
Abstract
The role of vitamin D (VitD) in calcium and bone homeostasis is well described. In the last years, it has been recognized that in addition to this classical function, VitD modulates a variety of processes and regulatory systems including host defense, inflammation, immunity, and repair. VitD deficiency appears to be frequent in industrialized countries. Especially patients with lung diseases have often low VitD serum levels. Epidemiological data indicate that low levels of serum VitD is associated with impaired pulmonary function, increased incidence of inflammatory, infectious or neoplastic diseases. Several lung diseases, all inflammatory in nature, may be related to activities of VitD including asthma, COPD and cancer. The exact mechanisms underlying these data are unknown, however, VitD appears to impact on the function of inflammatory and structural cells, including dendritic cells, lymphocytes, monocytes, and epithelial cells. This review summarizes the knowledge on the classical and newly discovered functions of VitD, the molecular and cellular mechanism of action and the available data on the relationship between lung disease and VitD status.
Similar content being viewed by others
VitD supplementation appears to be correlated with decreased total mortality [1]. In the early 1920s a group of scientists independently discovered that irradiating of certain foods with ultraviolet light renders them antirachitic [2, 3] and in 1922 Elmer V. McCollum identified an antirachitic substance in cod liver oil and called it "vitamin D" [4]. While the role of VitD in calcium and bone homeostasis has been well described, its activities on other physiological and pathophysiological processes have been recognized only in the last years. Epidemiological data suggest that several lung diseases, all inflammatory in nature, may be related to activities of VitD. VitD deficiency might have a role in the development of these diseases. The underlying mechanisms how VitD metabolisms could be linked to the pathophysiology of these diseases are often complex and not fully understood. This review summarizes the role of VitD in lung diseases.
Evolutionary aspects
VitD and its receptors are found throughout the animal kingdom and are often linked to bone and calcium metabolisms. The fact that precursors of VitD are found in ancient organisms like krill and phytoplankton that existed unchanged for at least 750 million years [5] highlights its importance in physiologic and homeostatic processes.
Variants of VitD and its receptors have been identified in higher terrestrial vertebrates like humans [6], rodents [7], birds [8], amphibia [9], reptiles [10], as well as in zebrafish [11]. These animals possess a calcified skeleton and depend on a functional VitD hormone system for calcium and phosphorus homeostasis. Surprisingly, functional VitD receptors (VDRs) have also been found in lampreys, an ancient vertebrate that lacks a calcified skeleton [12]. VDRs were also identified in animals with a naturally impoverished VitD status like the subterranean mole rat [13] and a frugivorous nocturnal mammal, the Egyptian fruit bat Cavaleros [14]. VitD precursors have been found in ancient organisms like phytoplankton and zooplankton, some of which exist unchanged for at least 750 million years [5, 15]. Functional VitD hydroxylases have also been characterized in bacteria like strains of actinomyces [16, 17] and streptomyces [18, 19]. The precursors of VitD in those organisms may function as a natural sunscreen to protect the host against UV-radiation, since the absorption spectra of pro-vitamin D and their photoproducts overlap with the absorption maxima of DNA, RNA, and proteins [20].
Role of VitD in bone metabolism
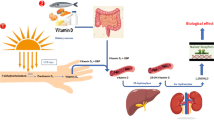
VitD, which is photosynthesized in the skin or has been derived from nutrition, is metabolized two times, before it mediates its calcemic effects by binding to the nuclear VitD receptor (VDR) [21, 22](Figure 1). The metabolizing enzymes belong to a group of cytochrome P450 hydroxylases, which can be found in eukaryotes, bacteria, fungi and plants. In the human liver, the first hydroxylation of VitD on C-25 is performed by mitochondrial 25-hydroxylase enzymes (gene names: CYP27A1 [23] and/or CYP2R1 [24]) that both belong to the cytochrome P450 family. The inactive 25-(OH)-vitamin D3 (25-(OH)D3) metabolite is further hydroxylated at position 1α by the mitochondrial cytochrome P450 enzyme 25-hydroxyvitamin-D-1α-hydroxylase (gene name: CYP27B1) and converted to the bioactive 1α,25-dihydroxyvitamin D(1,25-(OH)2D3). This latter step is mainly localized to the proximal kidney tubule [25], however, many other cell types, including lung epithelial cells, are capable to perform this reaction [26–29]. The serum concentration of 25-(OH)D3 reflects the organism's VitD supply [30]. In the blood, VitD and the inactive, relatively stable 25-(OH)D3 metabolite are bound in 99% to the vitamin D binding protein (DBP) [31]. DBP polymorphisms (Gc phenotype) are related to the DBP concentration and VitD status [32]. The 1α-hydroxylation of 25-(OH)D3 is upregulated by parathyroid hormone (PTH), calcitonin, low calcium- and phosphate levels as well as by estrogen, prolactin and growth hormone [33]. Calcitonin, cortisol, high phosphate levels and 25-(OH)D3 suppress the 25-hydroxyvitamin D-1α-hydroxylase activity [34]. 1,25-(OH)2D3 itself works as its own negative feedback regulator by induction of the expression of a 24-hydydroxylase (CYP24A1). Further, 1,25-(OH)2D3 decreases the production and secretion of PTH. PTH synthesis and secretion is induced by decreased serum calcium levels, which are detected by the calcium sensing receptor of the parathyroid gland. PTH effects renal tubular reabsorption of calcium, renal production of 1,25-(OH)2D3 and promotes osteoclastogenesis [35].

Metabolism and effects of VitD. VitD can be obtained from food or from synthesis in the skin under exposure to light. The precursor is hydroxylated cytochrome P450 25-hydroxylase enzymes CYP27A1 and/or CYP2R1 and subsequently by the cytochrome P450 enzyme 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1) and converted to the bioactive 1,25-(OH)2D3, which has role in Ca and bone metabolism and, in addition, in several other biological processes. Of note, bioactive 1,25-(OH)2D3 can also be generated in lung epithelia cells and monocytes/macrophages.
1,25-(OH)2D3 is essential for the development and maintenance of the growth plate, chondrocyte growth, and the mineralised bone [21]. 1,25-(OH)2D3 modulates the osteoclastogenesis by regulation of the receptor activator of nuclear factor kappa B (RANK), RANK ligand (RANKL) and the soluble receptor osteoprotegerin (OPG) [36]. It increases the expression of RANKL on the osteoblast surface, which supports maturation of progenitor and mature osteoclasts, and it inhibits OPG expression, which binds RANKL and prevents RANK mediated osteoclastogenesis [37].
VitD deficiency causes the development of an imbalanced calcium- and phosphate-homeostasis and the occurrence of the bone diseases osteopenia, osteoporosis, rickets, and osteomalacia with a subsequently increased fracture risk [38]. The 25-(OH)D3 serum concentration is directly associated with bone mineral densitys. VitD deficiency has several causes including inadequate sun exposure (and loss of functional capacity of the skin especially in the elderly), limited renal and hepatic function or insufficient intestinal resorption [39]. In VitD deficiency, the feedback on the PTH gene promoter is lacking resulting in parathyroid hyperplasia, hyperparathyroidism, and a mineralization defect of the bone.
1,25-(OH)2D3 regulates many target genes by binding to the VDR: approximately 3% of the mouse and human genome is regulated via the VitD pathway [40]. As non-genomic action of VitD in chondrocytes, it increases the membrane-lipid turnover, prostaglandin production and protease activity, leading to bone matrix modification and calcification. Additionally to the expression of VDR in bone and multiple tissues, the presence of 1α-hydroxylase in cells of several extrarenal tissues such as bone as well as skin, prostate, the respiratory and gastrointestinal tract, strongly suggest that VitD impacts on processes beyond the calcium and bone metabolism.
Role of VitD in immunity and host defense
More than a century ago (1849), the British physician C.J.B. Williams described the use of cod liver oil in the treatment of tuberculosis. He reported that among his tuberculosis patients, 206 out of 234 showed a "marked and unequivocal improvement" after treatment with cod liver oil [41]. Since then manifold functions of VitD have been discovered, indicating that VitD regulates many cellular processes and is potentially involved in the development of many diseases. Since the discovery of VDRs in a variety of cells of the adaptive immune system such as B- and T-lymphocytes [42, 43], there have been numerous reports about the immunomodulatory activities of VitD.
Cellular studies revealed that VitD modulates the activity of various defense and immune cells including monocytes, macrophages, lymphocytes, or epithelial cells:
-
Monocytes/macrophages: Low serum concentrations of VitD in patients with rickets correlate with decreased phagocytic activity of macrophages [44] that could be reversed by supplementation with 1,25-(OH)2D3[45]. Antimicrobial activity of macrophages against M. tuberculosis is increased in the presence of 25-(OH)D3 after stimulation with mycobacterial ligands. Mycobacterial activation of toll-like receptor-2 (TLR-2) leads to an increased expression of VDR and CYP27B that results in an increased conversion of 25-(OH)D3 to 1,25-(OH)2D3 and subsequent expression of the antimicrobial peptide cathelicidin via VDR [46, 47].
-
B lymphocytes: It has been shown that 1,25-(OH)2D3 plays a role in B cell homeostasis by the inhibition of proliferation and induction of apoptosis of activated B cells [48]. 1,25-(OH)2D3 inhibits the differentiation of B lymphocytes to plasma cells and memory B cells. These mechanisms may contribute to the pathogenesis of B-lymphocyte related diseases like systemic lupus erythematosus (SLE). Patients with SLE have significant lower serum concentration of both 25-(OH)D3 and 1,25-(OH)2D3[49, 50].
-
T lymphocytes: A well-established function of VitD within the adaptive immune system is its ability to modulate T lymphocyte proliferation and function. The biologically active 1,25-(OH)2D3 inhibits proliferation of TH lymphocytes [51] and shifts the expression of cytokines from a TH1 based response towards a TH2 based profile [52, 53]. Although 1,25-(OH)2D3 might be able to involve direct effects on T lymphocytes through the support of differentiation of regulatory T cells, current data indicate that 1,25-(OH)2D3 exerts its influence on the adaptive immune response by modulating the functions of dendritic cells (DCs). Regulatory T cells seem to be activated by VitD with skewing of the Th1/Th2 balance towards Th2 [54]. Of note, there is evidence for and against the role of VitD in Th2 biased diseases [55], which will be discussed in more detail in the asthma section below.
-
Dendritic cells: The response of DCs to 1,25-(OH)2D3 is restricted to myeloic DC, that express a different set of TLRs and cytokines than plasmacytoic DCs, which showed no tolerogenic response to 1,25-(OH)2D3[56]. 1,25-(OH)2D3 inhibits the maturation of DCs and enhances the expression of cytokines like IL-10, thereby 1,25-(OH)2D3 induces tolerance through the suppression of TH1 lymphocyte development and the induction of regulatory T cells [57].
-
Epithelial cells: Airway epithelial cell express enzymes of the VitD metabolism and are capable to convert the precursor 25-(OH)D3 into the active 1,25-(OH)2D3 from [29, 58]. They are an important source of 1,25-(OH)2D3 that induces the expression of cathelicidin or CD14 by cells of the innate immune system. 1,25-(OH)2D3 converted by airway epithelial cells is able to modulate the inflammatory profile after a viral infection by blocking the poly(I:C) induced chemokine and cytokine production while maintaining the antiviral activity [28, 59]. As epithelial cells are primary targets of respiratory pathogens and cathelicidin has antibacterial and antiviral activity, a seasonal decrease of VitD-dependent epithelial host defense could contribute to increased numbers of lower respiratory tract infection (RTI) during winter.
Roles of VitD in pulmonary diseases
VitD has complex effects on pulmonary cell biology and immunity with impact on inflammation, host defense, wound healing, repair, and other processes. While the knowledge on direct mechanistic links between VitD and lung diseases is limited, a number of epidemiological and experimental are available that highlight the relevance of this connection.
a) Asthma
A connection between VitD status and asthma has been considered since many years. VitD deficiency has been blamed as one cause of increased asthma prevalence in the last decades [60]. VDR variants were found to be associated with asthma in patient cohorts [61]. A recent clinical investigation showed that high VitD levels are associated with better lung function, less airway hyperresponsiveness and improved glucocorticoid response [62]. A population-based study suggested that lower VitD levels are associated with increased requirements for inhaled corticosteroids in children [63]. Vitamin D insufficiency is common in this children with mild-to-moderate persistent asthma and is associated with higher odds of severe exacerbation [64]. Epidemiologic studies have also shown that maternal VitD intake during pregnancy protects from wheezing in childhood [65, 66]. In contrast, also data exist that children whose mothers had high VitD levels in pregnancy had an increased risk of eczema and asthma [67], suggesting that the time point of Vit D supplementation seems to determine the susceptibility to atopic disease. On the experimental level in a murine asthma model, the VDR is necessary for the development of an allergic airway inflammation [68].
The underlying mechanisms how VitD modulates the pathogenesis of asthma are not clear. VitD may protect from developing respiratory infections that could serve as trigger for a deterioration of asthma [69]. VitD may also modulate the function of various immune cells as outlined above. Interestingly, application of VitD is potentially capable to overcome the poor glucocorticoid responsiveness in severe asthmatics by upregulation of IL-10 production from CD4+ T cells [70].
b) Chronic obstructive lung disease (COPD)
The connection between VitD status and COPD has attracted attention in the recent months. This is based on data from observational studies that determined levels of VitD in COPD patients. Black and colleagues examined data from the NHANES III data set (cross-sectional survey of 14091 adults in the US). After adjustment for potential confounders, a strong relationship between serum levels of VitD and lung function (FEV1 and FVC) was found [71]. Although a significant correlation with airway obstruction could not be found, the observed dose-response relationship may suggest a causal link [72]. A number of studies have reported on 25-(OH)D3 levels in COPD patients. Forli et al. found VitD deficiency (in this study defined as below 20 ng/ml) in more than 50% of a cohort waiting for lung transplantation [73]. In an outpatient study on patients with COPD in Denmark, 68% of the participants had osteoporosis or osteopenia [74]. A recent study showed that VitD deficiency is highly prevalent in COPD and correlates with variants in the VitD binding gene [75]. There are several factors that could account for VitD deficiency in COPD patients: Poor diet, a reduced capacity of aging skin for VitD synthesis, reduced outdoor activity and therefore sun exposure, an increased catabolism by glucocorticoids, impaired activation because of renal dysfunction, and a lower storage capacity in muscles or fat due to wasting [76]. Many steps of the VitD pathway (intake, synthesis, storage, metabolism) can potentially be disturbed in COPD patients.
A single nucleotide polymorphism (SNP) of the DBP was shown to be associated with a decreased risk of COPD by a mechanism that is unclear [77]. Similar SNPs in the gene coding for DBP may influence the level of circulating 25-(OH)D3 and 1,25-(OH)2D3 [32, 78]. Therefore it has been hypothesized that their protective role might be mediated by the bioavailability of 1,25-(OH)2D3 [79].
The mechanisms that link VitD biology with the development of COPD are largely speculative:
-
1)
The association of VitD deficiency and reduced lung function could depend on the calcemic effects of VitD. The vital capacity and total lung capacity was found to decline with an increasing number of thoracic vertebral fractures as a direct consequence of VitD deficiency [80]. Nuti et al. observed 3030 ambulatory COPD patients and found a strong association between COPD severity and fractures [81]. Kyphosis related to osteoporosis caused limitation in rib mobility and inspiratory muscle function and correlated with a reduction in FEV1 and FVC [82]. The altered properties of the thoracic skeleton could result in failure of the respiratory muscles contributing to the pathophysiology of COPD.
-
2)
VitD deficiency could result in altered host defense of the lung with subsequent growth of an abnormal flora that triggers inflammation. Acute exacerbations of COPD are an important cause of hospitalization and lead to a faster decline in FEV1 [83]. Exacerbations are triggered by viruses, bacteria, atypical strains, or a combination of these [84–87]. Potential bacterial pathogens are detected in about 50% of exacerbations. A therapeutic consequence would be the up-regulation of the innate immune defense system. Wang and colleagues demonstrated that genes coding for the antimicrobial peptide cathelicidin (LL-37/hCAP-18) are regulated by VDRE-containing promoters [88]. In cultured monocytes, a local increase of the 1,25D3-VDR complex stimulates the production of LL-37, resulting in an improved intracellular eradication of Mycobacterium tuberculosis [47]. The data demonstrated that the activation of TLRs on human monocytes triggers a microbicidal pathway that is dependent on both the endogenous production and action of 1,25-(OH)2D3 through the VDR.
-
3)
The effect of VitD on extracellular matrix homeostasis not only in bone tissue, but also within the lung may have a role in COPD development. Boyan et al. found VitD to be an autocrine regulator of extracellular matrix turnover and growth factor release via matrix metalloproteinases [89]. Matrix metalloproteinasis-9 (MMP-9) has been shown to be elevated in induced sputum of COPD patients and a causative role has been suggested in the development of COPD [90]. VitD also to attenuates TNF-alpha induced upregulation of MMP-9 in keratinocytes [91]. VitD deficiency may lead to a reduced attenuation of MMP-9 activity resulting in enhanced degradation of lung parenchyma.
Recently, it has been recognized that COPD is a systemic disease [92] with several closely related comorbidities [93]. Interestingly, VitD deficiency is associated with a equivalent spectrum of diseases including coronary heart disease, cancer, inflammatory disease and infection [76]. Comorbidities of COPD such as reduced bone mineral density and skeletal muscle weakness [94, 95] have been associated with low VitD serum concentrations.
c) Infection
Tuberculosis
A number of candidate polymorphisms of VitD receptor (VDR) and VitD binding protein (DBP) have been identified that modulate the development of tuberculosis [96]. The genotype tt (detected by Taq I digestion) is associated with decreased risk of tuberculosis. As described by Lewis et al. [97], larger studies are required to determine whether VDR polymorphisms play a role in genetic susceptibility to tuberculosis worldwide. In a recent meta-analysis, low serum levels of 25-(OH)D3 were associated with a higher risk of active tuberculosis. The pooled effect size was 0.68 with 95% CI 0.43 - 0.93. The authors concluded that the low VitD levels increase the risk of active tuberculosis [98]. There are several randomized, double-blind, placebo-controlled trials of VitD treatment in tuberculosis. In one study, 67 tuberculosis patients were randomized to receive VitD (0.25 mg/day) or placebo during the 6 initial week of Tb treatment [99]. A statistical significant difference in sputum conversion (i.e, the change of detectable to no detectable Mycobacteria in the sputum) was discovered in favor of the VitD group (100% vs. 76,7%; p = 0.002). Another trial was conducted in 192 healthy adult tuberculosis contacts in London, United Kingdom [100]. Participants were randomized to receive a single oral dose of 2.5 mg VitD or placebo and followed up at 6 weeks. VitD supplementation significantly enhanced the ability of participants' whole blood to restrict BCG-lux luminescence after 24 hours in vitro as compared with placebo, but did not affect antigen-stimulated IFN-gamma secretion after 96 hours. As the innate immune responses are mobilized more rapidly than acquired immune responses, the authors interpreted the 24- and 96-hour results as indicators of innate and acquired responses, respectively. They concluded that vitamin D supplementation may primarily enhance innate responses to mycobacterial infection. Wejse et al. included 365 tuberculosis patients starting anti-tuberculotic treatment in Guinea Bissau [101]. 281 patients completed the 12 month follow-up. The intervention was 100,000 IU cholecalciferol or placebo at inclusion and again at 5 and 8 months after start of treatment. Reduction in TBscore and sputum smear conversion rates did not differ among VitD and placebo treated patients. Taken those data together there seems to be a benefit of VitD in the treatment of tuberculosis but this could not be reproduced in the largest study so far.
Respiratory tract infections (RTI)
RTI are more common in the winter period than during summertime. Because the food intake of VitD is insufficient, sunlight exposure is the primary determinant of VitD status in humans, and seasonal differences in VitD level in human are well documented [76]. During the winter months, there is insufficient UV-B exposure to produce sufficient amounts of VitD. Wintertime VitD insufficiency may explain seasonal variation in influenza and other, mostly viral, RTIs [102]. Ginde et al. performed a secondary analysis of the Third National Health and Nutrition Examination Survey, hypothesizing an association between 25-(OH)D3 level and self-reported upper respiratory tract infections (URTI) in 18883 subjects [103]. After adjusting for season, body mass index, smoking history, asthma, and COPD, lower 25-(OH)D3 levels were independently associated with recent URTI. In patients with respiratory tract diseases (asthma and COPD) the association between 25-(OH)D3 level and URTI seemed to be even stronger (OR, 5.67 and 2.26, respectively). Avenell and colleagues used data from the RECORD trial (VitD in secondary prevention of osteoporotic fractures; n = 5292) [104]. In a "per protocol" analysis, a trend towards a benefit of VitD vs. placebo was detected, though not statistically significant. Despite the large number of patients in these studies, restrictions arise from the retrospective data analysis. A prospective cohort study included 800 young Finnish men serving on a military base [105]. Their serum 25-(OH)D3 was measured in the beginning of a 6 month observational period. Subjects with low 25-(OH)D3 levels had significantly more days of absence from duty due to respiratory infection than did control subjects (p = 0.004). In a case control study a total of 150 children (80 cases, 70 controls) was enrolled [106]. Low serum 25-(OH)D3 (≤ 22.5 nmol/l) was associated with a significantly higher odds ratio for having severe acute lower respiratory tract infections (p < 0.001). These studies support an role of VitD in the development of lung infection.
However, in a recent clinical trial, Li-Ng et al. randomized 162 adults to 50 μg VitD (2000 IU) daily or placebo for 12 weeks. Using a questionnaire they recorded the incidence and severity of upper RTI symptoms. Although VitD serum levels increased significantly in the VitD treated group (vs. no change in the placebo group), there was no benefit of VitD supplementation in decreasing the incidence or severity of symptomatic URTI [107]. This may be explained by the relatively low number of subjects. Furthermore, the time period of 12 weeks was probably too short to show any effect. Taken together, there is growing evidence for a protective role of VitD in the development of RTI but high quality randomized clinical trials within a sufficiently high number of patients and for a sufficient period of time are missing. In a recently published trial, the supplementation of 1500 E VitD per day resulted in deceases incidence of influenza A by 64% [69].
d) Cancer
A number of studies suggest that low levels of VitD are associated with an up to 50% increased risk of colon, prostate, or breast cancer [76, 108]. As an example, a recent nested case-control study showed that pre-diagnostic levels of VitD are inversely correlated with the risk of colon cancer [109]. For lung cancer, the picture is not clear at the present time. While TaqI polymorphism of the VDR gene appears to be a risk factor for lung cancer [110], low levels of VitD were only a cancer risk factor in subgroups, i.e., in women and young individuals [111]. In patients with diagnosed lung cancer, there was no main effect of VitD level on overall survival [112]. In preclinical animal models using carcinogen (NNK)-induced lung carcinogenesis, application of 1,25-(OH)2D3 resulted in decreased cancer growth [113].
Conclusions
VitD has a number of activities in addition to its effect on calcium and bone homeostasis and influences process such as immune regulation, host defense, inflammation, or cell proliferation. VitD deficiency is potentially involved in a number of lung disease. Several hurdles must be overcome to validate the benefit of VitD-based therapies: 1) Basic mechanisms are not clear and the involved molecular pathways are likely difficult to identify because VitD impacts on a variety of biological processes in parallel. 2) Conclusive data from interventional studies are missing for many disease entities. 3) Since VitD has been used for many years, the pharmaceutical industry might hesitate in starting a development program. Nevertheless, the data available indicate that VitD could be beneficial for the prevention or therapy of important lung diseases.
Abbreviations
- 1:
-
25-(OH)2D3: 1α: 25-dihydroxyvitamin D
- 25-(OH)D3:
-
D325-(OH)-vitamin D3
- TLR:
-
toll like receptor
- VitD:
-
vitamin D
References
Autier P, Gandini S: Vitamin D supplementation and total mortality: a meta-analysis of randomized controlled trials. Arch Intern Med. 2007, 167: 1730-1737. 10.1001/archinte.167.16.1730.
Goldblatt H, Soames KM: The Supplementary Value of Light Rays to a Diet Graded in its Content of Fat-Soluble Organic Factor. Biochem J. 1923, 17: 622-629.
Steenbock H: The Induction of Growth Promoting and Calcifying Properties in a Ration by Exposure to Light. Science. 1924, 60: 224-225. 10.1126/science.60.1549.224.
McCollum EV, Pitz W, Simmonds N, Becker JE, Shipley PG, Bunting RW: The effect of additions of fluorine to the diet of the rat on the quality of the teeth. 1925. Studies on experimental rickets. XXI. An experimental demonstration of the existence of a vitamin which promotes calcium deposition. 1922. The effect of additions of fluorine to the diet of the rat on the quality of the teeth. 1925. J Biol Chem. 2002, 277: E8-
Holick MF: Evolution and function of vitamin D. Recent Results Cancer Res. 2003, 164: 3-28.
Baker AR, McDonnell DP, Hughes M, Crisp TM, Mangelsdorf DJ, Haussler MR, et al: Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc Natl Acad Sci USA. 1988, 85: 3294-3298. 10.1073/pnas.85.10.3294.
Burmester JK, Wiese RJ, Maeda N, DeLuca HF: Structure and regulation of the rat 1,25-dihydroxyvitamin D3 receptor. Proc Natl Acad Sci USA. 1988, 85: 9499-9502. 10.1073/pnas.85.24.9499.
Lu Z, Hanson K, DeLuca HF: Cloning and origin of the two forms of chicken vitamin D receptor. Arch Biochem Biophys. 1997, 339: 99-106. 10.1006/abbi.1996.9864.
Li YC, Bergwitz C, Juppner H, Demay MB: Cloning and characterization of the vitamin D receptor from Xenopus laevis. Endocrinology. 1997, 138: 2347-2353. 10.1210/en.138.6.2347.
Laing CJ, Fraser DR: The vitamin D system in iguanian lizards. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology. 1999, 123: 373-379. 10.1016/S0305-0491(99)00081-4.
Ciesielski F, Rochel N, Mitschler A, Kouzmenko A, Moras D: Structural investigation of the ligand binding domain of the zebrafish VDR in complexes with 1alpha,25(OH)2D3 and Gemini: purification, crystallization and preliminary X-ray diffraction analysis. J Steroid Biochem Mol Biol. 2004, 89-90: 55-59. 10.1016/j.jsbmb.2004.03.109.
Whitfield GK, Dang HT, Schluter SF, Bernstein RM, Bunag T, Manzon LA, et al: Cloning of a functional vitamin D receptor from the lamprey (Petromyzon marinus), an ancient vertebrate lacking a calcified skeleton and teeth. Endocrinology. 2003, 144: 2704-2716. 10.1210/en.2002-221101.
Sergeev IN, Buffenstein R, Pettifor JM: Vitamin D receptors in a naturally vitamin D-deficient subterranean mammal, the naked mole rat (Heterocephalus glaber): biochemical characterization. Gen Comp Endocrinol. 1993, 90: 338-345. 10.1006/gcen.1993.1089.
Cavaleros M, Buffenstein R, Ross FP, Pettifor JM: Vitamin D metabolism in a frugivorous nocturnal mammal, the Egyptian fruit bat (Rousettus aegyptiacus). Gen Comp Endocrinol. 2003, 133: 109-117. 10.1016/S0016-6480(03)00150-3.
Copping AM: Origin of vitamin D in cod-liver oil: vitamin D content of zooplankton. Biochem J. 1934, 28: 1516-1520.
Sasaki J, Miyazaki A, Saito M, Adachi T, Mizoue K, Hanada K, et al: Transformation of vitamin D3 to 1 alpha,25-dihydroxyvitamin D3 via 25-hydroxyvitamin D3 using Amycolata sp. strains. Appl Microbiol Biotechnol. 1992, 38: 152-157. 10.1007/BF00174460.
Yasutake Y, Fujii Y, Cheon WK, Arisawa A, Tamura T: Crystallization and preliminary X-ray diffraction studies of vitamin D3 hydroxylase, a novel cytochrome P450 isolated from Pseudonocardia autotrophica. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2009, 65: 372-375. 10.1107/S1744309109007829.
Sasaki J, Mikami A, Mizoue K, Omura S: Transformation of 25- and 1 alpha-hydroxyvitamin D3 to 1 alpha, 25-dihydroxyvitamin D3 by using Streptomyces sp. strains. Appl Environ Microbiol. 1991, 57: 2841-2846.
Sawada N, Sakaki T, Yoneda S, Kusudo T, Shinkyo R, Ohta M, et al: Conversion of vitamin D3 to 1alpha,25-dihydroxyvitamin D3 by Streptomyces griseolus cytochrome P450SU-1. Biochem Biophys Res Commun. 2004, 320: 156-164. 10.1016/j.bbrc.2004.05.140.
MacLaughlin JA, Anderson RR, Holick MF: Spectral character of sunlight modulates photosynthesis of previtamin D3 and its photoisomers in human skin. Science. 1982, 216: 1001-1003. 10.1126/science.6281884.
St-Arnaud R: The direct role of vitamin D on bone homeostasis. Arch Biochem Biophys. 2008, 473: 225-230. 10.1016/j.abb.2008.03.038.
Janssens W, Lehouck A, Carremans C, Bouillon R, Mathieu C, Decramer M: Vitamin D beyond bones in chronic obstructive pulmonary disease: time to act. Am J Respir Crit Care Med. 2009, 179: 630-636. 10.1164/rccm.200810-1576PP.
Sawada N, Sakaki T, Ohta M, Inouye K: Metabolism of vitamin D(3) by human CYP27A1. Biochem Biophys Res Commun. 2000, 273: 977-984. 10.1006/bbrc.2000.3050.
Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW: Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci USA. 2004, 101: 7711-7715. 10.1073/pnas.0402490101.
Negri AL: Proximal tubule endocytic apparatus as the specific renal uptake mechanism for vitamin D-binding protein/25-(OH)D3 complex. Nephrology (Carlton ). 2006, 11: 510-515. 10.1111/j.1440-1797.2006.00704.x.
Cross HS, Kallay E, Lechner D, Gerdenitsch W, Adlercreutz H, Armbrecht HJ: Phytoestrogens and vitamin D metabolism: a new concept for the prevention and therapy of colorectal, prostate, and mammary carcinomas. J Nutr. 2004, 134: 1207S-1212S.
Penna G, Adorini L: 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol. 2000, 164: 2405-2411.
Hansdottir S, Monick MM, Hinde SL, Lovan N, Look DC, Hunninghake GW: Respiratory epithelial cells convert inactive vitamin D to its active form: potential effects on host defense. J Immunol. 2008, 181: 7090-7099.
Yim S, Dhawan P, Ragunath C, Christakos S, Diamond G: Induction of cathelicidin in normal and CF bronchial epithelial cells by 1,25-dihydroxyvitamin D(3). J Cyst Fibros. 2007, 6: 403-410. 10.1016/j.jcf.2007.03.003.
Holick MF: Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009, 19: 73-78. 10.1016/j.annepidem.2007.12.001.
Kochupillai N: The physiology of vitamin D: current concepts. Indian J Med Res. 2008, 127: 256-262.
Lauridsen AL, Vestergaard P, Hermann AP, Brot C, Heickendorff L, Mosekilde L, et al: Plasma concentrations of 25-hydroxy-vitamin D and 1,25-dihydroxy-vitamin D are related to the phenotype of Gc (vitamin D-binding protein): a cross-sectional study on 595 early postmenopausal women. Calcif Tissue Int. 2005, 77: 15-22. 10.1007/s00223-004-0227-5.
Kann P: [Vitamin D and osteoporosis. Pathogenesis--therapy]. Dtsch Med Wochenschr. 1994, 119: 1479-1485. 10.1055/s-2008-1058862.
Zhong Y, Armbrecht HJ, Christakos S: Calcitonin, a regulator of the 25-hydroxyvitamin D3 1alpha-hydroxylase gene. J Biol Chem. 2009, 284: 11059-11069. 10.1074/jbc.M806561200.
Anderson PH, O'Loughlin PD, May BK, Morris HA: Quantification of mRNA for the vitamin D metabolizing enzymes CYP27B1 and CYP24 and vitamin D receptor in kidney using real-time reverse transcriptase- polymerase chain reaction. J Mol Endocrinol. 2003, 31: 123-132. 10.1677/jme.0.0310123.
Hofbauer LC, Heufelder AE: Role of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin in bone cell biology. J Mol Med. 2001, 79: 243-253. 10.1007/s001090100226.
Kochupillai N: The physiology of vitamin D: current concepts. Indian J Med Res. 2008, 127: 256-262.
Holick MF, Chen TC: Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr. 2008, 87: 1080S-1086S.
Lips P: Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implications. Endocr Rev. 2001, 22: 477-501. 10.1210/er.22.4.477.
Uitterlinden AG, Fang Y, van Meurs JB, Pols HA, van Leeuwen JP: Genetics and biology of vitamin D receptor polymorphisms. Gene. 2004, 338: 143-156. 10.1016/j.gene.2004.05.014.
Williams CJB: Cod-liver Oil in Phthisis. Lond J Med. 1849, 1: 1-18. 10.1136/bmj.s2-1.1.1.
Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC: 1,25-dihydroxyvitamin D3 receptors in human leukocytes. Science. 1983, 221: 1181-1183. 10.1126/science.6310748.
Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC: 1 alpha,25-Dihydroxyvitamin D3-binding macromolecules in human B lymphocytes: effects on immunoglobulin production. J Immunol. 1986, 136: 2734-2740.
Stroder J, Kasal P: Evaluation of phagocytosis in rickets. Acta Paediatr Scand. 1970, 59: 288-292. 10.1111/j.1651-2227.1970.tb09005.x.
Bar-Shavit Z, Noff D, Edelstein S, Meyer M, Shibolet S, Goldman R: 1,25-dihydroxyvitamin D3 and the regulation of macrophage function. Calcif Tissue Int. 1981, 33: 673-676. 10.1007/BF02409507.
Stenger S, Modlin RL: Control of Mycobacterium tuberculosis through mammalian Toll-like receptors. Curr Opin Immunol. 2002, 14: 452-457. 10.1016/S0952-7915(02)00355-2.
Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al: Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006, 311: 1770-1773. 10.1126/science.1123933.
Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE: Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol. 2007, 179: 1634-1647.
Adams JS, Liu PT, Chun R, Modlin RL, Hewison M: Vitamin D in defense of the human immune response. Ann N Y Acad Sci. 2007, 1117: 94-105. 10.1196/annals.1402.036.
Adorini L, Penna G: Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008, 4: 404-412. 10.1038/ncprheum0855.
Lemire JM, Adams JS, Kermani-Arab V, Bakke AC, Sakai R, Jordan SC: 1,25-Dihydroxyvitamin D3 suppresses human T helper/inducer lymphocyte activity in vitro. J Immunol. 1985, 134: 3032-3035.
Lemire JM, Archer DC, Beck L, Spiegelberg HL: Immunosuppressive actions of 1,25-dihydroxyvitamin D3: preferential inhibition of Th1 functions. J Nutr. 1995, 125: 1704S-1708S.
Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O'Garra A: 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol. 2001, 167: 4974-4980.
Smolders J, Thewissen M, Peelen E, Menheere P, Cohen Tervaert JW, Damoiseaux J, et al: Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. PLoS ONE. 2009, 4: e6635-10.1371/journal.pone.0006635.
Ginde AA, Sutherland ER: Vitamin D in asthma: panacea or true promise?. J Allergy Clin Immunol. 2010, 126: 59-60. 10.1016/j.jaci.2010.05.030.
Penna G, Amuchastegui S, Giarratana N, Daniel KC, Vulcano M, Sozzani S, et al: 1,25-Dihydroxyvitamin D3 selectively modulates tolerogenic properties in myeloid but not plasmacytoid dendritic cells. J Immunol. 2007, 178: 145-153.
Penna G, Adorini L: 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol. 2000, 164: 2405-2411.
Hansdottir S, Monick MM, Hinde SL, Lovan N, Look DC, Hunninghake GW: Respiratory epithelial cells convert inactive vitamin D to its active form: potential effects on host defense. J Immunol. 2008, 181: 7090-7099.
Hansdottir S, Monick MM, Lovan N, Powers L, Gerke A, Hunninghake GW: Vitamin D decreases respiratory syncytial virus induction of NF-kappaB-linked chemokines and cytokines in airway epithelium while maintaining the antiviral state. J Immunol. 2010, 184: 965-974. 10.4049/jimmunol.0902840.
Litonjua AA, Weiss ST: Is vitamin D deficiency to blame for the asthma epidemic?. J Allergy Clin Immunol. 2007, 120: 1031-1035. 10.1016/j.jaci.2007.08.028.
Poon AH, Laprise C, Lemire M, Montpetit A, Sinnett D, Schurr E, et al: Association of vitamin D receptor genetic variants with susceptibility to asthma and atopy. Am J Respir Crit Care Med. 2004, 170: 967-973. 10.1164/rccm.200403-412OC.
Sutherland ER, Goleva E, Jackson LP, Stevens AD, Leung DY: Vitamin D Levels, Lung Function and Steroid Response in Adult Asthma. Am J Respir Crit Care Med. 2010, 181: 699-704. 10.1164/rccm.200911-1710OC.
Brehm JM, Celedon JC, Soto-Quiros ME, Avila L, Hunninghake GM, Forno E, et al: Serum vitamin D levels and markers of severity of childhood asthma in Costa Rica. Am J Respir Crit Care Med. 2009, 179: 765-771. 10.1164/rccm.200808-1361OC.
Brehm JM, Schuemann B, Fuhlbrigge AL, Hollis BW, Strunk RC, Zeiger RS, et al: Serum vitamin D levels and severe asthma exacerbations in the Childhood Asthma Management Program study. J Allergy Clin Immunol. 2010, 126: 52-58. 10.1016/j.jaci.2010.03.043.
Camargo CA, Rifas-Shiman SL, Litonjua AA, Rich-Edwards JW, Weiss ST, Gold DR, et al: Maternal intake of vitamin D during pregnancy and risk of recurrent wheeze in children at 3 y of age. Am J Clin Nutr. 2007, 85: 788-795.
Devereux G, Litonjua AA, Turner SW, Craig LC, McNeill G, Martindale S, et al: Maternal vitamin D intake during pregnancy and early childhood wheezing. Am J Clin Nutr. 2007, 85: 853-859.
Gale CR, Robinson SM, Harvey NC, Javaid MK, Jiang B, Martyn CN, et al: Maternal vitamin D status during pregnancy and child outcomes. Eur J Clin Nutr. 2008, 62: 68-77. 10.1038/sj.ejcn.1602680.
Wittke A, Weaver V, Mahon BD, August A, Cantorna MT: Vitamin D receptor-deficient mice fail to develop experimental allergic asthma. J Immunol. 2004, 173: 3432-3436.
Urashima M, Segawa T, Okazaki M, Kurihara M, Wada Y, Ida H: Randomized trial of vitamin D supplementation to prevent seasonal influenza A in schoolchildren. Am J Clin Nutr. 2010, 91: 1255-1260. 10.3945/ajcn.2009.29094.
Xystrakis E, Kusumakar S, Boswell S, Peek E, Urry Z, Richards DF, et al: Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. J Clin Invest. 2006, 116: 146-155. 10.1172/JCI21759.
Black PN, Scragg R: Relationship between serum 25-hydroxyvitamin d and pulmonary function in the third national health and nutrition examination survey. Chest. 2005, 128: 3792-3798. 10.1378/chest.128.6.3792.
Wright RJ: Make no bones about it: increasing epidemiologic evidence links vitamin D to pulmonary function and COPD. Chest. 2005, 128: 3781-3783. 10.1378/chest.128.6.3781.
Forli L, Halse J, Haug E, Bjortuft O, Vatn M, Kofstad J, et al: Vitamin D deficiency, bone mineral density and weight in patients with advanced pulmonary disease. J Intern Med. 2004, 256: 56-62. 10.1111/j.1365-2796.2004.01337.x.
Jorgensen NR, Schwarz P, Holme I, Henriksen BM, Petersen LJ, Backer V: The prevalence of osteoporosis in patients with chronic obstructive pulmonary disease: a cross sectional study. Respir Med. 2007, 101: 177-185. 10.1016/j.rmed.2006.03.029.
Janssens W, Bouillon R, Claes B, Carremans C, Lehouck A, Buysschaert I, et al: Vitamin D Deficiency is Highly Prevalent in COPD and Correlates with Variants in the Vitamin D Binding Gene. Thorax. 2010, 65: 215-20. 10.1136/thx.2009.120659.
Holick MF: Vitamin D deficiency. N Engl J Med. 2007, 357: 266-281. 10.1056/NEJMra070553.
Schellenberg D, Pare PD, Weir TD, Spinelli JJ, Walker BA, Sandford AJ: Vitamin D binding protein variants and the risk of COPD. Am J Respir Crit Care Med. 1998, 157: 957-961.
Taes YE, Goemaere S, Huang G, Van PI, De BD, Verhasselt B, et al: Vitamin D binding protein, bone status and body composition in community-dwelling elderly men. Bone. 2006, 38: 701-707. 10.1016/j.bone.2005.10.006.
Janssens W, Lehouck A, Carremans C, Bouillon R, Mathieu C, Decramer M: Vitamin D Beyond Bones in Chronic Obstructive Pulmonary Disease: Time to Act. Am J Respir Crit Care Med. 2009, 179: 630-636. 10.1164/rccm.200810-1576PP.
Leech JA, Dulberg C, Kellie S, Pattee L, Gay J: Relationship of lung function to severity of osteoporosis in women. Am Rev Respir dis. 1990, 141: 68-71.
Nuti R, Siviero P, Maggi S, Guglielmi G, Caffarelli C, Crepaldi G, et al: Vertebral fractures in patients with chronic obstructive pulmonary disease: the EOLO Study. Osteoporos Int. 2009, 20: 989-998. 10.1007/s00198-008-0770-4.
Schlaich C, Minne HW, Bruckner T, Wagner G, Gebest HJ, Grunze M, et al: Reduced pulmonary function in patients with spinal osteoporotic fractures. Osteoporos Int. 1998, 8: 261-267. 10.1007/s001980050063.
Niewoehner DE: The impact of severe exacerbations on quality of life and the clinical course of chronic obstructive pulmonary disease. Am J Med. 2006, 119: 38-45. 10.1016/j.amjmed.2006.08.006.
Camargo CA, Ginde AA, Clark S, Cartwright CP, Falsey AR, Niewoehner DE: Viral pathogens in acute exacerbations of chronic obstructive pulmonary disease. Intern Emerg Med. 2008, 3: 355-359. 10.1007/s11739-008-0197-0.
Mogulkoc N, Karakurt S, Isalska B, Bayindir U, Celikel T, Korten V, et al: Acute purulent exacerbation of chronic obstructive pulmonary disease and Chlamydia pneumoniae infection. Am J Respir Crit Care Med. 1999, 160: 349-353.
Lieberman D, Lieberman D, Printz S, Ben-Yaakov M, Lazarovich Z, Ohana B, et al: Atypical pathogen infection in adults with acute exacerbation of bronchial asthma. Am J Respir Crit Care Med. 2003, 167: 406-410. 10.1164/rccm.200209-996OC.
Papi A, Luppi F, Franco F, Fabbri LM: Pathophysiology of exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006, 3: 245-251. 10.1513/pats.200512-125SF.
Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, et al: Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol. 2004, 173: 2909-2912.
Boyan BD, Wong KL, Fang M, Schwartz Z: 1alpha,25(OH)2D3 is an autocrine regulator of extracellular matrix turnover and growth factor release via ERp60 activated matrix vesicle metalloproteinases. J Steroid Biochem Mol Biol. 2007, 103: 467-472. 10.1016/j.jsbmb.2006.11.003.
Culpitt SV, Rogers DF, Traves SL, Barnes PJ, Donnelly LE: Sputum matrix metalloproteases: comparison between chronic obstructive pulmonary disease and asthma. Respir Med. 2005, 99: 703-710. 10.1016/j.rmed.2004.10.022.
Bahar-Shany K, Ravid A, Koren R: Upregulation of MMP-9 production by TNFalpha in keratinocytes and its attenuation by vitamin D. J Cell Physiol. 2010, 222: 729-737.
Sin DD, Man SF: Systemic inflammation and mortality in chronic obstructive pulmonary disease. Can J Physiol Pharmacol. 2007, 85: 141-147. 10.1139/Y06-093.
Walter RE, Wilk JB, Larson MG, Vasan RS, Keaney JF, Lipinska I, et al: Systemic inflammation and COPD: the Framingham Heart Study. Chest. 2008, 133: 19-25. 10.1378/chest.07-0058.
Gosselink R, Troosters T, Decramer M: Peripheral muscle weakness contributes to exercise limitation in COPD. Am J Respir Crit Care Med. 1996, 153: 976-980.
Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, Staehelin HB, Bazemore MG, Zee RY, et al: Effect of Vitamin D on falls: a meta-analysis. Jama. 2004, 291: 1999-2006. 10.1001/jama.291.16.1999.
Leandro AC, Rocha MA, Cardoso CS, Bonecini-Almeida MG: Genetic polymorphisms in vitamin D receptor, vitamin D-binding protein, Toll-like receptor 2, nitric oxide synthase 2, and interferon-gamma genes and its association with susceptibility to tuberculosis. Braz J Med Biol Res. 2009, 42: 312-322. 10.1590/S0100-879X2009000400002.
Lewis SJ, Baker I, Davey SG: Meta-analysis of vitamin D receptor polymorphisms and pulmonary tuberculosis risk. Int J Tuberc Lung Dis. 2005, 9: 1174-1177.
Nnoaham KE, Clarke A: Low serum vitamin D levels and tuberculosis: a systematic review and meta-analysis. Int J Epidemiol. 2008, 37: 113-119. 10.1093/ije/dym247.
Nursyam EW, Amin Z, Rumende CM: The effect of vitamin D as supplementary treatment in patients with moderately advanced pulmonary tuberculous lesion. Acta Med Indones. 2006, 38: 3-5.
Martineau AR, Wilkinson RJ, Wilkinson KA, Newton SM, Kampmann B, Hall BM, et al: A single dose of vitamin D enhances immunity to mycobacteria. Am J Respir Crit Care Med. 2007, 176: 208-213. 10.1164/rccm.200701-007OC.
Wejse C, Gomes VF, Rabna P, Gustafson P, Aaby P, Lisse IM, et al: Vitamin D as Supplementary Treatment for Tuberculosis - A Double-blind Randomized Placebo-controlled Trial. Am J Respir Crit Care Med. 2009, 179: 843-50. 10.1164/rccm.200804-567OC.
Cannell JJ, Vieth R, Umhau JC, Holick MF, Grant WB, Madronich S, et al: Epidemic influenza and vitamin D. Epidemiol Infect. 2006, 134: 1129-1140. 10.1017/S0950268806007175.
Ginde AA, Mansbach JM, Camargo CA: Association between serum 25-hydroxyvitamin D level and upper respiratory tract infection in the Third National Health and Nutrition Examination Survey. Arch Intern Med. 2009, 169: 384-390. 10.1001/archinternmed.2008.560.
Avenell A, Cook JA, Maclennan GS, Macpherson GC: Vitamin D supplementation to prevent infections: a sub-study of a randomised placebo-controlled trial in older people (RECORD trial, ISRCTN 51647438). Age Ageing. 2007, 36: 574-577. 10.1093/ageing/afm091.
Laaksi I, Ruohola JP, Tuohimaa P, Auvinen A, Haataja R, Pihlajamaki H, et al: An association of serum vitamin D concentrations < 40 nmol/L with acute respiratory tract infection in young Finnish men. Am J Clin Nutr. 2007, 86: 714-717.
Wayse V, Yousafzai A, Mogale K, Filteau S: Association of subclinical vitamin D deficiency with severe acute lower respiratory infection in Indian children under 5 y. Eur J Clin Nutr. 2004, 58: 563-567. 10.1038/sj.ejcn.1601845.
Li-Ng M, Aloia JF, Pollack S, Cunha BA, Mikhail M, Yeh J, et al: A randomized controlled trial of vitamin D3 supplementation for the prevention of symptomatic upper respiratory tract infections. Epidemiol Infect. 2009, 137: 1-9. 10.1017/S0950268809002404.
Garland CF, Gorham ED, Mohr SB, Garland FC: Vitamin D for cancer prevention: global perspective. Ann Epidemiol. 2009, 19: 468-483. 10.1016/j.annepidem.2009.03.021.
Jenab M, Bueno-de-Mesquita HB, Ferrari P, van Duijnhoven FJ, Norat T, Pischon T, et al: Association between pre-diagnostic circulating vitamin D concentration and risk of colorectal cancer in European populations:a nested case-control study. Bmj. 2010, 340: b5500-10.1136/bmj.b5500.
Dogan I, Onen HI, Yurdakul AS, Konac E, Ozturk C, Varol A, et al: Polymorphisms in the vitamin D receptor gene and risk of lung cancer. Med Sci Monit. 2009, 15: BR232-BR242.
Kilkkinen A, Knekt P, Heliovaara M, Rissanen H, Marniemi J, Hakulinen T, et al: Vitamin D status and the risk of lung cancer: a cohort study in Finland. Cancer Epidemiol Biomarkers Prev. 2008, 17: 3274-3278. 10.1158/1055-9965.EPI-08-0199.
Heist RS, Zhou W, Wang Z, Liu G, Neuberg D, Su L, et al: Circulating 25-hydroxyvitamin D, VDR polymorphisms, and survival in advanced non-small-cell lung cancer. J Clin Oncol. 2008, 26: 5596-5602. 10.1200/JCO.2008.18.0406.
Mernitz H, Smith DE, Wood RJ, Russell RM, Wang XD: Inhibition of lung carcinogenesis by 1alpha,25-dihydroxyvitamin D3 and 9-cis retinoic acid in the A/J mouse model: evidence of retinoid mitigation of vitamin D toxicity. Int J Cancer. 2007, 120: 1402-1409. 10.1002/ijc.22462.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) to R.B. (Ba 1641/12 and SFB/TR 22 (A8)) and the Kompetenznetz Asthma/COPD (Competence Network for Asthma/COPD funded by the Federal Ministry of Education and research (FKZ 01GI0881-0888 (SP4/12) to RB.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RB developed the concept of the review, all authors contributed in writing and reviewing the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Herr, C., Greulich, T., Koczulla, R.A. et al. The role of vitamin D in pulmonary disease: COPD, asthma, infection, and cancer. Respir Res 12, 31 (2011). https://doi.org/10.1186/1465-9921-12-31
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1465-9921-12-31