
Abstract
Background
Chronic obstructive pulmonary disease (COPD) is characterized by incompletely reversible airflow obstruction associated with inflammation in which monocytes/macrophages are the predominant inflammatory cells. The only known genetic factor related to COPD is inherited PiZZ deficiency of α1-antitrypsin (AAT), an inhibitor of serine proteases.
Methods
We investigated the basal and LPS-stimulated release of pro-inflammatory molecules from blood monocytes isolated from age and gender matched healthy (n = 30) and COPD (n = 20) individuals with and without AAT deficiency.
Results
After 18 h of cell culture the basal release of MMP-9 was 2.5-fold, p < 0.02 greater, whereas IL-8 was 1.8-fold (p < 0.01) lower from COPD patient monocytes than from controls. LPS-stimulated release of IL-6 and MCP-1 was greater from COPD patient's monocytes relative to controls, while activation of control cells resulted in enhanced secretion of ICAM-1 and MMP-9 compared to COPD patients. Independent of disease status, monocytes from PiZZ AAT carriers released less TNFα (by 2.3-fold, p < 0.03).
Conclusions
The basal and LPS-stimulated secretion of specific pro-inflammatory molecules from circulating monocytes differs between healthy and COPD subjects. These findings may be valuable for further studies on the mechanisms involved in recruitment and activation of inflammatory cells in COPD.
Similar content being viewed by others

Background
Chronic obstructive pulmonary disease (COPD) is a complex disease caused by various genetic and environmental risk factors acting singly or in concert. Severe alpha1-antitrypsin (AAT) deficiency, which results from a PiZZ genotype, is a well-known genetic risk factor associated with the development of early onset COPD [1]. Although it has been shown in a large number of AAT deficient individuals that smokers suffer more severe pulmonary impairment at an early age compared to non-smokers [2], the development of COPD in AAT deficient subjects, even among current or ex-smokers, is not universal [3]. Tobacco smoking represents the most important environmental risk factor for respiratory diseases and it is the first risk factor for COPD [4, 5]. However, only about 20% of individuals who are smokers and have the normal PiMM genotype of AAT will develop COPD [3]. These findings lead to the idea that a combination of environmental factors, such as smoking, air pollution, childhood respiratory infections and latent adenoviral infections, and/or genetic factors, such as AAT-deficiency, are likely the essential contributors to the development of COPD.
Although inflammation has always been considered a prominent feature in COPD, there is little work describing the initiating events, regulatory mechanisms and subsequent underlying cellular and molecular processes that lead to inflammation, tissue damage and remodeling in COPD. Current studies show that neutrophils, eosinophils, lymphocytes and macrophages are involved in the inflammatory process of COPD [6]. Various inflammatory mediators derived from these inflammatory cells are suggested to contribute to the chronic inflammation and cause lung tissue damage [7]. Neutrophils are short-lived cells that are recruited from the circulation to the airways and are found in increased numbers in the airways of smokers and patients with COPD [8]. Several studies have demonstrated the differential roles of pro-inflammatory mediators such as cytokines, chemokines, integrin receptors and endothelial markers in controlling neutrophil sequestration and migration [9, 10]. The neutrophil cell products have been shown directly to cause all of the pathological features of COPD [11].
Monocyte recruitment to the lungs is also an important step in COPD [6]. Monocytes release a variety of macromolecules and low molecular weight products that mediate inflammation and repair. Expression of the genes for these secretory products and induction of their release depends on local signals in their microenvironment [12]. Different cytokines, chemokines and monocyte-specific adhesion molecules are known to be involved in monocyte activation, binding to stimulated endothelium and in further migration within tissue. It has been reported that in patients with COPD the migration of monocytes to growth-related protein is significantly increased compared to monocytes from healthy volunteers [13]. It was also found that there are significantly higher levels of monocyte chemoattractant protein-1 (MCP-1) and growth-related protein in the sputum in COPD patients compared to healthy volunteers and smokers [13].
Blood monocytes from the circulating pool migrate through the blood vessel walls into various organs and then differentiate into macrophages. Macrophages are the predominant defence cell in the normal lung and are increased during conditions associated with chronic inflammation. For example, in bronchoalveolar-lavage of patients with COPD, macrophage numbers are found to be increased by 5 to 10 times [14]. Macrophages are suggested to play a role in driving inflammatory process by release of chemotactic factors and recruitment of neutrophils [15]. A more recent theory suggests that alveolar macrophage-derived metalloproteinases mediate inflammation by releasing TNF-α from macrophages with subsequent neutrophil influx, endothelial activation and tissue breakdown caused by neutrophil derived proteinases [16]. Unique mechanisms for leukocyte migration from the bloodstream to the lung and activation have been proposed based on the profile of the adhesion molecules, cytokines and chemokines involved [17].
In this study we investigated pro-inflammatory molecular release and expression of transcription factor NFκB by blood monocytes isolated from individuals with COPD and healthy controls with and without severe PiZZ alpha1-antitrypsin (AAT) deficiency under basal conditions and after stimulation with LPS. By using this experimental approach we aimed to determine the differences in pro-inflammatory molecular profile among monocytes isolated from COPD patients and healthy controls, and also to investigate if pro-inflammatory molecular release is influenced by inherited deficiency of AAT.
Materials and methods
Subjects
The studied group consisted of 20 healthy PiMM AAT adults (11 males and 9 females, aged 53 ± 9.6), 10 asymtomatic PiZZ AAT adults (5 males and 5 females, aged 53 ± 9.6) and 20 COPD patients, among them 10 patients with PiZZ AAT (5 males and 5 females, aged 47.4 ± 11) and 10 patients with PiMM AAT (5 males and 5 females, aged 59.4 ± 6.7). COPD was diagnosed according to the NHLBI/WHO Workshop guidelines [18]. The PiZZ and PiMM COPD patients had a forced expiratory volume in one second (FEV1) ≤ 80% of that predicted and the FEV1/Forced vital capacity ratio (FVC) ≤ 70%, while asymptomatic PiZZ individuals had a FEV1 ≥ 80% and the FEV1/FVC ratio ≥ 70%. All COPD patients included in the study were in a stable, non-exacerbated phase of the disease. The exclusion criteria were liver diseases, vasculitic or other extra-pulmonary diseases. The control subjects showed no evidence of any disease and had no respiratory symptoms; none of them was on medication, and all had PiMM variant and normal plasma concentration of AAT. The PiZZ individuals were recruited from the Swedish AAT Deficiency Register. The PiMM COPD individuals were outpatients at the Department of Respiratory Medicine, University Hospital, Malmo. The healthy volunteers were recruited from the hospital staff and their relatives. All the individuals gave a signed, informed consent to take part in this study, which has been approved by the research ethical committee of Lund University, Sweden. Patient characteristics are given in Table 1.
Lung function tests
Lung-function tests included FEV1 and FVC. The measurements were performed in accordance with European recommendations [19]. The results of FEV1 and FVC are expressed as percent of predicted values according to Berglund and co-workers [20]. The lung function tests were performed after inhalation of bronchodilatator terbutaline (1 mg).
Isolation and culture of monocytes
Monocytes were isolated from the whole blood by the Ficoll-Hypaque procedure as previously detailed [21]. Cell purity was > 95% as determined on an AC900EO Auto Counter (Swelab Instruments, AB). Cell viability was analysed by 0.4% trypan blue staining. Monocytes were plated at a density of 4 × 106 cells/ml into plastic dishes. After removal of non-adhering cells, the monocytes were cultured in RPMI 1640 (Gibco, Life Technologies, Paisley, Scotland) supplemented with 2 mM N-acetyl-L-alanyl-L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1% nonessential amino acid, 2% sodium pyruvate and 20 mM Hepes (Fluka, Chemie AG) without serum at 37°C in a 5%CO2. Monocytes were cultured alone or in the presence of lipopolisaccharide (LPS) 1 μg/ml for 18 h. Minimum triplicate of repeats were undertaken for each condition.
Assays of Inflammatory markers
Monocyte culture supernatants were analyzed to determine tumor necrosis factor-alpha (TNF-α), monocyte chemoattractant protein (MCP-1), intracellular adhesion molecule 1 (ICAM-1), human interleukin-1β (IL-1β), -6 (IL-6) and -8 (IL-8), gelatinase B (MMP-9) and sE-selectin by using sandwich enzyme immunoassay kits (R&D Systems Europe Ltd, Abingdon, UK) according to the manufacturer's instructions. Absorbance was measured spectrophotometrically at 450 nm using microplate reader (Labsystems). The minimum detectable levels of MMP-9 and sE-selectin were less than 0.156 ng/ml and 0,1 ng/ml, respectively.
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extracts from monocytes were prepared exclusively as described previously [22]. Electrophoretic mobility shift assay (EMSA) was used to detect the binding of specific DNA sequence by NFκB p50 transcription factor complexes. The Gelshift™ Kit for the detection of NFκB p50 transcription factor was used (Geneka Biotechnology, Inc, USA) according to the manufacturer's recommendations. The sequences of the double-stranded oligonucleotide probe are as follows: 5'-GCC-ATG-G GG-GGA-TCC-CCG-AAG-TCC-3' and 3'-CGG-TAC-C CC-CCT-AGG-GGC-TTC-AGG-5' (consensus binding sequence is underlined). Unlabeled competitor and mutant oligonucleotides were added in 50 × excess to confirm the specificity of the binding reactions. In addition, rabbit polyclonal antibody against NFκB was used in the competition reactions to determine the specificity of the assay. In addition, nuclear extracts were analyzed by using ELISA based NFκB p65/NF B p50 transcription factor assay kit (Trans AM™, Active Motif, Europe) according to manufactures instructions. Detection limit was <0.5 μg cell extract/well.
Statistical analysis
Statistical analysis was performed with use of SPSS software (version 11.0 for Windows, SPSS Inc. Chicago, USA). The differences in measured parameters between study groups were analyzed for their statistical significance with the independent-samples two-sided t test and One-way analysis of variance (ANOVA) combined with a multiple comparison procedure (Scheffe multiple range test). Significance was determined at the 5% level. Data are expressed as median ± SD.
Results
In this study we investigated the release of pro-inflammatory molecules from monocytes isolated from both PiZZ AAT-deficiency and wild type PiMM-AAT groups of COPD patients (n = 20) and in controls (n = 30) who were carefully sex-, age-, and AAT-phenotype matched. Care was also taken to avoid confounding effects of smoking and therefore only a minority of subjects analysed were current smokers. The characteristics of the studied individuals are summarised in Table 1.
Release of Inflammatory molecular species from monocytes of COPD patients- vs controls
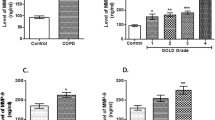
ELISA assays were performed on blood monocyte-conditioned medium (18 h culture) to monitor secretion of pro-inflammatory molecular species (Table 2). Monocytes obtained from COPD patients, independent of AAT-genotype, released greater amounts of MMP-9 (2.5-fold, p < 0.028) but lower amounts of IL-8 (1.8-fold, p < 0.013) compared to those obtained from controls. No significant difference was observed in other measured molecules in culture media from controls and COPD-monocytes (Table 2). In separate experiments we measured secretion of the same pro-inflammatory molecules from LPS-exposed monocytes. After LPS-stimulation, monocytes from COPD patients showed significantly greater release of IL-6 (1.9-fold, p < 0.01) and MCP-1 (2.6-fold, p < 0.01) than LPS-activated monocytes from control individuals (Figure 1A). In contrast, ICAM-1 levels were higher in LPS-activated monocytes (by 4.9-fold, p < 0.001) from control individuals than from COPD patient-monocytes (Figure 1B). The up-regulation of other pro-inflammatory markers in LPS-stimulated monocytes was found to be of the same magnitude in both groups (data not shown).

LPS-stimulated release of pro-inflammatory molecule from COPD patient monocytes relative to controls. Isolated blood monocytes from COPD patients (n = 20) and controls (n = 30) were activated with LPS (1 μg/ml) for 18 h. Pro-inflammatory molecular release was measured by ELISA methods. Monocytes from patients with COPD released greater levels of IL-6 and MCP-1, relative to controls (A), while the activation of control cells resulted in an enhanced secretion of ICAM-1 and MMP-9 (B).
Release of inflammatory molecular species from monocytes isolated from PiZZ and PiMM AAT individuals
To evaluate whether AAT-genotype has any influence on monocyte release of pro-inflammatory molecules, we compared isolated monocytes from individuals with wild type M and deficiency Z AAT, independent of disease state. Monocytes isolated from M-AAT carriers were found to release more TNFα (2.3-fold, p < 0.03) while the mean levels of other measured molecules showed no significant differences between Z-and M-AAT groups (Table 3). It should be noted that exposure of Z-AAT monocytes to LPS resulted in significantly greater release of IL-8 (3.3-fold, p < 0.01) compared to M-AAT monocytes (data not shown).
NFκ-B activation in monocytes isolated from controls and COPD individuals
The expression and release of a variety of pro-inflammatory molecules release are mediated by activation of transcription factor NFκ-B [23]. We compared NF-κB expression in COPD and healthy monocytes by using both EMSA and ELISA based assays. As shown in figure 2, NF-κB activity was increased by about 23.5% (not statistically significant) in monocytes from COPD patients compared to monocytes from controls. Similarly, NF-κB activity was found to be slightly, but not significantly (by about 33%) higher in monocytes from Z-AAT than M-AAT carriers.

Monitoring of NFκB activation in monocytes from patients with COPD and controls by ELISA-based TransAM kitAMCell nuclear extracts were prepared from monocytes isolated from COPD patients and controls with and without PiZZ AAT deficiency. NFκB activity assay was performed in a 96-well plates with 10 μg of cell extract per well. Absorbance was measured by spectrophotometry at 405 nm. A positive control performed by using a nuclear extract derived from Jurkat cells. This extract is optimized to give a strong signal when used at 2.5 μg/well. Specificity controls were performed by adding a molar excess (20 pmol/well] of mutant NFκB oligonucleotide [the positive signal remained unaffected] and wild-type NFκB oligonucleotide (a signal was abolished). The bars represent mean values of three independent measurements ± SD.
Discussion
There is accumulating evidence that the mechanisms involved in leukocyte recruitment to the lung are dependent upon the stimulus and the site of migration. For example, monocyte recruitment to mouse alveoli was found to be dependent upon CD18 and intercellular adhesion molecule-1 under basal conditions, but following bacterial endotoxin stimulation, CD11a and vascular cell adhesion molecule-1 were also involved, indicating a switch of mechanism [16]. Release from circulating monocytes of higher quantities of specific cytokines, chemokines and MMPs may also contribute to cell migration. It has been suggested that activated peripheral blood monocytes more easily enter the lung and/or stimulate immune activation when present in the lung [24].
The concept that stable COPD is characterized by a systemic inflammation is supported by reports showing increased circulating levels of IL-6 and raised counts of activated leukocytes [25]. To test the hypothesis that circulating monocytes in stable COPD patients have enhanced propensities to release certain pro-inflammatory molecules, we designed a study in which we compared isolated monocytes from COPD patients and controls. In addition, the influence of LPS-stimulation on pro-inflammatory responses of these monocytes in vitro was studied. Circulating monocyte activities could be influenced by variations in patient characteristics, such as the degree of lung function impairment, the presence of acute exacerbations, cigarette smoking, and corticosteroid treatment. We were careful to ensure that groups of individuals were well matched in terms of these characteristics and that blood was collected from patients when they were in a stable clinical state. Therefore, we infer that the differences observed in basal monocyte behaviour and in response to LPS represent stable COPD.
Results from this study show that monocytes isolated from COPD patients release significantly more MMP-9 but less IL-8 compared to controls. LPS-stimulation of these cells results in a larger enhancement of IL-6 and MCP-1 release from COPD-monocytes while monocytes isolated from healthy individuals release higher levels of ICAM-1. The in vivo significance of the observed variability in monocyte release of specific pro-inflammatory molecules is not known, but basal differences between monocytes isolated from control and COPD individuals in the secretion of pro-inflammatory markers as well as differences in release of specific molecules upon activation might be important parameters in the chronic inflammatory processes in COPD.
Circulating monocytes generally are known to synthesise significant amounts of MMPs, including matrilysin (MMP-7), collagenase 1 (MMP-1) and gelatinase B (MMP-9). Interestingly, expression of serine proteinases and MMP-7 is suggested to be limited to a subset of "pro-inflammatory" monocytes (15% of total) [26, 27]. Most MMPs are only expressed upon differentiation of monocytes to macrophages and are not expressed, or are expressed at nearly undetectable levels, in normal, healthy tissues. In contrast, MMP expression is seen in any repair or remodelling process in diseased or inflamed tissue, and in essentially any cell type grown in culture. Although the qualitative pattern and quantitative levels of MMPs vary among cells, it is generally accepted that activated cells express more MMPs. Our finding that monocytes isolated from patients with stable COPD release more MMP-9 favours recent theory supporting the potential role of high levels of MMPs (including MMP-9) in amplifying inflammation in COPD. MMP-9 has also been shown to be markedly over-expressed by alveolar macrophages from patients with COPD [28].
IL-8, a selective chemoattractant of neutrophils that may be secreted by macrophages, neutrophils, and airway epithelial cells, is present at high concentrations in induced sputum and BAL fluid of patients with COPD [29]. IL-8 may play a primary role in the activation of both neutrophils and eosinophils in the airways of COPD patients and may serve as a marker in evaluating the severity of airway inflammation [30]. It was also reported that IL-8 is released from active monocytes and induces neutrophil and basophil chemotaxis [29, 31]. We found that monocytes isolated from COPD patients release significantly less IL-8 than monocytes from control subjects. These results lead to the conclusion that chemotactic activity of circulating monocytes in COPD patients in the stable clinical state is not increased, but rather suppressed. Although it has been found that high doses of corticosteroids cannot reduce the inflammatory response, concentrations of IL-8, or proteases in the sputum of the patients with COPD [32], observed lower release of IL-8 from COPD patient blood monocytes might be related to corticosteroid treatment. We also find that the LPS-activated COPD monocytes release more IL-6 and MCP-1 and less ICAM-1 than monocytes isolated from healthy individuals. The significance of these differences in IL-8, IL-6 and MCP-1 release by monocytes from COPD and healthy subjects remains to be determined, but may arise from different mechanisms dependent on the basal status of monocytes.
We compared levels of pro-inflammatory molecules released by monocytes obtained from PiZZ and PiMM AAT individuals independent of disease status. Our results show that M-AAT monocytes release significantly more only TNFα, while LPS-activation results in a larger release of IL-8 from Z-AAT compared to M-AAT monocytes. The profile of release of other measured molecules was similar for both subject groups. Our findings that LPS-activation of monocytes isolated from PiZZ AAT results in a larger release of IL-8 suggest that these monocytes might have higher chemoattractant activity than those isolated from PiMM AAT individuals. Recently it has been reported that chemotactic activity from COPD patient sputum of AAT-deficiency is increased compared to matched control patients with COPD [33].
The genes for a number of cytokines involved in airway inflammation in COPD are regulated by nuclear factor-κB (NF κB), which in non-stimulated cells is localised in cytoplasm but upon stimulation translocates into the nucleus and binds to specific κB elements in the promoter of target genes [34]. In this study we evaluated whether NFκB expression is modified in monocytes isolated from stable COPD patients compared to controls. Although no statistically significant difference was found in NFκB expression between the groups analyzed, monocytes isolated from COPD patients showed consistently higher NFκB activation (by about 22.5%) than controls. In addition, monocytes isolated from PiZZ AAT carriers, compared to PiMM-AAT, also showed higher levels of NFκB (by 33%). These findings suggest that NFκB might be involved in the activation of circulating monocytes in COPD patients, particularly with severe PiZZ AAT deficiency.
Conclusions
Although further clinical studies are necessary to determine the significance and factors influencing the phenomena described here, this study represents the first attempt to compare the pro-inflammatory properties of circulating monocytes isolated from stable COPD patients and controls. Pro-inflammatory molecules, such as endotoxin and lipopolysaccharide (LPS), are known to play a role in the up-regulating inflammation [35]. However, other factors such as cellular migration, infiltration and activation state, specific cytokines and chemokines, and the initiation and regulatory mechanisms are likely to play major roles COPD pathology. Our findings that circulating monocytes isolated from COPD patients show a profile of released pro-inflammatory molecules and response to LPS stimulus distinct from control monocytes offer a guide to future investigation of the complex mechanisms involved in chronic inflammation in COPD.
Abbreviations
- AAT:
-
alpha-1-antitrypsin
- PiZZ:
-
homozygous AAT-deficiency variant
- PiMM:
-
wilde type AAT variant
- COPD:
-
chronic obstructive pulmonary disease
- MMP:
-
matrix metalloproteinase
- MCP:
-
monocyte chemoattractant protein
- CRP:
-
C-reactive protein
- FEV1 :
-
forced expiratory volume in 1 second
- FVC:
-
forced vital capacity
- ACE:
-
angiotensin converting enzyme
- LPS:
-
lipopolysaccharide
References
Joos L, Pare PD, Sandford AJ: Genetic risk factors of chronic obstructive pulmonary disease. Swiss Med Wkly 2002, 132:27–37.
Janus ED, Phillips NT, Carrell RW: Smoking, lung function, and alpha 1-antitrypsin deficiency. Lancet 1985, 1:152–154.
Siafakas NM, Tzortzaki EG: Few smokers develop COPD. Why? Respir Med 2002, 96:615–624.
Mannino DM: COPD: epidemiology, prevalence, morbidity and mortality, and disease heterogeneity. Chest 2002,5(Suppl):121S-126S.
Hill AT, Campbell EJ, Bayley DL, Hill SL, Stockley RA: Evidence for excessive bronchial inflammation during an acute exacerbation of chronic obstructive pulmonary disease in patients with alpha[1]-antitrypsin deficiency [PiZ]. Am J Respir Crit Care Med 1999, 160:1968–1975.
Corrigan CJ, Kay AB: The roles of inflammatory cells in the pathogenesis of asthma and of chronic obstructive pulmonary disease. Am Rev Respir Dis 1991, 143:1165–1168.
Chung KF: Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl 2001, 34:50–59.
Stockley RA: Neutrophils and the pathogenesis of COPD. Chest 2002, 121:151–155.
Hubbard RC, Fells G, Gadek J, Pacholok S, Humes J, Crystal RG: Neutrophil accumulation in the lung in 1-antitrypsin deficiency: spontaneous release of leukotriene B4 by alveolar macrophages. J Clin Invest 1991, 88:891–897.
Cavarra E, Martorana PA, Gambelli F, de Santi M, van Even P, Lungarella G: Neutrophil recruitment into the lungs is associated with increased lung elastase burden, decreased lung elastin, and emphysema in alpha 1 proteinase inhibitor-deficient mice. Lab Invest 1996, 75:273–280.
Nadel JA: Role of neutrophil elastase in hypersecretion during COPD exacerbations, and proposed therapies. Chest 2000, 117:386–389.
Hume DA, Ross IL, Himes SR, Sasmono RT, Wells CA, Ravasi TJ: The mononuclear phagocyte system revisited. Leukoc Biol 2002, 72:621–627.
Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE: Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax 2002, 57:590–595.
Barnes PJ: Chronic obstructive pulmonary disease. N Engl J Med 2000, 343:269–280.
Shapiro SD: The macrophage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999, 160:29–32.
Maus U, Huwe J, Ermert L, Ermert M, Seeger W, Lohmeyer J: Molecular pathways of monocyte emigration into the alveolar air space of intact mice. Am J Respir Crit Care Med 2002, 165:95–100.
Wagner JG, Roth RA: Neutrophil migration mechanisms, with an emphasis on the pulmonary vasculature. Pharmacol Rev 2000, 52:349–374.
xx x: NHLBI/WHO workshop summary: global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001, 163:1256–1276.
Viegi G, Pedreschi M, Pistelli F, Di Pede F, Baldacci S, Carrozzi L, Giuntini C: Prevalence of airways obstruction in a general population: European Respiratory Society vs American Thoracic Society definition. Chest 2000, 117:339–345.
Berglund E: The pathophysiology of chronic respiratory failure. Scand J Respir Dis Suppl 1970, 72:13–18.
Janciauskiene S, Moraga F, Lindgren S: C-terminal fragment of alpha1-antitrypsin activates human monocytes to a pro-inflammatory state through interactions with the CD36 scavenger receptor and LDL receptor. Atherosclerosis 2001, 158:41–51.
Dichtl W, Moraga F, Ares MP, Crisby M, Nilsson J, Lindgren S, Janciauskiene S: The carboxyl-terminal fragment of alpha1-antitrypsin is present in atherosclerotic plaques and regulates inflammatory transcription factors in primary human monocytes. Mol Cell Biol Res Commun 2000, 4:50–61.
Renard P, Raes M: The proinflammatory transcription factor NFkappaB: a potential target for novel therapeutical strategies. Cell Biol Toxicol 1999, 15:341–344.
Coffey MJ, Phare SM, Peters-Golden M, Huffnagle GB: Regulation of 5-lipoxygenase metabolism in mononuclear phagocytes by CD4 T lymphocytes. Exp Lung Res 1999, 25:617–629.
Wanner A, Boushey H, Lee TH, Perruchoud AP: Inflammation in chronic obstructive pulmonary disease: Proceedings from the Transatlantic Airway Conference. Am J Respir Crit Care Med 1999, 160:1–79.
Parks WC, Shapiro SD: Matrix metalloproteinases in lung biology. Respir Res 2001, 2:10–19.
Shapiro SD: Elastolytic metalloproteinases produced by human mononuclear phagocytes. Potential roles in destructive lung disease. Am J Respir Crit Care Med 1994, 150:160–164.
Russell RE, Thorley A, Culpitt SV, Dodd S, Donnelly LE, Demattos C, Fitzgerald M, Barnes PJ: Alveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, cysteine, and serine proteases. Am J Physiol Lung Cell Mol Physiol 2002, 283:867–873.
Beeh KM, Kornmann O, Buhl R, Culpitt SV, Giembycz MA, Barnes PJ: Neutrophil Chemotactic Activity of Sputum From Patients With COPD: Role of Interleukin 8 and Leukotriene B[4]. Chest 2003, 123:1240–1247.
Beeh KM, Beier J, Kornmann O, Mander A, Buhl R: Long-term repeatability of induced sputum cells and inflammatory markers in stable, moderately severe COPD. Chest 2003, 123:778–783.
Marsh CB, Gadek JE, Kindt GC, Moore SA, Wewers MD: Monocyte Fc gamma receptor cross-linking induces IL-8 production. J Immunol 1995, 155:3161–3167.
Culpitt SV, Maziak W, Loukidis S, Nightingale JA, Matthews JL, Barnes PJ: Effect of high dose inhaled steroid on cells, cytokines, and proteases in induced sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999, 160:1635–1639.
Woolhouse IS, Bayley DL, Stockley RA: Sputum chemotactic activity in chronic obstructive pulmonary disease: effect of alpha[1]-antitrypsin deficiency and the role of leukotriene B[4] and interleukin 8. Thorax 2002, 57:709–714.
Li X, Stark GR: NFkappaB-dependent signaling pathways. Exp Hematol 2002, 30:285–296.
Ginsburg I: The role of bacteriolysis in the pathophysiology of inflammation, infection and post-infectious sequelae. APMIS 2002, 110:753–770.
Acknowledgments
The authors wish to thank Ewa Szemberg Rindahl for sample collection and preparation. This work was supported by grants from Astra Zeneca, Lund University [Sweden] and the Swedish Heart Lung foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Aldonyte, R., Jansson, L., Piitulainen, E. et al. Circulating monocytes from healthy individuals and COPD patients. Respir Res 4, 11 (2003). https://doi.org/10.1186/1465-9921-4-11
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1465-9921-4-11