
Abstract
Background
The androgen receptor (AR) is a member of the nuclear receptor (NR) superfamily of ligand-inducible DNA transcription factors, and is the major mediator of male sexual development, prostate growth and the pathogenesis of prostate cancer. Cell and gene specific regulation by the AR is determined by availability of and interaction with sets of key accessory cofactors. Ski-interacting protein (SKIP; SNW1, NCOA62) is a cofactor shown to interact with several NRs and a diverse range of other transcription factors. Interestingly, SKIP as part of the spliceosome is thought to link mRNA splicing with transcription. SKIP has not been previously shown to interact with the AR.
Results
The aim of this study was to investigate whether SKIP interacts with the AR and modulates AR-dependent transcription. Here, we show by co-immunoprecipitation experiments that SKIP is in a complex with the AR. Moreover, SKIP increased 5α-dihydrotestosterone (DHT) induced N-terminal/C-terminal AR interaction from 12-fold to almost 300-fold in a two-hybrid assay, and enhanced AR ligand-independent AF-1 transactivation. SKIP augmented ligand- and AR-dependent transactivation in PC3 prostate cancer cells. Live-cell imaging revealed a fast (half-time=129 s) translocation of AR from the cytoplasm to the nucleus upon DHT-stimulation. Förster resonance energy transfer (FRET) experiments suggest a direct AR-SKIP interaction in the nucleus upon translocation.
Conclusions
Our results suggest that SKIP interacts with AR in the nucleus and enhances AR-dependent transactivation and N/C-interaction supporting a role for SKIP as an AR co-factor.
Similar content being viewed by others


Background
SNW/SKIP proteins, which include human Ski-interacting protein (SKIP; SNW1, NCOA62) and its yeast homologue the essential splicing factor Prp45 (Pr e-mRNA Processing 45) [1], are phylogenetically highly conserved [2], and important during early development [3, 4]. SKIP was identified by its interaction with the proto-oncogenes, v-Ski and c-Ski [5]. In addition to pre-mRNA splicing in the spliceosome [6], SKIP appears to have multiple other functions in transcription [2]. It acts as a transcriptional co-regulator of a number of key cellular signalling molecules, such as of CREB binding protein (CBP)/ p300, the nuclear co-repressor (N-CoR) and silencing mediator for retinoic acid and thyroid hormone receptors (SMRT) [7]. It also interacts with a large range of DNA binding proteins, including Smad2 and 3 proteins of the TGF-β pathway [8], and proteins involved in MyoD and Notch signalling [2] and may be involved in Wnt signalling [9]. A recent study suggests SKIP regulates the cell cycle arrest factor p21 (Cip1) and subsequent effects on p53-dependent DNA cell damage [10]. This appeared to involve recruitment of SKIP to the p21 promoter where SKIP plays a critical role for splicing and p21 gene expression, suggesting a role of SKIP in cancer cell apoptosis.
The fundamental effects of SKIP in transcriptional regulation are supported by its co-regulatory effect on nuclear hormone receptors, including estrogen receptor [11], the Vitamin D receptor (VDR) and Retinoid X Receptor (RXR) [12–15], which it antagonistically regulates in association with SIRT1 [16]. More recently SKIP was shown to associate with P-TEFb, c-myc and Menin to act on the HIV-1 promoter [17, 18].
The androgen receptor (AR) is a member of the nuclear hormone receptor superfamily that regulates male sexual development, and is a major player in the pathogenesis and progression of prostate cancer [19–22]. Upon binding to its natural ligands testosterone and 5α-dihydrotestosterone (DHT), the AR becomes phosphorylated and translocates into the nucleus, where it binds as a homodimer to canonical nuclear receptor inverted repeat DNA response elements to activate target gene transcription [23]. A comprehensive ChIP-on-chip analysis of AR-responsive elements (ARE) on chromosomes 21 and 22 however suggests that the majority of the 90 identified binding sites are non-canonical, with isolated half-sites, head-to-head, head-to-tail and direct repeat AREs [24].
The AR contains distinct structural domains also found in other nuclear receptor superfamily members. The amino (N)-terminal transactivation domain, which is also termed ligand-independent activation-function-1 domain (AF-1), is followed by a DNA-binding domain (DBD) that is linked via a hinge region to a carboxy (C)-terminal ligand-binding domain (LBD), which contains the activation function-2 domain (AF-2) [25, 26]. Ligand binding induces interaction between the N-terminal and C-terminal domains of AR [27, 28]. This interdomain rearrangement slows ligand dissociation and AR degradation [29]. Moreover, mutations in the AR AF-2 domain, which are associated with partial or complete androgen insensitivity syndrome, also abrogate N/C-interaction in vitro, suggesting that N/C-interaction is functionally important in vivo[27]. The AR intrinsic N-terminal FXXLF (residues 23–27) and WXXLF (residues 433–437) motifs form amphipathic α-helices that stabilise the N/C-interaction, by binding to a hydrophobic pocket at the C-terminus [30]. Importantly, the intrinsic (F/W)XXLF motifs compete with the similar, extrinsic LXXLL-motif from p160-family type I co-regulators for the hydrophobic pocket on the AR C-terminus [31]. Examples for type 1 co-regulators are steroid receptor co-activators (SRC1 and SRC3), transcriptional intermediary factor 2 (TIF2) and amplified in breast cancer-1 (AIB1) [25, 32].
While AF-1 mediates hormone-independent constitutive transactivation when artificially isolated, the AF-2 is inactive in the absence of hormone, but required for strong, ligand-dependent activity in androgen-dependent prostate cancer cells [33–36]. This may be due to the fact that even in the absence of ligand the extended AF-1 fragment (including the DBD) assumes a similar reticular or speckled distribution to nuclear foci, as the full-length receptor with its co-regulators, while the isolated AF-2 is distributed homogenously in the nucleoplasm, even if ligand is added [37]. This speckle-pattern distribution is typical for markers of various nuclear compartments, such as speckles, nucleoli and cajal bodies [38, 39]. Nuclear speckles are sites of pre-mRNA splicing and SKIP was found among other splicing proteins enriched in nuclear speckles [40].
In this study, we demonstrate that SKIP interacts with the AR. We show that SKIP acts in multiple ways, by augmenting AR AF-1-dependent activity as a classical type I co-activator, while it also enhanced AR N/C-interaction and AR-dependent transcription in prostate cancer cells.
Results
Mammalian one-hybrid data show that SKIP augments ligand-independent AR transcription
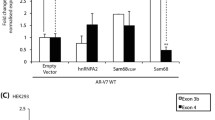
SKIP modulates the transcriptional activity of a variety of nuclear hormone receptors. We therefore tested, whether SKIP can also act as a co-regulator of androgen receptor (AR), which is critically involved in prostate cancer [19, 22]. To address, whether SKIP influences AR-mediated transactivation, we used a mammalian one-hybrid assay, which assesses the transcriptional co-activating property of a candidate protein (e.g. SKIP) on a potential interaction partner (e.g. AR) fused to the GAL4 DNA binding domain. The presence of the co-activator increases binding of the fusion protein to GAL4-binding sites on the reporter plasmid, which then drives luciferase transcription [41]. We expressed in HEK293 cells a fragment comprising the N-terminal ligand-independent activation-function-1 (AF-1) domain of AR (residues 1–555) fused to the GAL4 DNA binding domain (AR1-555) with and without SKIP. In the absence of the AR1-555 fusion protein, overexpression of SKIP did not induce reporter gene expression compared to control cells co-transfected with empty vectors (Figure 1). Consistent with the ligand-independent transcriptional activity of the AR AF-1 domain [42], transfection of the AR1-555 fusion protein induced a 10-fold increase in basal, ligand-independent luciferase activity compared to control levels. This AR-AF-1 transcriptional activity was increased ~5-fold by overexpression of SKIP (Figure 1). These data suggest a co-stimulatory activity of SKIP by a functional interaction with the N-terminal ligand-independent activation-function-1 (AF-1) domain of AR.

SKIP augments AR-AF-1 dependent transactivation. Mammalian one-hybrid analysis in HEK293 cells that were transiently co-transfected with the GAL45E1bTATA-luciferase reporter (250 ng/well), with AR 1–555 N-terminal domain in pM-GAL4DBD expression vector (125 ng) or empty pM-GAL4DBD vector (125 ng), as well as with SKIP-pCGN (100 ng) or empty pCGN (100 ng). To allow for comparison to the N/C-interaction transfection experiments (Figure 2) the empty vector pVP16AD containing the VP16 transactivation domain was also transfected (125 ng). Data are relative to the control sample (no SKIP, no AR-AF1) as means ± SEM calculated from four independent luciferase reporter experiments, as described in Materials and Methods. Statistically significant difference between indicated means was determined using Student’s t-test.
SKIP facilitates interaction of AR N- and C-termini
AR transcriptional activity is modulated by ligand-mediated interdomain interaction of its N- and C-termini [27, 30]. To address, whether SKIP facilitates AR N- and C-terminal, ligand-dependent interaction, the mammalian two-hybrid system was used [43, 44]. In the mammalian two-hybrid system, potentially interacting protein fragments are fused to either the GAL4 DNA binding domain (GAL4-DBD) or the V16 transactivation domain. Interaction of the fused proteins brings the GAL4-DBD and V16 domain into a complex, which increases transcription of luciferase from a GAL4 responsive promoter. Residues 1–538 of AR were fused to the VP16 activation domain and C-terminal AR-LBD residues 642–917 to the GAL4 DNA binding domain (AR642-917) to detect N/C-terminal interaction. Modulation of this interaction by SKIP would alter reporter gene expression in HEK293.
In the absence of SKIP, the AR N- and C-terminal, ligand-dependent interaction resulted in a significant increase in relative luciferase activity by 12-fold (Figure 2 column 4). SKIP also enhanced ligand-dependent activation of the AR-LBD 642–917 construct by 9-fold (Figure 2 column 7). Overexpression of SKIP markedly increased ligand-dependent transcriptional activity mediated by the AR N- and C-terminal constructs to ~300-fold over control levels (Figure 2 column 8 compared with column 4). Thus SKIP facilitates the ligand-induced, interdomain interaction of the N- and C-termini of AR or processes downstream of this rearrangement, augmenting AR transcriptional activity by an order of magnitude.

SKIP increases N- and C-terminal interaction of AR. Mammalian two-hybrid analysis in HEK293 cells that were transiently co-transfected with the GAL45E1bTATA-luciferase reporter (250 ng/well), and as indicated, GAL4-DBD-fusion constructs and VP16-fusion constructs of the C- or N-terminus of AR (GAL4DBD-AR 642–917 (125 ng each) and pVP16-AR1-538 (125 ng), respectively) with SKIP-pCGN (100 ng) or empty pCGN (100 ng). Reporter activity was determined in cells with (black bars) and without (grey bars) 1 nM DHT stimulation overnight for 16 hours. Response of cells that did not co-express SKIP (columns 1–4), was markedly lower than that of cells expressing SKIP (columns 5–8). Note the discontinuous y-axis for the relative activity. Data are relative to the control sample (empty vectors without DHT stimulation) as means ± SEM calculated from five independent luciferase reporter experiments, as described in Materials and Methods. Statistically significant difference between indicated means was determined using Student’s t-test.
SKIP increases AR-dependent transcription in prostate cancer cell lines
To provide further evidence that SKIP is a co-activator of AR function, the effects of SKIP overexpression on AR–dependent transcription were assessed using the androgen-responsive mouse mammary tumour virus (MMTV) MMTV-luc and the prostate-specific antigen (PSA) PSA-luc luciferase reporter plasmids in a prostate cancer cell line. We used the AR-negative PC3 cell line, where we overexpressed WT AR. In these cells SKIP overexpression augmented basal ligand-independent MMTV reporter activity ~6-fold. Additional stimulation with the AR ligand DHT (0.01 to 10 nM) increased activity ~18-fold over control levels (no SKIP, empty pSG5 vector only, no DHT), without any apparent dose-dependence (Figure 3). On the other hand, SKIP alone induced only a 2-fold increase in basal PSA-reporter activity. This was however markedly and dose-dependently increased by DHT (8-60- fold over control levels), as compared to PC3-cells without co-transfected SKIP (up to 10-fold over control levels).

SKIP augments AR-dependent transcription in prostate cancer cells. Luciferase reporter transactivation assays were performed in PC3 prostate cancer cells using transiently transfected MMTV- and PSA-luciferase reporters (250 ng/well) with SKIP-pSG5 expression plasmid (200 ng) or empty vector pSG5 (200 ng) as indicated. As PC3 cells have no endogenous AR expression, pCMV-AR WT expression plasmid (25 ng/well) was also transfected in these cells. Data are relative to the no SKIP and no ligand control, which was set as one and expressed as mean ± SEM calculated from 3 independent experiments.
These results suggest that SKIP augments DHT-induced MMTV- and PSA-reporter activity several-fold, with a response pattern that depends on the reporter gene construct.
SKIP co-immunoprecipitates with androgen receptor
SKIP has been shown to interact with a number of nuclear hormone receptors via its helical repeat domain [2]. We therefore investigated, whether SKIP is also in a complex with AR in a co-immunoprecipitation experiment (Figure 4).

SKIP and androgen receptor co-immunoprecipitate. Co-immunoprecipitation experiments in COS-7 cells co-transfected with full-length AR WT plasmid and HA-SKIP-plasmid as indicated and described in Materials and Methods. AR-bound and HA-positive (left) or SKIP-bound and AR-positive (N20-antibody) proteins (right) are shown. rIgG stands for rabbit IgG and represents a negative control. Experiments were carried out in presence or absence of 1 nM DHT, as indicated. Lanes 5 and 10 show blotted lysates as input controls for the immunoprecipitation. Approximate molecular weights are as indicated on the left. Arrow shows AR-fragment positive controls, while arrow-head points to HA-SKIP positive controls. Blots are representative of three independent repeats.
We co-expressed AR with SKIP in COS-7 cells and precipitated bound proteins from the cell lysates using anti-AR or anti-HA antibodies (Figure 4). HA tagged SKIP proteins (~62 kD) were specifically detected in AR-immunoprecipitates in similar amounts, irrespective of whether cells were DHT treated (Figure 4 lanes 1 and 2). Consistent with AR co-immunoprecipitating HA-SKIP, the inverse experiment showed that AR protein (~110 kD) was specifically detected in HA-SKIP immunoprecipitates (Figure 4 lane 6 and 7).
In conclusion, these data suggest that AR and SKIP interact in a specific complex.
FRET experiments reveal interaction of AR and SKIP in the nucleus
Finally, we wanted to monitor AR and SKIP interaction in intact living cells and provide evidence for their direct interaction, using Förster/fluorescence resonance energy transfer (FRET) imaging. We transiently co-expressed AR, which was C-terminally tagged with ECFP (AR-ECFP) as fluorescent donor, and SKIP, N-terminally tagged with EYFP (EYFP-SKIP) as an acceptor in BHK cells (Figure 5). The EYFP-SKIP construct localized to small nuclear structures of various sizes (Figure 5A; Additional file 1). For our FRET experiments, we selected for those cells that contained larger SKIP-positive nuclear structures. Note, that even under serum starvation, cells exhibited various degrees of nuclear co-localization of AR-ECFP and EYFP-SKIP (data not shown). Using donor-dequenching FRET experiments on maximally DHT stimulated BHK cells, we measured a high FRET efficiency of 57±3%, as compared to 11±2% background (Figure 5B).

AR interacts with SKIP after nuclear translocation on SKIP-positive nuclear speckles. (A,B) Donor dequenching and (C-E) sensitised acceptor emission FRET imaging data of AR-ECFP and EYFP-SKIP co-expressed in BHK cells. (A) BHK cells were transiently transfected with AR-ECFP and EYFP-SKIP. Before being fixed with 4% PFA, cells were stimulated for 2.5 min with 10 nM DHT at room temperature. The acceptor was then bleached and the increase of the donor-fluorescence was monitored in the donor channel, using confocal imaging. (B) The average FRET efficiency of AR-ECFP/EYFP-SKIP (8 images, 24 speckles), E%=57±3% (mean±SEM) was significantly higher (P<0.001, 2-tailed Student’s t-test), than that of the control FRET-pair, ECFP/EYFP-SKIP, E%=11±2% (mean±SEM) (3 images, 9 speckles) on EYFP-SKIP positive nuclear speckles. This is in line with a strong interaction between AR and SKIP on these structures. (C) Representative time series of serum starved BHK cells, transiently transfected with AR-ECFP and EYFP-SKIP that were stimulated with 10 nM DHT (+DHT). Using confocal imaging, nuclear translocation of AR-ECFP and FRET-changes (FRET2) were measured over time. Time points are shown in FRET2 images. The approximate boundary of the nucleus is indicated in the first AR-ECFP image. (D) The nuclear translocation index of data in C was calculated using the largest speckle (arrow in first image) and a region in the cytoplasm (asterisk) (apparent translocation half-time, τ0.5=213±9 s). (E) The change of the FRET2 signal (FRET2) and the FRET2 signal normalized with the unprocessed acceptor signal (FRET2/Ac) were averaged over the whole nucleus for each time point. Both FRET signals reached their maximum at ~ 129 s after DHT addition, corresponding to ~ 32% translocation. Subsequently the FRET signal decreased, as the fluorescence of EYFP-SKIP disappeared.
In order to gain further insight into the temporal characteristics of this interaction, we followed the DHT induced nuclear translocation of AR-ECFP to EYFP-SKIP positive structures in live BHK cells. We used sensitised acceptor emission FRET imaging with fully crosstalk corrected FRET images [45]. When stimulated with DHT (Figure 5C), AR-ECFP accumulated on EYFP-SKIP positive structures of the nucleus within 3 minutes (Figure 5D), with concomitant increase in the FRET signal (Figure 5E; Additional file 2). On individual SKIP-positive nuclear structures, the FRET signal first increased at the periphery and then progressed towards the centre. Thereafter, the FRET signal decreased, due to a drop in the EYFP-SKIP signal, which was not due to photobleaching caused by progressive scanning of the sample (data not shown).
Additional file 2:The video shows the donor- (left, green), calculated FRET- (middle, colour coded), and the acceptor-images (right, yellow). DHT was added in frame 1 (time given in middle panel) at 10 nM. The donor images show a DHT induced increase in AR-ECFP translocation from the cytoplasm to the EYFP-SKIP positive nuclear speckles. This leads to a transient increase in FRET on the speckles, suggesting an increased number of AR/SKIP interactions. The FRET signal subsequently decreased, as the signal of EYFP-SKIP diminished. (MOV 1 MB)
In conclusion, FRET-experiments provide additional evidence that a direct and transient interaction of AR and SKIP occurs within the nucleus, which increases within minutes after DHT-stimulated nuclear translocation of AR.
Discussion
In this study we provide evidence that SKIP acts as a co-regulator of AR transcription. Using mammalian one- and two-hybrid-experiments, we showed that SKIP augments AR AF-1 ligand-independent transcriptional activity and AR ligand-dependent AF-1 and AF-2 (AR N/ C-terminal) transcriptional interaction. Moreover, SKIP augmented AR-dependent transcription with two ARE-containing reporters in prostate cancer PC3 cells. Immunoprecipitation and FRET-experiments showed that AR interacts with SKIP and that the AR translocates to the nucleus within minutes after ligand stimulation to interact with SKIP.
FRET detects proximities of 3–7 nm for ECFP/EYFP-FRET pairs [46], which is a distance that can in most cases be regarded to report on direct interactions, considering that the size of the fluorescent proteins is already 2 nm × 4 nm [47]. The observed high FRET efficiency between AR-ECFP/ EYFP-SKIP on the speckle patterned SKIP-positive structures is therefore in agreement with a direct interaction of these proteins.
Our FRET data are supported by our one- and two-hybrid data, which suggest that SKIP can activate both the N- and C-terminus of AR. Therefore, FRET-fluorophores on the C-terminus of AR and N-terminus of SKIP may come into very close proximity, despite the relatively large size of both proteins. The strong increase of FRET upon DHT-stimulated nuclear translocation of AR-ECFP to EYFP-SKIP positive structures that resemble nuclear speckles, can be explained by the fact that an excess of the acceptor, EYFP-SKIP, leads to high FRET-levels in a molecular complex, where the donor-acceptor-ratio is 1: >1 [48].
Nuclear speckles form a functional compartment within the nucleus that include splicing factors. Active gene transcription coupled with pre-mRNA splicing is thought to occur at the periphery of the splicing factor compartment [49–51]. It was therefore intriguing to see that the FRET between SKIP and AR increased from the outside to the inside of the speckle-like SKIP-positive structures. However, as fluorescently tagged proteins had to be overexpressed, care must be taken when relating the magnitude of the FRET with the affinity of the two proteins under physiological conditions. On the other hand, overexpression does not seem to induce random interaction in our experiments, as the interaction only significantly increased a) after DHT-stimulation and b) involving a translocation from the cytoplasm to the nucleus. We cannot say, whether AR is in addition targeted to other nuclear compartments.
It is of interest that the AR AF-1 domain has been shown to interact within nuclear speckles with another splicing factor ANT-1 (homologous to yeast splicing factor Prp6p) [52]. This distribution resembles the AR-SKIP co-localization that we have observed (Figure 5). Thus AR interaction with SKIP and SKIP enhancement of AR AF-1 transcriptional activity is similar to AR/ ANT-1-dependent augmentation of transcription and splicing. Therefore, further studies will be required to determine if SKIP, like ANT-1 may recruit AR into a transcription-splicing coupled machinery.
Interestingly, the SKIP-EYFP signal disappeared completely ~16 minutes after DHT mediated translocation of AR to the nucleus, suggesting AR-mediated degradation or masking of SKIP. Loss of the SKIP-EYFP signal started soon after the maximum FRET was reached, raising the possibility that a regulated degradation is triggered at high AR concentrations. The AR has been previously linked to proteasome-mediated degradation, by its interaction with ubiquitin protease USP10 [53], the ubiquitin ligase ARNIP [54] and sensitivity of AR transactivation to proteasome inhibitors [55].
AR transcriptional activity is dependent on its N-terminal and C-terminal domains, which if artificially isolated can act in a ligand-independent (AF-1) and ligand-dependent (AF-2) manner, respectively [23, 34]. Immunoprecipitation and FRET-data allow us to explain the transactivation data by the direct interaction of SKIP with AR, however details of the exact mechanism are still unclear. We showed that the AR N-terminal domain was sufficient to engage SKIP to increase transactivation. It is possible that in a transcriptionally active complex, such as that induced by the N-terminal AR fragment, SKIP is engaged in the recruitment of general co-regulators such as N-CoR and p300 [7]. In line with the latter interpretation, it was previously found that AR ligand-dependently interacts with the nuclear receptor co-repressor, N-CoR [56], which also interacts with SKIP [7].
Moreover, our two-hybrid data indicate that SKIP bi-functionally interacts with the N- and C-termini of AR, or at least facilitates this N/C- interaction. Interestingly, Saitoh et al. [37] observed something similar for the effect of the co-regulator CBP, which is also required for nuclear foci or speckle formation. In addition, SRC1 also modulates N/C-terminal interactions [57], however, this and its co-regulator activities are handled by distinct parts of the protein with an apparently higher significance for its LXXLL-motif independent interaction [58]. Moreover, co-regulators were shown to modulate the N/C-interaction, in a cell type dependent manner. Correspondingly, their effect on the N/C-interaction did not always correlate with their effects on AR-mediated transactivation or cell growth, which may highlight the importance of specific microenvironments that influence this interaction [57]. This may be explained by the assumption that the AF-1 may be the major transactivator under normal physiological conditions, while type I co-regulators come into play, if their concentration is high, such as observed in recurrent prostate cancer [28].
Conclusions
In conclusion, our data suggest SKIP as a novel AR co-activator that enhances its AF-1 function and interdomain-interaction, as well as AR-dependent transcription. The diverse network of interactions shown for SKIP, in particular with nuclear hormone receptors, explains its impact on several signalling pathways involved in growth and development, including possibly in AR-dependent prostate cancer cell growth.
Methods
Reporter gene assays
HEK293 cells (origin: H. sapiens: embryonal kidney cells); PC3 (origin: H. sapiens: prostate cancer cells) were maintained in medium with 10% foetal calf serum and plated 24–36 hours before transfection into 24 well plates at a density of 2×104 cells (HEK293) or 2.5×104 cells (PC3) per well so that at time of transfection cells were about 75% confluent. Cells were transfected overnight with Fugene 6 (Roche) following the manufacturer’s instructions, using 1.5 μl Fugene per 0.6 μg of total plasmid DNA per well. The ligand DHT in DMEM/F12 for HEK293 cells of RPMI for PC3 cells was added to medium supplemented with 2% charcoal stripped FCS for 24 hours. Medium was removed and cells were washed once with ice cold PBS and then lysed with 2× Promega lysis buffer. Luciferase assays were performed in triplicate with the Firefly luciferase assay kit (Promega) and measured on a luminometer (Berthold LB953 Autolumat), as described previously [7]. The PC3 cell line expresses no endogenous AR [59].
Co-immunoprecipitation and western blotting
Co-immunoprecipitations were performed as follows. COS-7 cells (origin: Cercopithecus aethiops, African green monkey, derived from CV-1 cells by SV40 mediated immortalization [60]) cultured under standard conditions in 10 cm culture dish (NUNC) were co-transfected using Lipofectamine™ 2000 (Invitrogen) with pCMV-AR and/or SKIP-pCGN plasmid DNA (10 μg each) and cultured in the presence or absence of 1 nM DHT. Cells were harvested 48 h after transfection and were lysed in 0.5 ml of cold RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 0.5% deoxcholic acid, 1% Triton X-100) containing 1 mM PMSF and proteinase inhibitors (Roche). Lysates were sonicated for 30 seconds on ice and centrifuged to remove cell debris then pre-clarified by incubation with 40 μl of 50% protein G-Sepharose beads (Amersham) and 1 μg of rabbit IgG (Dako) at 4°C for 1 hour with slow rotation to reduce non-specific binding. After pelleting the beads, the lysates were incubated with rabbit anti-AR antibody, N20 (Santa Cruz Biotechnology), and rabbit anti-HA antibody (Santa Cruz Biotechnology), respectively, at 4°C overnight, and followed by incubating with 20 μl of 50% protein G-Sepharose beads at 4°C for 1 hour to recover AR or HA-SKIP containing protein complexes. The immunoprecipitates were washed 5 times with cold RIPA buffer and resuspended in 20 μl of sample buffer before subjecting to western blotting with antibodies to AR or HA-SKIP. Briefly, parts of the immunoprecipitated sample was resolved on an 8% SDS-PAGE, blotted and probed for HA-tagged SKIP using anti-HA antibody. The other parts were separated on gels, blotted and probed with anti-AR antibody, N20 to detect an AR N-terminal fragment. The probed blots were incubated with anti-rabbit IgG HRP (Amersham) and imaged using ECL (Amersham).
Plasmid constructs
SKIP-pSG5, SKIP-pM, SKIP-pCGN were previously described [7, 8]. All mammalian one and two-hybrid plasmids (pM-GAL4DBD and pVP16AD; Clontech State, USA) and the GAL45E1bTATA-luciferase reporter [61] were previously described [7]. The MMTV-luciferase reporter MMTV-luc was provided by Dr. R.M. Evans, Salk Institute, La Jolla, CA and the PSA-luciferase reporter plasmid (pGL3-PSA540-enhancer, PSA-luc) provided by Bristol-Myers Squibb (Princeton, NJ) as previously described [62]. The vector pM-GAL4DBD-AR-AF1 expressed the activation function 1 fragment of AR (residues 1–555). The vectors pM-GAL4DBD-AR642-917 and pVP16-AR1-538 were as described [41]. pCMV-AR WT-pCMV3.1 plasmid for co-immunoprecipitation experiment was previously described [63, 64]. All AR constructs are derived from human AR. EYFP-SKIP human Skip cDNA was amplified by PCR using primer pairs (5′-TTT GAA TTC ATG GCG CTC ACC AGC TTT TTA CCT GC-3′ and 5′-TTT GTC GAC TAT TCC TTC CTC CTC TTC TTG CC-3′), phosphorylated by T4 polynucleotide kinase, and cloned into the SmaI site of pEYFP-C1 (Clontech). The AR-pECFP-N1 plasmid was as previously described [37].
Donor dequenching and sensitised acceptor FRET imaging experiments
FRET experiments were performed in BHK-21 cells (origin: Mesocricetus auratus, golden hamster) that were cultured and transfected as previously described. BHK cells were chosen as they were easy to transfect and showed good expression of the constructs. Donor dequenching experiments were carried out, using a LSM 510 Meta confocal microscope (Zeiss). Fluorescent 12 bit images were recorded in the donor- (ex 405 nm, em 530–600 nm) and acceptor- (ex 514 nm, em 530–600 nm) channels before and after bleaching of EYFP (at 514 nm with 100% laser transmission at 50% output in a ROI circumscribing nuclear speckles using 200 iterations). Average bleaching of the EYFP-signal was 98.3±0.5% (sem). The average apparent donor dequenching FRET-efficiency was calculated on individual EYFP-SKIP positive speckles, as: E=1-Dbefore/Dafter, where Dbefore/after are the background corrected average donor channel signals before and after bleaching, respectively. On each recorded image 3 different SKIP-positive speckles were analysed. Sensitized acceptor emission FRET imaging was carried out essentially as described [45]. However, due to the inherently large difference in donor and acceptor expression, only the FRET index image, FRET2 [65], was calculated for each time point. The colour-lookup-table was assigned to the FRET2-images using Image J. The video showing the donor, FRET and acceptor-images was generated using Image J and Quick Time Pro. The translocation index, TL, was calculated on the indicated regions using the intensity of the AR-ECFP on the speckle, S, and the AR-ECFP intensity of a reference region in the cytoplasm, C: TL=S/C. For graphical representation and curve fitting, this value was then normalized using the TL at time 0 and at the last time point 988 s: TLnorm=(TL-TL0)/(TL988-TL0). To determine the translocation half-time, τ0.5, where 50% translocation occurred, a monoexponential function was fitted: TLnorm=1-exp(−t/τ), with τ0.5= − τ ln0.5.
References
Figueroa JD, Hayman MJ: The human Ski-interacting protein functionally substitutes for the yeast PRP45 gene. Biochem Biophys Res Commun. 2004, 319 (4): 1105-1109. 10.1016/j.bbrc.2004.05.096.
Folk P, Puta F, Skruzny M: Transcriptional coregulator SNW/SKIP: the concealed tie of dissimilar pathways. Cell Mol Life Sci. 2004, 61 (6): 629-640. 10.1007/s00018-003-3215-4.
Kostrouchova M, Housa D, Kostrouch Z, Saudek V, Rall JE: SKIP is an indispensable factor for Caenorhabditis elegans development. Proc Natl Acad Sci USA. 2002, 99 (14): 9254-9259. 10.1073/pnas.112213799.
Negeri D, Eggert H, Gienapp R, Saumweber H: Inducible RNA interference uncovers the Drosophila protein Bx42 as an essential nuclear cofactor involved in Notch signal transduction. Mech Dev. 2002, 117 (1–2): 151-162.
Dahl R, Wani B, Hayman MJ: The Ski oncoprotein interacts with Skip, the human homolog of Drosophila Bx42. Oncogene. 1998, 16 (12): 1579-1586. 10.1038/sj.onc.1201687.
Albers M, Diment A, Muraru M, Russell CS, Beggs JD: Identification and characterization of Prp45p and Prp46p, essential pre-mRNA splicing factors. RNA. 2003, 9 (1): 138-150. 10.1261/rna.2119903.
Leong GM, Subramaniam N, Issa LL, Barry JB, Kino T, Driggers PH, Hayman MJ, Eisman JA, Gardiner EM: Ski-interacting protein, a bifunctional nuclear receptor coregulator that interacts with N-CoR/SMRT and p300. Biochem Biophys Res Commun. 2004, 315 (4): 1070-1076. 10.1016/j.bbrc.2004.02.004.
Leong GM, Subramaniam N, Figueroa J, Flanagan JL, Hayman MJ, Eisman JA, Kouzmenko AP: Ski-interacting protein interacts with Smad proteins to augment transforming growth factor-beta-dependent transcription. J Biol Chem. 2001, 276 (21): 18243-18248. 10.1074/jbc.M010815200.
Wang Y, Fu Y, Gao L, Zhu G, Liang J, Gao C, Huang B, Fenger U, Niehrs C, Chen YG: Xenopus skip modulates Wnt/beta-catenin signaling and functions in neural crest induction. J Biol Chem. 2010, 285 (14): 10890-10901. 10.1074/jbc.M109.058347.
Chen Y, Zhang L, Jones KA: SKIP counteracts p53-mediated apoptosis via selective regulation of p21Cip1 mRNA splicing. Genes Dev. 2011, 25 (7): 701-716. 10.1101/gad.2002611.
Edwards DP, Wardell SE, Boonyaratanakornkit V: Progesterone receptor interacting coregulatory proteins and cross talk with cell signaling pathways. J Steroid Biochem Mol Biol. 2002, 83 (1–5): 173-186.
Baudino TA, Kraichely DM, Jefcoat SC, Winchester SK, Partridge NC, MacDonald PN: Isolation and characterization of a novel coactivator protein, NCoA-62, involved in vitamin D-mediated transcription. J Biol Chem. 1998, 273 (26): 16434-16441. 10.1074/jbc.273.26.16434.
Zhang C, Baudino TA, Dowd DR, Tokumaru H, Wang W, MacDonald PN: Ternary complexes and cooperative interplay between NCoA-62/Ski-interacting protein and steroid receptor coactivators in vitamin D receptor-mediated transcription. J Biol Chem. 2001, 276 (44): 40614-40620. 10.1074/jbc.M106263200.
Zhang C, Dowd DR, Staal A, Gu C, Lian JB, van Wijnen AJ, Stein GS, MacDonald PN: Nuclear coactivator-62 kDa/Ski-interacting protein is a nuclear matrix-associated coactivator that may couple vitamin D receptor-mediated transcription and RNA splicing. J Biol Chem. 2003, 278 (37): 35325-35336. 10.1074/jbc.M305191200.
Barry JB, Leong GM, Church WB, Issa LL, Eisman JA, Gardiner EM: Interactions of SKIP/NCoA-62, TFIIB, and retinoid X receptor with vitamin D receptor helix H10 residues. J Biol Chem. 2003, 278 (10): 8224-8228. 10.1074/jbc.C200712200.
Kang MR, Lee SW, Um E, Kang HT, Hwang ES, Kim EJ, Um SJ: Reciprocal roles of SIRT1 and SKIP in the regulation of RAR activity: implication in the retinoic acid-induced neuronal differentiation of P19 cells. Nucleic Acids Res. 2010, 38 (3): 822-831. 10.1093/nar/gkp1056.
Bres V, Gomes N, Pickle L, Jones KA: A human splicing factor, SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1 Tat. Genes Dev. 2005, 19 (10): 1211-1226. 10.1101/gad.1291705.
Bres V, Yoshida T, Pickle L, Jones KA: SKIP interacts with c-Myc and Menin to promote HIV-1 Tat transactivation. Mol Cell. 2009, 36 (1): 75-87. 10.1016/j.molcel.2009.08.015.
Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M: Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009, 138 (2): 245-256. 10.1016/j.cell.2009.04.056.
Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC: Molecular cell biology of androgen receptor signalling. Int J Biochem Cell Biol. 2010, 42 (6): 813-827. 10.1016/j.biocel.2009.11.013.
Heinlein CA, Chang C: Androgen receptor in prostate cancer. Endocr Rev. 2004, 25 (2): 276-308. 10.1210/er.2002-0032.
Gelmann EP: Molecular biology of the androgen receptor. J Clin Oncol. 2002, 20 (13): 3001-3015. 10.1200/JCO.2002.10.018.
Dehm SM, Tindall DJ: Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol. 2007, 21 (12): 2855-2863. 10.1210/me.2007-0223.
Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M: A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007, 27 (3): 380-392. 10.1016/j.molcel.2007.05.041.
Heinlein CA, Chang C: Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002, 23 (2): 175-200. 10.1210/er.23.2.175.
Gao W, Bohl CE, Dalton JT: Chemistry and structural biology of androgen receptor. Chem Rev. 2005, 105 (9): 3352-3370. 10.1021/cr020456u.
Langley E, Kemppainen JA, Wilson EM: Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J Biol Chem. 1998, 273 (1): 92-101. 10.1074/jbc.273.1.92.
He B, Wilson EM: The NH(2)-terminal and carboxyl-terminal interaction in the human androgen receptor. Mol Genet Metab. 2002, 75 (4): 293-298. 10.1016/S1096-7192(02)00009-4.
Zhou ZX, Lane MV, Kemppainen JA, French FS, Wilson EM: Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol Endocrinol. 1995, 9 (2): 208-218. 10.1210/me.9.2.208.
He B, Kemppainen JA, Wilson EM: FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem. 2000, 275 (30): 22986-22994. 10.1074/jbc.M002807200.
He B, Bowen NT, Minges JT, Wilson EM: Androgen-induced NH2- and COOH-terminal Interaction Inhibits p160 coactivator recruitment by activation function 2. J Biol Chem. 2001, 276 (45): 42293-42301. 10.1074/jbc.M107492200.
McKenna NJ, Lanz RB, O’Malley BW: Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999, 20 (3): 321-344. 10.1210/er.20.3.321.
Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, Chambon P: The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell. 1989, 59 (3): 477-487. 10.1016/0092-8674(89)90031-7.
Danielian PS, White R, Lees JA, Parker MG: Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992, 11 (3): 1025-1033.
Dehm SM, Tindall DJ: Ligand-independent androgen receptor activity is activation function-2-independent and resistant to antiandrogens in androgen refractory prostate cancer cells. J Biol Chem. 2006, 281 (38): 27882-27893. 10.1074/jbc.M605002200.
A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999, 97 (2): 161-163.
Saitoh M, Takayanagi R, Goto K, Fukamizu A, Tomura A, Yanase T, Nawata H: The presence of both the amino- and carboxyl-terminal domains in the AR is essential for the completion of a transcriptionally active form with coactivators and intranuclear compartmentalization common to the steroid hormone receptors: a three-dimensional imaging study. Mol Endocrinol. 2002, 16 (4): 694-706. 10.1210/me.16.4.694.
Handwerger KE, Gall JG: Subnuclear organelles: new insights into form and function. Trends Cell Biol. 2006, 16 (1): 19-26. 10.1016/j.tcb.2005.11.005.
Lamond AI, Spector DL: Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol. 2003, 4 (8): 605-612. 10.1038/nrm1172.
Mintz PJ, Patterson SD, Neuwald AF, Spahr CS, Spector DL: Purification and biochemical characterization of interchromatin granule clusters. EMBO J. 1999, 18 (15): 4308-4320. 10.1093/emboj/18.15.4308.
Need EF, Scher HI, Peters AA, Moore NL, Cheong A, Ryan CJ, Wittert GA, Marshall VR, Tilley WD, Buchanan G: A novel androgen receptor amino terminal region reveals two classes of amino/carboxyl interaction-deficient variants with divergent capacity to activate responsive sites in chromatin. Endocrinology. 2009, 150 (6): 2674-2682. 10.1210/en.2008-1181.
Simental JA, Sar M, Lane MV, French FS, Wilson EM: Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991, 266 (1): 510-518.
Fields S, Song O: A novel genetic system to detect protein-protein interactions. Nature. 1989, 340 (6230): 245-246. 10.1038/340245a0.
Chien CT, Bartel PL, Sternglanz R, Fields S: The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc Natl Acad Sci USA. 1991, 88 (21): 9578-9582. 10.1073/pnas.88.21.9578.
Abankwa D, Vogel H: A FRET map of membrane anchors suggests distinct microdomains of heterotrimeric G proteins. J Cell Sci. 2007, 120 (Pt 16): 2953-2962.
Patterson GH, Piston DW, Barisas BG: Forster distances between green fluorescent protein pairs. Anal Biochem. 2000, 284 (2): 438-440. 10.1006/abio.2000.4708.
Vogel SS, Thaler C, Koushik SV: Fanciful FRET. Sci STKE. 2006, 2006 (331): re2-
Berney C, Danuser G: FRET or no FRET: a quantitative comparison. Biophys J. 2003, 84 (6): 3992-4010. 10.1016/S0006-3495(03)75126-1.
Melcak I, Cermanova S, Jirsova K, Koberna K, Malinsky J, Raska I: Nuclear pre-mRNA compartmentalization: trafficking of released transcripts to splicing factor reservoirs. Mol Biol Cell. 2000, 11 (2): 497-510.
Misteli T: Cell biology of transcription and pre-mRNA splicing: nuclear architecture meets nuclear function. J Cell Sci. 2000, 113 (Pt 11): 1841-1849.
Spector DL, Lamond AI: Nuclear speckles. Cold Spring Harb Perspect Biol. 2011, 3 (2):
Zhao Y, Goto K, Saitoh M, Yanase T, Nomura M, Okabe T, Takayanagi R, Nawata H: Activation function-1 domain of androgen receptor contributes to the interaction between subnuclear splicing factor compartment and nuclear receptor compartment. Identification of the p102 U5 small nuclear ribonucleoprotein particle-binding protein as a coactivator for the receptor. J Biol Chem. 2002, 277 (33): 30031-30039. 10.1074/jbc.M203811200.
Faus H, Meyer HA, Huber M, Bahr I, Haendler B: The ubiquitin-specific protease USP10 modulates androgen receptor function. Mol Cell Endocrinol. 2005, 245 (1–2): 138-146.
Beitel LK, Elhaji YA, Lumbroso R, Wing SS, Panet-Raymond V, Gottlieb B, Pinsky L, Trifiro MA: Cloning and characterization of an androgen receptor N-terminal-interacting protein with ubiquitin-protein ligase activity. J Mol Endocrinol. 2002, 29 (1): 41-60. 10.1677/jme.0.0290041.
Lin HK, Altuwaijri S, Lin WJ, Kan PY, Collins LL, Chang C: Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. J Biol Chem. 2002, 277 (39): 36570-36576. 10.1074/jbc.M204751200.
Wu Y, Kawate H, Ohnaka K, Nawata H, Takayanagi R: Nuclear compartmentalization of N-CoR and its interactions with steroid receptors. Mol Cell Biol. 2006, 26 (17): 6633-6655. 10.1128/MCB.01534-05.
Hsu CL, Chen YL, Ting HJ, Lin WJ, Yang Z, Zhang Y, Wang L, Wu CT, Chang HC, Yeh S: Androgen receptor (AR) NH2- and COOH-terminal interactions result in the differential influences on the AR-mediated transactivation and cell growth. Mol Endocrinol. 2005, 19 (2): 350-361.
Bevan CL, Hoare S, Claessens F, Heery DM, Parker MG: The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol Cell Biol. 1999, 19 (12): 8383-8392.
Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW: Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest Urol. 1979, 17 (1): 16-23.
Jensen FC, Girardi AJ, Gilden RV, Koprowski H: Infection of Human and Simian Tissue Cultures with Rous Sarcoma Virus. Proc Natl Acad Sci USA. 1964, 52: 53-59. 10.1073/pnas.52.1.53.
Rubino D, Driggers P, Arbit D, Kemp L, Miller B, Coso O, Pagliai K, Gray K, Gutkind S, Segars J: Characterization of Brx, a novel Dbl family member that modulates estrogen receptor action. Oncogene. 1998, 16 (19): 2513-2526. 10.1038/sj.onc.1201783.
Butler LM, Centenera MM, Neufing PJ, Buchanan G, Choong CS, Ricciardelli C, Saint K, Lee M, Ochnik A, Yang M: Suppression of androgen receptor signaling in prostate cancer cells by an inhibitory receptor variant. Mol Endocrinol. 2006, 20 (5): 1009-1024. 10.1210/me.2004-0401.
Irvine RA, Ma H, Yu MC, Ross RK, Stallcup MR, Coetzee GA: Inhibition of p160-mediated coactivation with increasing androgen receptor polyglutamine length. Hum Mol Genet. 2000, 9 (2): 267-274. 10.1093/hmg/9.2.267.
Buchanan G, Yang M, Harris JM, Nahm HS, Han G, Moore N, Bentel JM, Matusik RJ, Horsfall DJ, Marshall VR: Mutations at the boundary of the hinge and ligand binding domain of the androgen receptor confer increased transactivation function. Mol Endocrinol. 2001, 15 (1): 46-56. 10.1210/me.15.1.46.
Gordon GW, Berry G, Liang XH, Levine B, Herman B: Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys J. 1998, 74 (5): 2702-2713. 10.1016/S0006-3495(98)77976-7.
Acknowledgements
DA was a fellow of the Swiss National Science Foundation (PA00A-111446) and GML was a NHMRC Clinical Career Development Awardee during this study. The Adelaide Prostate Cancer Research Centre is supported by an establishment grant from the Prostate Cancer Foundation of Australia [ID 2011/0452]. WT and LB were supported by NHMRC project grant #627185.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors’ contributions
DA, SMM, NM, CSC, MY, LMB, GML conceived and designed the experiments; DA, NM, CSC, MY, LMB, GB, GML analyzed the data; DA, SMM, NM, CSC, MY, LB, WDT, NU, MJH, GML contributed reagents/materials/analysis tools; DA, GB, GML wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12858_2012_417_MOESM1_ESM.pdf
Additional file 1:Confocal imaging data of EYFP-SKIP showing its diverse speckled distribution patterns in BHK cells.(PDF 683 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Abankwa, D., Millard, S.M., Martel, N. et al. Ski-interacting protein (SKIP) interacts with androgen receptor in the nucleus and modulates androgen-dependent transcription. BMC Biochem 14, 10 (2013). https://doi.org/10.1186/1471-2091-14-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2091-14-10