linearity and reproducibility of proteins in a segregated fashion according to protein abundance. For example, visual inspection of Figure1 suggests that perhaps the SI N measure yields more reproducible counts for high abundance proteins, with a corresponding decrease in reproducibility as the abundance drops. Arguably, in many studies, such low abundance proteins are of the greatest interest; hence, it may be worthwhile to investigate in a systematic fashion the extent to which either the linearity or the reproducibility of a given spectral counting measure varies as a function of protein abundance.
Conclusions
Quantifying protein amounts in mass spectrometry by spectral counting is a simple and robust method for measuring the relative change of protein amounts across different samples; however, many different algorithms exist for assigning a score to each identified protein. Using crux spectral-counts, we compared and contrasted four spectral counting methods with respect to their reproducibility across replicates and their linear response relative to protein abundance. Crux provides a flexible, easy to use open source tool for performing protein quantification using spectral counting.
Availability and requirements
Project name: Crux tandem mass spectrometry analysis software
Project home page: http://noble.gs.washington.edu/proj/crux
Operating systems: Linux, MacOS, Windows + Cygwin
Programming language: C++
Other requirements: Crux has no requirements to install the binary version under Linux or MacOS. On Windows, Crux requires Cygwin. To compile Crux requires a c++ compiler, cmake, and Subversion.
License: Apache
Any restrictions to use by non-academics: None
References
Wang M, You J, Bemis KG, Tegeler TJ, Brown DP: Label-free mass spectrometry-based protein quantification technologies in proteomic analysis. Brief Funct Genomic Proteomic 2008, 7(5):329–339. 10.1093/bfgp/eln031
Searle BC, Tabb DL, Falkner JA, Kowalak JA, Meyer-Arendt K, Rudnick PA, Seymour SL, Lane WS: iPRG2009 study: testing for differences between complex samples in proteomics datasets. Poster at ABRF2009 2009, 28(1):83–89.
Griffin NM, Yu J, Long F, Oh P, Shore S, Li Y, Koziol JA, Schnitzer JE: Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat Biotechnol 2010, 28: 83–89. 10.1038/nbt.1592
Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M: Exponentially Modified Protein Abundance Index (emPAI) for Estimation of Absolute Protein Amount in Proteomics by the Number of Sequenced Peptides per Protein. Mol Cell Proteomics 2005, 4(9):1265–1272. 10.1074/mcp.M500061-MCP200
Paoletti AC, Parmely TJ, Tomomori-Sato C, Sato S, Zhu D, Conaway RC, Conaway JW, Florens L, Washburn MP: Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc Nat Acad Sci USA 2006, 103(50):18928–18933. 10.1073/pnas.0606379103
Zhang Y, Wen Z, Washburn MP, Florens L: Refinements to Label Free Proteome Quantitation: How to Deal with Peptides Shared by Multiple Proteins. Anal Chem 2010, 82(6):2272–2281. 10.1021/ac9023999
Keller A, Eng J, Zhang N, Li X, Aebersold R: A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol 2005, 1: 2005.0017.
Neilson KA, Ali NA, Muralidharan S, Mirzaei M, Mariani M, Assadourian G, Lee A, van Sluyter SC, Haynes PA: Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11(4):535–553. 10.1002/pmic.201000553
Searle BC: Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 2010, 10(6):1265–1269. 10.1002/pmic.200900437
Braisted J, Kuntumalla S, Vogel C, Marcotte E, Rodrigues A, Wang R, Huang ST, Ferlanti E, Saeed A, Fleischmann R, Peterson S, Pieper R: The APEX Quantitative Proteomics Tool: Generating protein quantitation estimates from LC-MS/MS proteomics results. BMC Bioinformatics 2008, 9: 529. 10.1186/1471-2105-9-529
Shinoda K, Tomita M, Ishihama Y: emPAI Calc-for the estimation of protein abundance from large-scale identification data by liquid chromatography-tandem mass spectrometry. Bioinformatics 2010, 26(4):576–577. 10.1093/bioinformatics/btp700
Heinecke NL, Pratt BS, Vaisar T, Becker L: PepC: proteomics software for identifying differentially expressed proteins based on spectral counting. Bioinformatics 2010, 26(12):1574–1575. 10.1093/bioinformatics/btq171
Park CY, Klammer AA, Käll L, MacCoss MP, Noble WS: Rapid and accurate peptide identification from tandem mass spectra. J Proteome Res 2008, 7(7):3022–3027. 10.1021/pr800127y
Käll L, Storey JD, MacCoss MJ, Noble WS: Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J Proteome Res 2008, 7: 29–34. 10.1021/pr700600n
Spivak M, Weston J, Tomazela D, MacCoss MJ, Noble WS: Direct maximization of protein identifications from tandem mass spectra. Mol Cell Proteomics 2012, 11(2):M111.012161. [PMC3277760] [PMC3277760] 10.1074/mcp.M111.012161
Klammer AA, Park CY, Noble WS: Statistical calibration of the Sequest XCorr function. J Proteome Res 2009, 8(4):2106–2113. 10.1021/pr8011107
Hsieh E, Hoopmann M, Maclean B, Maccoss M: Comparison of database search strategies for high precursor mass accuracy MS/MS data. J Proteome Res 2009.
McIlwain S, Draghicescu P, Singh P, Goodlett DR, Noble WS: Detecting cross-linked peptides by searching against a database of cross-linked peptide pairs. J Proteome Res 2010, 9(5):2488–2495. [PMC20349954] [PMC20349954] 10.1021/pr901163d
Jones AR, Eisenacher M, Mayer G, Kohlbacher O, Siepen J, Hubbard SJ, Selley JN, Searle BC, Shofstahl J, Seymour SL, Julian R, Binz PA, Deutsch EW, Hermjakob H, Reisinger F, Griss J, Vizcaíno JA, Chambers M, Pizarro A, Creasy D: The mzIdentML data standard for mass spectrometry-based proteomics results. Mol Cell Proteomics 2012, 11(7):M111.014381. 10.1074/mcp.M111.014381
Zhang B, Chambers MC, Tabb DL: Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J Proteome Res 2007, 6(9):3549–3557. 10.1021/pr070230d
Colaert N, Vandekerckhove J, Gavaert K, Martens L: A comparison of MS2-based label-free quantitative proteomic techniques with regards to accuracy and precision. Proteomics 2011, 11(6):1110–1113. 10.1002/pmic.201000521
Acknowledgements
NIH awards R01 EB007057, P41 GM103533 and R01 DC03829. The authors acknowledge Karl Schweighofer for his input on the crux spectral-counts tool and the anonymous reviewers for many helpful suggestions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The chicken and mouse samples were provided by ER’s lab, and the LC-MS/MS data were collected by members of the MM lab. MB prepared and collected the UPS1 + C. elegans dilution sample datasets. MM wrote the initial code for crux spectral-counts and the initial draft of the manuscript. SM finished the coding of crux spectral-counts and the final draft with WSN. All authors revised and approved the final manuscript.
Electronic supplementary material
12859_2012_5738_MOESM1_ESM.pdf
Additional file 1:Supplementary Information. Supplementary Tables 1 and 2 and Suplementary Figures 1 and 2 are provided as quantify-supplement.pdf. (PDF 13 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
McIlwain, S., Mathews, M., Bereman, M.S. et al. Estimating relative abundances of proteins from shotgun proteomics data. BMC Bioinformatics 13, 308 (2012). https://doi.org/10.1186/1471-2105-13-308
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-13-308