
Abstract
Atopic asthma is a chronic inflammatory pulmonary disease characterised by recurrent episodes of wheezy, laboured breathing with an underlying Th2 cell-mediated inflammatory response in the airways. It is currently treated and, more or less, controlled depending on severity, with bronchodilators e.g. long-acting beta agonists and long-acting muscarinic antagonists or anti-inflammatory drugs such as corticosteroids (inhaled or oral), leukotriene modifiers, theophyline and anti-IgE therapy. Unfortunately, none of these treatments are curative and some asthmatic patients do not respond to intense anti-inflammatory therapies. Additionally, the use of long-term oral steroids has many undesired side effects. For this reason, novel and more effective drugs are needed. In this review, we focus on the CD4+ Th2 cells and their products as targets for the development of new drugs to add to the current armamentarium as adjuncts or as potential stand-alone treatments for allergic asthma. We argue that in early disease, the reduction or elimination of allergen-specific Th2 cells will reduce the consequences of repeated allergic inflammatory responses such as lung remodelling without causing generalised immunosuppression.
Similar content being viewed by others

Introduction
Asthma is a serious chronic inflammatory lung disease characterised by recurrent episodes of wheezy laboured breathing with prolonged expiration accompanied by dry coughing and viscous mucus. These symptoms result from bronchoconstriction, bronchial mucosal thickening by oedema, eosinophilic infiltration, bronchial wall remodelling and excessive mucus production with plugging of the conducting airways in the lungs. These airway changes lead to increased bronchial hyperreactivity to a variety of allergic and non-allergic stimuli. Obstruction is usually reversible, either spontaneously or in response to appropriate therapy. Asthma affects approximately 300 million people worldwide and can be fatal. Atopic or allergic asthma generally occurs in childhood or young adulthood (under the age of 40) in about 70-80% of cases and is caused by common allergens e.g. pollens, house dust, animal dander, inhalants, foods, drugs and occupationally encountered dust. Atopic asthma is characterised by detectable allergen-specific IgE and a positive skin test upon allergen provocation. The most severe chronic refractory asthma accounts for 5-10% of adults with asthma and is characterised by persistent symptoms and frequent exacerbations, despite treatment with high dose inhaled and/or oral corticosteroids and inhaled β2 adrenoceptor agonists. These patients are at greater risk of fatal and near-fatal exacerbations and display serious unremitting symptoms, resulting in a considerable impact on quality of life, disproportionate use of health care resources and adverse effects from regular systemic steroid use.
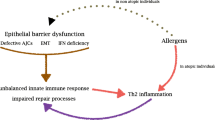
The allergic immune response is a complex process beginning with the activation of allergen-specific Th2 cells by antigen presenting cells (APCs) followed by their proliferation, cytokine production, helper functions and the emergence of memory cells (Figure 1). The resulting pathophysiological response includes lung eosinophilic inflammation, oedema, smooth muscle contraction and increased mucus production, resulting in airway obstruction and eventual lung damage. Numerous experimental models and clinical studies support a central role of allergen-specific Th2 cells in pathophysiological responses [1–4]. Although much is known about the pathogenesis of the disease, the mechanisms underlying Th2 cell differentiation and perpetuation remain unclear. Allergen-specific memory Th2 cells take up long-term residence within experimental mice after recovering from a single episode allergic asthma [5] illustrated by the maintenance of elevated serum allergen-specific IgG1 and persistent inflammatory chronic lung infiltrates. Asthma exacerbations are induced by respiratory tract allergen challenge leading to pathology resembling patients [6–8]. A reduction or elimination of specific Th2 responses permits the treatment of disease without causing generalised immunosuppression and makes it a prime target for disease abrogation. Although current asthma therapies (especially inhaled corticosteroids and β2-agonists) efficiently control the disease, development of novel drugs is crucial for disease control in patients with severe, corticosteroid-insensitive asthma, as well as for improvement of existing therapies in terms of a more favourable side effect profile [9]. Additionally, the use of highly active drugs that reduce disease in the early stages may obviate the need for high dose steroids later on and may reduce the potential for unremitting, steroid-resistant disease. Current asthma therapies do not cure the disease and symptoms return soon after treatment is terminated. Treatment in the late stages of chronic, severe, unremitting allergic asthma may be too late. It is therefore, important to start treatment early to reduce disease. In the early stages of disease, allergen-specific Th2 memory cells appear to play an important role in initiating the immune response against the offending allergen. Eliminating these pathogenic cells at an early stage may lead to complete disease remission. There is a myriad of strategies to eliminate Th2 memory cells that are promising. This review focuses on these targets during the evolution of the Th2-mediated allergic immune response from allergen presentation to activation and survival of Th2 memory cells (Figure 1).

Helper Th2 cells play a central role in allergic asthma and could be targeted through individual allergic immune processes. (1) Allergen handling and presentation by activated APC to naïve CD4+ T cells induces their activation. (2) Activated naïve CD4+ T cells differentiate to Th2 cells, or (3) possibly to other types of helper T cells e.g. Th1, Th17 or Treg cells. (4) Secondary exposure to allergen leads to Th2 cell activation, (5) as well as their migration into the lungs. (6) Activated Th2 cell-mediated asthma is caused in part by the secretion of interleukins e.g. IL-4, IL-5 and IL-13. These cytokines stimulate B cell activation and IgE secretion. Th2 cell cytokines and IgE activate cells of the innate immune system e.g. eosinophils, mast cells, etc. causing the release of vasoactive, pro-inflammatory mediators, smooth muscle contraction, mucus hypersecretion, oedema and, eventually, airway remodelling. (7) Homeostasis and survival of memory T cells in the lymph nodes and lungs perpetuates disease. Interruption of these molecular and cellular targets may reduce symptoms and pathological consequences of allergic asthma.
Improvement of existing anti-Th2 cell therapies
Inhaled and oral corticosteroids, leukotriene modifiers, theophyline, anti-IgE and specific allergen immunotherapy (AI) are well-established treatments for asthma [10]. Of these therapies, only AI specifically targets Th2 cells [11]. AI is thought to function by either skewing the allergic Th2 response towards Th1 immunity or generating regulatory T cells (Tregs) [12, 13]. While the mechanism remains controversial, AI is effective in a subset of patients. Classical immunotherapy or "allergy shots" in the last years is evolving towards non-injectable forms like subcutaneous and sublingual immunotherapy [13, 14]. Progress in AI focuses on the dose and nature of the allergens, with higher allergen doses improving AI effectiveness [15] and chemically modified allergens (allergoids) increasing efficacy [14, 16]. The production of recombinant allergens of common allergens from DNA sequences that can be mutated, fragmented or chimerised leads to efficient hypoallergenic mixtures of allergens for treatment [14, 17, 18]. Additionally important is the ability of producing T cell epitopes without B cell epitopes, which reduces adverse reactions [12, 16, 17] or new technologies like covalently linked T cell epitopes [14], DNA vaccines encoding allergens [19], production of fusion proteins to increase allergen presentation [20], or expression of recombinant allergens in lactic bacteria able to colonise the gut [21]. Equally promising is the production of random peptide libraries to determine structural equivalents, so called mimotopes [14], producing shorter peptides [16, 22], though patients may develop de novo IgE antibodies against the treatment peptide. Although some novel adjuvants such as monophosphoryl lipid A from Salmonella minnesota [12, 23] or heat killed or live Mycobacterium tuberculosis did not meet expectations in clinical trials [14], other adjuvants like fusion proteins with bacterial surface layer components [14] and cytosine-guanine dinucleotides (CpG) oligonucleotides (CpG-ODNs) [9, 12, 23], as well as routes of allergen delivery, in oral microencapsulated forms [24] or embedded in nanoparticles [23], are being explored.
Strategies to modulate antigen presentation and Th2 cell activation
Dendritic cells (DCs) expressing CD11c+CD11b+ [25], CD16+ [26], CD141+ [27] or CD8α [28] predispose to allergic asthma. Sputum and bronchial biopsies of asthmatic patients contain higher DC numbers in comparison to healthy individuals [29, 30] and are increased after allergen exposure [31]. Asthmatic DCs differ in cytokine, prostaglandin (PG), and chemokine synthesis and costimulatory molecule expression compared to healthy controls [32–34]. In addition, allergen-pulsed DCs from asthmatic patients, but not healthy controls, preferentially stimulate T- cells to produce IL-4 [35]. DCs from asthmatics produce high amounts of PGE2 [34], which decreases IL-12 [36] and increases CCL17 and CCL22 production [37] from DCs causing the polarisation of DCs, which promote Th2 cell differentiation and recruitment. Recently, thymic stromal lymphopoietin (TSLP) has emerged as a key mediator, which promotes DC-induced Th2 differentiation through the interaction of OX40:OX40L [38, 39]. Inhibition of DC-mediated antigen presentation represents a suitable treatment option for allergic diseases. While DCs are the most potent APCs, other cells also contribute to antigen presentation and may provide useful targets. Table 1 illustrates the cell type, target and mechanism of action for compounds and biologicals that reduce antigen presentation to Th2 cells and their subsequent activation.
Interference with Th2 differentiation and activation
Antigen presentation induces clonal expansion and differentiation of naïve Th cells into mature Th1, Th2, Th17 or inducible Tregs [reviewed in [40]]. Th2 cell polarisation is mediated by transcription factors, including GATA-3, which are crucial for Th2 lineage commitment. Initial signals that drive Th2 differentiation induce expression of the GATA-3 [41], which mediates Th2 differentiation by inducing chromatin remodelling of Th2 gene loci, direct transactivation of Th2 gene expression and inhibition of IFNγ expression [42]. Furthermore, GATA-3 expression must be sustained to maintain a Th2 phenotype [42, 43]. Beside other important factors, microRNAs have recently emerged as regulators of gene expression during differentiation and function [reviewed in [44, 45]]. Numerous microRNAs play important roles in asthma [46] and selective inhibition of these molecules can be utilised to specifically target development of Th2 cells. Examples of other signal transduction pathway targets and their inhibitors are listed in Tables 2 and 3. Unfortunately, most of these targets are not selectively expressed in Th2 cells and their inhibitors have broad immunosuppressive effects.
Modulation of effector cytokines
The interplay between cells and cytokines involved in Th2-mediated disease is complex. Th2 cells secrete and express a variety of cytokines and receptors [40]. In the past decade, mAbs targeting the most prominent Th2 cytokines, IL-4, IL-5 and IL-13 have had variable success in clinical trials and the perception is that effectiveness will be improved by inhibiting two or all of them simultaneously. Furthermore, additional cytokines including IL-9 and IL-31 are secreted by Th2 cells and might represent novel or additive targets. Moreover, cytokines secreted by other cells such as Th1, Th17 and Tregs may suppress Th2 cell function. Importantly, augmenting suppressive effects and inhibiting disease-promoting effects of T cells may lead to new compounds. Table 4 illustrates examples of cytokines secreted by Th2 cells, have direct effects on Th2 differentiation or are involved in differentiation of other helper T cell subtypes that could inhibit Th2 cells.
Interference of Th2 cell homing and adhesion
Chemokine-chemokine receptors (CKRs) are a complex system of 42 molecules and 19 receptors that orchestrate leukocyte migration in physiologic and pathologic conditions [47]. Among CKRs, CCR4, CCR8, CXCR4 and CCR3 appear to be selectively expressed on Th2 lymphocytes [48, 49] making them potentially important specific Th2 cell targets. CCR4 regulates chemotaxis of Th2 cells and its ligands CCL17 and CCL22 are elevated in allergic asthma [50, 51]. Hence, selective CCR4 antagonists, such as bipiperidinyl carboxylic acid amides, or antibodies directed against CCR4 ligands could be promising treatments [9, 50, 51]. However, CCR4 is also expressed on Tregs and cells with either Th1 or Th2 potential [52] leading to CCR4 inhibitors causing immunosuppressive effects. CCR8 expression also appears to be increased in lung and airway Th2 cells in asthmatic patients [53]. Airway eosinophilia and airway hyperresponsiveness (AHR), however, are not diminished in CCR8-/- mice [54] and adoptively transferred Th2 cells not expressing CCR8 accumulate in the lungs [55]. Despite these contrasting results, several CCR8 agonists [56] and antagonists [57] are in development and might help to clarify the role of CCR8 in disease pathogenesis. CXCR4 is also involved in Th2 cell migration into the lungs [58] and treatment of allergic mice with selective CXCR4 inhibitors significantly reduces AHR and inflammatory responses [59, 60], supporting the further development of CXCR4 antagonists for asthma treatment. CCR3, which regulates eosinophil and mast cell accumulation into the lungs [61], is expressed on Th2 lymphocytes [62]. CCR3 inhibition is a promising Th2 cell target that reduces innate and adaptive allergic inflammation [63]. TPI ASM8 is a compound that contains modified antisense oligonucleotides targeting CCR3 and the common beta chain of the receptors of GM-CSF, IL-5 and IL-13, decreases airway inflammation in humans after allergen exposure and is under clinical evaluation [64]. Other CKRs that appear to regulate CD4+ T cell homing to the lungs in asthma include CCR5, CCR6, CCR7 and CXCR3 [65–67]. CCR7 is a CKR expressed on a large number of naïve and memory T cells [47] and therefore does not represent suitable target. Expression of CCR5, CCR6 and CXCR3 is related to Th1 (CXCR3 and CCR5) [48, 49] or Th17 (CCR6) cells [68]. Thus, it is possible that CKR agonists, rather than antagonists, might inhibit Th2 cells in asthma. Importantly, the chemokine system is highly redundant with promiscuous chemokine-CKR interactions, suggesting that a single chemokine or CKR could have compensatory mechanisms leading to unexpected side effects. Moreover, blocking of a single chemokine or CKR might also not have an effect due to this redundancy.
CRTH2 is a mediator involved in the migration and activation of basophils, eosinophils and Th2 cells [69, 70]. CRTH2 inhibition leads to attenuated airway hyperreactivity and inflammation in animal models [71]. Ramatroban, a dual thrombroxane A2 receptor (TP) and CRTH2 receptor antagonist, suppresses eosinophil chemotaxis in vitro and in vivo and is approved for the treatment of allergic rhinitis in Japan [72]. Numerous other CRTH2 antagonists, such as 4-aminotetrahyrochinoline derivatives or indoleacetic acid derivatives, are currently under development [69, 70, 72] and OC000459 is in clinical trials for the treatment of allergic asthma (ClinicalTrials.gov identifier: NCT01057927, NCT00890877). The CRTH2 receptor is a DP2 receptor. Biological effects of PGD2 and PGH2 are mediated by D prostanoid receptor 1 (DP1) and CRTH2 (DP2). PGD2 activates DP1, thereby affecting NK cells and their cytokine production into a profile more favourable for Th2 skewing [73]. PGH2 is implicated in the accumulation of CRTH2+ cells at sites of inflammation [74]. Additionally, as discussed above, PGE2 polarises DCs to promote Th2 cell differentiation and recruitment [34, 36, 37]. These effects of PGE2 seem to be mediated by PGE2 receptor type 2 (EP2) and type 4 (EP4) [75]. Therefore, PGs and CRTH2 appear to be promising Th2 cell-specific targets.
While homing receptors are important for Th2 cell migration, several adhesion molecules also play a role. For example, intercellular adhesion molecule (ICAM)-1 and ICAM-2 play important roles in T cell migration in the lungs [76] and ICAM-1 deficiency reduces leukocyte infiltration into the airways, as well as IL-4 and IL-5 concentration in bronchoalveolar lavage fluid [77]. Additionally, VCAM-1 plays a role in eosinophil migration and activation in addition to T cell trafficking [78]. There are no clinical data to date for mAbs against ICAM-1 or VCAM-1 in the treatment of asthma. Other potential adhesion targets include VLA-4 (α4ß1 integrin) [79] or P-, E- and L-selectins [80]. Natalizumab blocks both α4ß1 and α4ß7 integrins, but was discontinued due to severe side -effects [81]. Novel α4 integrin mAb LLP2A reduces AHR and inflammation in mouse allergic asthma [82]. Unfortunately, initial results with VLA-4 antagonist GW559090 were disappointing [83], but newer and safer alternative VLA-4 antagonists are in development [84–86]. Lastly, a pan-selectin inhibitor is currently in phase IIa clinical trials for COPD, might also be promising for asthma [81]. None of these adhesion molecules is selectively expressed on Th2 cells.
The anticoagulant heparin has anti-inflammatory properties that inhibit leukocyte extravasation [87]. IVX-0142 is a heparin-derived hypersulfated disaccharide that appears to be well-tolerated and shows a trend towards attenuation of asthmatic responses, but does not affect AHR [88]. Additional studies are needed to evaluate effects of these molecules on Th2 cells.
Inhibition of long-lived Th2 memory cells
It is possible that long-lived Th2 memory cells establish anti-apoptotic mechanisms for long-term maintenance, which when inhibited may result in cell death. Interfering with mechanisms for their longevity in the lungs may eliminate Th2 cells. Corticosteroids [89], calcineurin inhibitors [90] and the cysteine leukotriene receptor antagonist montelukast [89] have pro-apoptotic effects on activated T cells, one of the many mechanisms that lead to their effectiveness in asthma. CX3CR1 seems to provide a survival signal for lung Th2 and Th1 cells, which when inhibited reduces allergic inflammation [67]. T cells from p53-deficient mice have decreased apoptosis and increased Th2 differentiation [91], cytoxic lymphocyte antigen-4 (CTLA-4) promotes T cell apoptosis [92, 93] and CTLA-4-deficient Th cells are directed towards Th2 differentiation [94]. Additionally, the ratio of anti-apoptotic protein Bcl-2 over pro-apoptotic protein Bax in peripheral blood lymphocytes of asthmatic patients is increased in comparison to healthy controls [95]. Interestingly, Th2 cells express less Fas ligand (FasL) and are more resistant to apoptosis than other Th subtypes [96, 97]. Moreover, the Th2 cytokine IL-4 reduces FasL, while Th1 cytokines IFNγ, TGFβ and IL-2 increase FasL expression [89]. Regulation of FasL plays an important role because FasL-expressing T cells are pivotal during the resolution of airway inflammation [98] and intratracheal delivery of DCs co-transfected with FasL and allergen genes before allergen challenge-induced T cell apoptosis and decreased airway inflammation in mice [99]. Induction of Fas expression on Th2 cells might be a possible treatment approach that would decrease their survival in the lungs despite the fact that Th2 cells are somewhat resistant to Fas-induced apoptosis. An additional important pathway for apoptosis in T cells involves granzyme B, which is critical for activation-induced cell death [100]. Inhibition of granzyme B rescues Th2 cells from apoptosis [100], suggesting that selective activation of granzyme B in Th2 cells might be a novel target. Another possibility is that increased apoptosis of Tregs and their protection from apoptosis might be a method of treating disease but there is little information related to cell death of Tregs in allergic diseases and it is possible that dysregulated apoptosis of Tregs may contribute to allergic asthma [90].
New categories of targets: Statins and Rho kinases; TIM proteins; Galectins; Siglecs; Arginases; Histone deacetylase inhibitors; Pathogens and Toll-like receptors
Statins are a class of cholesterol lowering drugs that also possess anti-inflammatory and immune properties [101, 102]. Simvastatin, Lovastatin and Pravastatin reduced eosinophilia and Th2 cytokines in animal models of asthma [103–105]. Clinical trials evaluating Simvastatin (NCT00792337), Lovastatin (NCT00689806) and Atorvastatin (NCT00463827), are ongoing or completed, but data are not yet available. Some statins exert their action through regulation of Rho kinases [106], which are expressed at high levels in airway smooth muscle and regulate their contractility [107], but inhibition appears to impair lymphocyte cytokine secretion [108].
The genes for the T cell immunoglobulin domain and mucin domain (TIM) proteins are encoded in the T cell and airway phenotype regulator region on chromosome 11 [109]. Initial results indicate that although TIM-1 is involved in Th2 cell differentiation and is associated with Th2-mediated diseases [110], it also regulates Th17 and Treg development. Furthermore, TIM proteins are expressed by other immune- cell types [111]. Because TIM proteins do not exclusively regulate Th2 cells, they are less useful as targets than originally anticipated.
Galectins are β-galactoside-binding proteins that bind to glycan residues on the surface of mammalian cells [112]. Examples are Galectin-3 and -9, which appear to have numerous functions in T cell activation, differentiation and apoptosis [112]. Airway inflammation and challenge is decreased in Galectin-3 knockout mice [113] and intranasal administration of a plasmid encoding Galectin-3 abates chronic airway inflammation in a murine model of asthma [114]. Galectin-9 binds to TIM-3, which is expressed on Th1 cells and is important for protective immunity against microbes [111] and intravenous administration of Galectin-9 suppresses AHR and airway inflammation in a mouse model of asthma [115].
Siglecs are sialic acid-recognising Ig-superfamily lectins [116]. CD33-related Siglecs, which in humans include Siglec-3 and Siglecs-5 through -11, are predominantly found on human leukocytes and involved in innate immunity [116, 117]. Mouse Siglec-F, the equivalent of human Siglec-8, is expressed on eosinophils and regulates their apoptosis [118]. Blocking Siglec-F function with a mAb reduces airway and lung eosinophilia in mice [119]. Although Siglec-8 is a promising target directed against eosinophils, human T lymphocytes express little or no siglec molecules [120] and do not appear to be a candidate for inhibiting Th2 cells.
Arginase I and II are cationic amino acid transporters involved in the metabolism of basic amino acids expressed in inflammatory lesions of patients with allergic asthma [121–123]. Arginase gene expression and enzyme activity are enhanced by IL-4 and IL-13 [122, 123]. Inhibition of arginase I by RNA interference suppresses IL-13-mediated AHR in a murine model of asthma [124] and inhalation of an arginase inhibitor decreases AHR and airway inflammation in a guinea pig model of asthma [122]. Conversely, deletion of arginase in macrophages impairs their ability to suppress Th2-dependent inflammation and fibrosis [125]. Further research is needed in order to clarify the role of arginases in Th2 immunity.
Histone deacetylases (HDAC) appear to play an important role in cytokine transcription [126]. Corticosteroid signalling requires HDAC2 to suppress inflammatory gene products and HDAC2 activity is diminished in corticosteroid-resistance [9]. HDAC inhibitor Trichostatin A reduces allergic airway inflammation by decreasing expression of the Th2 cytokines, IL-4, IL-5 and IgE [127]. In contrast, HDAC1 appears to be a negative regulator of Th2 cytokine expression [128]. Chromatin modification enzymes might be potential targets for inhibition of Th2-mediated diseases.
Many microbials or their proteins inhibit Th2 immune responses in murine models of asthma by polarising towards Th1 immunity [129–131] or by generating suppressive Tregs [132, 133]. Interestingly, microbial agents have both time- and dose-dependent effects on allergic asthma [134, 135]. Certain allergens such as dust mite Der p2 and Der f2, bind LPS and are related to the MD-2 protein of the LPS-binding component of the TLR4 signalling complex [136, 137], which might influence the induction of Th2 responses demonstrating a potential for microbials augmenting rather than reducing Th2 responses. Alternatively, bacterial DNA or chemically synthesised de novo unmethylated CpG are immunostimulatory ligands that bind to TLR9 and induce strong Th1 immune responses [reviewed in [138]]. Such bacterial and synthetic DNA immunostimulatory oligonucleotides (ISS-ODNs) containing CpG motifs suppress Th2 responses during the sensitisation phase or immediately before challenge in experimental asthma [reviewed in [139]]. They have therapeutic and prophylactic properties [140], including suppression of DC migration and co-stimulatory molecule expression and inhibition of IgE-dependent Th2 cytokine release from mast cells and basophils [141, 142]. Moreover, ISS-ODNs added to allergen immunotherapy significantly reduce clinical symptoms in patients with asthma [143].
Conclusions
Th2 cells and/or their secreted effector molecules mediate the immune response to allergens and are triggered by exposure to specific allergens leading to allergic asthma. Thus, inhibiting or eliminating Th2 cells is a beneficial strategy for treating asthma as long as generalised immunosuppression is avoided. Additionally, it is especially important to consider targeting Th2 cells early in disease because when disease is chronic additional factors may cause perpetuation. Although there are a myriad of potential Th2 targets (Figure 2), the optimal, most effective anti-Th2 cell target for the clinic remains elusive.

Promising cellular and molecular target candidates for inhibiting memory Th2 cells in allergic asthma are grouped according to the subchapters of the text. Targets in the inner circle (dash line) were tested in clinical trials, whereas other targets are still in pre-clinical stages. Treatment targets are divided into the following categories: Red colour indicates target inhibition; Blue colour indicates target activation; and Green colour indicates targets that can be inhibited or activated to inhibit Th2 cells. For details, please refer to the text. Abbreviations: DC - dendritic cell; FasL - Fas ligand; IL - interleukin; IL-25R - interleukin 25 receptor; IL-33R - interleukin 33 receptor; miRNA - microRNA; Mφ - macrophage; PDE - phosphodiesterase; STAT - Signal transducer and activator of transcription; Th1 - helper T cell type 1; Th2 - helper T cell type 2; TLR - Toll like receptor; Treg - regulatory T cell; TSLP - thymic stromal lymphopoietin.
Aside from anti-IgE therapy for severe asthma, there are no major new drugs for the treatment of asthma in the last 20 years. The latest research in allergic asthma that has elucidated key factors governing Th2 immunity and identified potential targets is predominantly from animal models. Now, the challenge is to discover candidates, which best translate from animal models to patients. However, choosing the most effective new drug target candidate is especially difficult because human data is often lacking or incomplete. Additionally, the use of accurate, predictive biomarkers to evaluate Th2-modulating drugs such as FEV1, Quality of Life, reduction in steroid use, decrease allergen-induced late phase response and others are important to ensure that the efficacy/adverse effect profiles are standardised and enable easier decision-making for the best candidates. We would argue that the most promising new compounds for the clinic are those in which proof of concept in patients is established e.g. anti-IL-13 antibodies and CRTH2 antagonists. Other candidates currently tested in the clinic are anti-IL-5, anti-IL-4 and anti-IL-9 compounds and CCR4 antagonists. However, based on available human data, we suggest that the epithelial cell-derived cytokines TSLP, IL-25 and IL-33, which drive Th2 responses are the most promising candidates, with TLSP the clear frontrunner.
Steroids are efficient for treating asthma because they inhibit numerous pro-inflammatory responses and induce numerous anti-inflammatory pathways. Thus, targeting a single mediator may not suffice for the treatment of allergic asthma because of the redundant immune and inflammatory pathways involved upon allergen challenge. Thus, we suggest that targeting more than one molecule simultaneously using dual specific antibody/protein platforms to engineer new drugs will be the next major approach in drug discovery. However, while this approach creates a scenario in which numerous targets can be combined, the caveat is that optimal candidates must be carefully chosen. Another important consideration for the therapeutic strategy for allergic asthma is that drugs may need to be developed for specific subtypes of disease in which particular cellular and molecular pathways drive the disease. One example is the anti-IL-5 mAb, which is only effective in asthmatics with very high sputum and lung eosinophil numbers. This example suggests that it is beneficial to better categorise patients and consider personalised medicine based on a clear classification of disease.
These are exciting times for Th2 cell immunology as the results of basic research are defining key molecular and cellular components in the response to allergens. This information is already being converted to targets that are being tested in the clinic. Currently, irrespective of approach, we consider that a successful strategy for the treatment of allergic asthma will include a selective inhibition of Th2 cells with the ultimate aim of eliminating allergen-specific Th2 immune responses. We anticipate that new candidates will be approved in the near future and offer treatment options for patients suffering with asthma and other allergic diseases.
Abbreviations
- AHR:
-
Airway hyperresponsiveness
- AI:
-
Allergen immunotherapy
- APC:
-
Antigen presenting cell
- Bcl:
-
B cell lymphoma
- Th:
-
CD4+ T helper
- CKR:
-
Chemokine receptor
- CRTH2:
-
Chemoattractant receptor-homologous molecule expressed on TH2 cells
- CpG:
-
Cytosine-guanine dinucleotides
- CpG-ODN:
-
Cytosine-phosphate-guanine oligonucleotides
- CTLA-4:
-
Cytoxic lymphocyte antigen- 4
- DC:
-
Dendritic cell
- DP1:
-
D prostanoid receptor 1
- EGF:
-
Epidermal growth factor
- ERK:
-
Extracellular signal regulated kinase
- FasL:
-
Fas ligand
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- HDAC:
-
Histone deacetylases
- Ig:
-
Immunoglobulin
- ISS-ODNs:
-
Immunostimulatory oligodeoxynucleotides
- ICAM:
-
Intercellular adhesion molecule
- IFN:
-
Interferon
- IL:
-
Interleukin
- JAK:
-
Janus kinase
- JNK:
-
Jun kinase
- mAb:
-
monoclonal antibody
- MAPK:
-
Mitogen-activated protein kinases
- PDE:
-
Phosphodiesterase
- PI3K:
-
Phosphoinositide 3-kinase
- PD-1:
-
Programmed death-1
- PG:
-
Prostaglandin
- Siglec:
-
Sialic acid binding Ig-like lectins
- STAT:
-
Signal transducer and activator of transcription
- SOCS:
-
Suppressor of cytokine signalling
- Treg:
-
regulatory T cell
- TSLP:
-
Thymic stromal lymphopoietin
- TLR:
-
Toll-like receptor
- TGF:
-
Transforming growth factor
- TNF:
-
Tumour necrosis factor
- VCAM:
-
Vascular cell adhesion molecule
- VLA:
-
Very late antigen.
References
Garlisi CG, Falcone A, Kung TT, Stelts D, Pennline KJ, Beavis AJ, Smith SR, Egan RW, Umland SP: T cells are necessary for Th2 cytokine production and eosinophil accumulation in airways of antigen-challenged allergic mice. Clin Immunol Immunopathol. 1995, 75: 75-83. 10.1006/clin.1995.1055.
Leigh R, Ellis R, Wattie JN, Hirota JA, Matthaei KI, Foster PS, O'Byrne PM, Inman MD: Type 2 cytokines in the pathogenesis of sustained airway dysfunction and airway remodeling in mice. Am J Respir Crit Care Med. 2004, 169: 860-867. 10.1164/rccm.200305-706OC.
Larche M, Robinson DS, Kay AB: The role of T lymphocytes in the pathogenesis of asthma. J Allergy Clin Immunol. 2003, 111: 450-463. 10.1067/mai.2003.169.
Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB: Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992, 326: 298-304. 10.1056/NEJM199201303260504.
Mojtabavi N, Dekan G, Stingl G, Epstein MM: Long-Lived Th2 Memory in Experimental Allergic Asthma. J Immunol. 2002, 169: 4788-4796.
Blackburn MR, Lee CG, Young HW, Zhu Z, Chunn JL, Kang MJ, Banerjee SK, Elias JA: Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J Clin Invest. 2003, 112: 332-344.
Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD: Interleukin-13: central mediator of allergic asthma. Science. 1998, 282: 2258-2261.
Zhu Z, Lee CG, Zheng T, Chupp G, Wang J, Homer RJ, Noble PW, Hamid Q, Elias JA: Airway inflammation and remodeling in asthma. Lessons from interleukin 11 and interleukin 13 transgenic mice. Am J Respir Crit Care Med. 2001, 164: S67-70.
Adcock IM, Caramori G, Chung KF: New targets for drug development in asthma. Lancet. 2008, 372: 1073-1087. 10.1016/S0140-6736(08)61449-X.
Bateman ED, Hurd SS, Barnes PJ, Bousquet J, Drazen JM, FitzGerald M, Gibson P, Ohta K, O'Byrne P, Pedersen SE, et al: Global strategy for asthma management and prevention: GINA executive summary. Eur Respir J. 2008, 31: 143-178. 10.1183/09031936.00138707.
Caramori G, Groneberg D, Ito K, Casolari P, Adcock IM, Papi A: New drugs targeting Th2 lymphocytes in asthma. J Occup Med Toxicol. 2008, 3 (Suppl 1): S6-10.1186/1745-6673-3-S1-S6.
James LK, Durham SR: Update on mechanisms of allergen injection immunotherapy. Clin Exp Allergy. 2008, 38: 1074-1088. 10.1111/j.1365-2222.2008.02976.x.
Esch RE: Sublingual immunotherapy. Curr Opin Otolaryngol Head Neck Surg. 2008, 16: 260-264. 10.1097/MOO.0b013e3282fc706f.
Rolland JM, Gardner LM, O'Hehir RE: Allergen-related approaches to immunotherapy. Pharmacol Ther. 2009, 121: 273-284. 10.1016/j.pharmthera.2008.11.007.
Reefer AJ, Carneiro RM, Custis NJ, Platts-Mills TA, Sung SS, Hammer J, Woodfolk JA: A role for IL-10-mediated HLA-DR7-restricted T cell-dependent events in development of the modified Th2 response to cat allergen. J Immunol. 2004, 172: 2763-2772.
Niederberger V: Allergen-specific immunotherapy. Immunol Lett. 2009, 122: 131-133. 10.1016/j.imlet.2008.11.012.
Saltoun C, Avila PC: Advances in upper airway diseases and allergen immunotherapy in 2007. J Allergy Clin Immunol. 2008, 122: 481-487. 10.1016/j.jaci.2008.06.027.
Valenta R, Niederberger V: Recombinant allergens for immunotherapy. J Allergy Clin Immunol. 2007, 119: 826-830. 10.1016/j.jaci.2007.01.025.
Qiu J, Li GP, Liu ZG, Ran PX, Zhong NS: DNA vaccine encoding Der p2 allergen down-regulates STAT6 expression in mouse model of allergen-induced allergic airway inflammation. Chin Med J (Engl). 2006, 119: 185-190.
Crameri R, Fluckiger S, Daigle I, Kundig T, Rhyner C: Design, engineering and in vitro evaluation of MHC class-II targeting allergy vaccines. Allergy. 2007, 62: 197-206.
Schabussova I, Wiedermann U: Lactic acid bacteria as novel adjuvant systems for prevention and treatment of atopic diseases. Curr Opin Allergy Clin Immunol. 2008, 8: 557-564. 10.1097/ACI.0b013e328317b88b.
Larche M: Peptide immunotherapy for allergic diseases. Allergy. 2007, 62: 325-331. 10.1111/j.1398-9995.2006.01309.x.
Holgate ST, Polosa R: Treatment strategies for allergy and asthma. Nat Rev Immunol. 2008, 8: 218-230. 10.1038/nri2262.
Marazuela EG, Prado N, Moro E, Fernandez-Garcia H, Villalba M, Rodriguez R, Batanero E: Intranasal vaccination with poly(lactide-co-glycolide) microparticles containing a peptide T of Ole e 1 prevents mice against sensitization. Clin Exp Allergy. 2008, 38: 520-528. 10.1111/j.1365-2222.2007.02922.x.
Medoff BD, Seung E, Hong S, Thomas SY, Sandall BP, Duffield JS, Kuperman DA, Erle DJ, Luster AD: CD11b+ myeloid cells are the key mediators of Th2 cell homing into the airway in allergic inflammation. J Immunol. 2009, 182: 623-635.
Rivas-Carvalho A, Meraz-Rios MA, Santos-Argumedo L, Bajana S, Soldevila G, Moreno-Garcia ME, Sanchez-Torres C: CD16+ human monocyte-derived dendritic cells matured with different and unrelated stimuli promote similar allogeneic Th2 responses: regulation by pro- and anti-inflammatory cytokines. Int Immunol. 2004, 16: 1251-1263. 10.1093/intimm/dxh127.
Yerkovich ST, Roponen M, Smith ME, McKenna K, Bosco A, Subrata LS, Mamessier E, Wikstrom ME, Le Souef P, Sly PD, et al: Allergen-enhanced thrombomodulin (blood dendritic cell antigen 3, CD141) expression on dendritic cells is associated with a TH2-skewed immune response. J Allergy Clin Immunol. 2009, 123: 209-216. 10.1016/j.jaci.2008.09.009. e204
Hammad H, de Vries VC, Maldonado-Lopez R, Moser M, Maliszewski C, Hoogsteden HC, Lambrecht BN: Differential capacity of CD8+ alpha or CD8- alpha dendritic cell subsets to prime for eosinophilic airway inflammation in the T-helper type 2-prone milieu of the lung. Clin Exp Allergy. 2004, 34: 1834-1840. 10.1111/j.1365-2222.2004.02133.x.
Bertorelli G, Bocchino V, Zhou X, Zanini A, Bernini MV, Damia R, Di Comite V, Grima P, Olivieri D: Dendritic cell number is related to IL-4 expression in the airways of atopic asthmatic subjects. Allergy. 2000, 55: 449-454. 10.1034/j.1398-9995.2000.055005449.x.
Moller GM, Overbeek SE, Van Helden-Meeuwsen CG, Van Haarst JM, Prens EP, Mulder PG, Postma DS, Hoogsteden HC: Increased numbers of dendritic cells in the bronchial mucosa of atopic asthmatic patients: downregulation by inhaled corticosteroids. Clin Exp Allergy. 1996, 26: 517-524. 10.1111/j.1365-2222.1996.tb00571.x.
Bratke K, Lommatzsch M, Julius P, Kuepper M, Kleine HD, Luttmann W, Christian Virchow J: Dendritic cell subsets in human bronchoalveolar lavage fluid after segmental allergen challenge. Thorax. 2007, 62: 168-175. 10.1136/thx.2006.067793.
Chen XQ, Yang J, Hu SP, Nie HX, Mao GY, Chen HB: Increased expression of CD86 and reduced production of IL-12 and IL-10 by monocyte-derived dendritic cells from allergic asthmatics and their effects on Th1- and Th2-type cytokine balance. Respiration. 2006, 73: 34-40. 10.1159/000087457.
Hammad H, Charbonnier AS, Duez C, Jacquet A, Stewart GA, Tonnel AB, Pestel J: Th2 polarization by Der p 1--pulsed monocyte-derived dendritic cells is due to the allergic status of the donors. Blood. 2001, 98: 1135-1141. 10.1182/blood.V98.4.1135.
Long JA, Fogel-Petrovic M, Knight DA, Thompson PJ, Upham JW: Higher prostaglandin e2 production by dendritic cells from subjects with asthma compared with normal subjects. Am J Respir Crit Care Med. 2004, 170: 485-491. 10.1164/rccm.200311-1595OC.
Hammad H, Lambrecht BN, Pochard P, Gosset P, Marquillies P, Tonnel AB, Pestel J: Monocyte-derived dendritic cells induce a house dust mite-specific Th2 allergic inflammation in the lung of humanized SCID mice: involvement of CCR7. J Immunol. 2002, 169: 1524-1534.
Kalinski P, Schuitemaker JH, Hilkens CM, Kapsenberg ML: Prostaglandin E2 induces the final maturation of IL-12-deficient CD1a+CD83+ dendritic cells: the levels of IL-12 are determined during the final dendritic cell maturation and are resistant to further modulation. J Immunol. 1998, 161: 2804-2809.
McIlroy A, Caron G, Blanchard S, Fremaux I, Duluc D, Delneste Y, Chevailler A, Jeannin P: Histamine and prostaglandin E up-regulate the production of Th2-attracting chemokines (CCL17 and CCL22) and down-regulate IFN-gamma-induced CXCL10 production by immature human dendritic cells. Immunology. 2006, 117: 507-516. 10.1111/j.1365-2567.2006.02326.x.
Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, Qin FX, Yao Z, Cao W, Liu YJ: TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005, 202: 1213-1223. 10.1084/jem.20051135.
Shi L, Leu SW, Xu F, Zhou X, Yin H, Cai L, Zhang L: Local blockade of TSLP receptor alleviated allergic disease by regulating airway dendritic cells. Clin Immunol. 2008, 129: 202-210. 10.1016/j.clim.2008.07.015.
Zhu J, Paul WE: CD4 T cells: fates, functions, and faults. Blood. 2008, 112: 1557-1569. 10.1182/blood-2008-05-078154.
Zhang DH, Cohn L, Ray P, Bottomly K, Ray A: Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem. 1997, 272: 21597-21603. 10.1074/jbc.272.34.21597.
Yamashita M, Ukai-Tadenuma M, Miyamoto T, Sugaya K, Hosokawa H, Hasegawa A, Kimura M, Taniguchi M, DeGregori J, Nakayama T: Essential role of GATA3 for the maintenance of type 2 helper T (Th2) cytokine production and chromatin remodeling at the Th2 cytokine gene loci. J Biol Chem. 2004, 279: 26983-26990. 10.1074/jbc.M403688200.
Pai SY, Truitt ML, Ho IC: GATA-3 deficiency abrogates the development and maintenance of T helper type 2 cells. Proc Natl Acad Sci USA. 2004, 101: 1993-1998. 10.1073/pnas.0308697100.
Bi Y, Liu G, Yang R: MicroRNAs: novel regulators during the immune response. J Cell Physiol. 2009, 218: 467-472. 10.1002/jcp.21639.
Lodish HF, Zhou B, Liu G, Chen CZ: Micromanagement of the immune system by microRNAs. Nat Rev Immunol. 2008, 8: 120-130. 10.1038/nri2252.
Garbacki N, Di Valentin E, Huynh-Thu VA, Geurts P, Irrthum A, Crahay C, Arnould T, Deroanne C, Piette J, Cataldo D, Colige A: MicroRNAs profiling in murine models of acute and chronic asthma: a relationship with mRNAs targets. PLoS One. 2011, 6: e16509-10.1371/journal.pone.0016509.
Bonecchi R, Galliera E, Borroni EM, Corsi MM, Locati M, Mantovani A: Chemokines and chemokine receptors: an overview. Front Biosci. 2009, 14: 540-551.
Annunziato F, Cosmi L, Galli G, Beltrame C, Romagnani P, Manetti R, Romagnani S, Maggi E: Assessment of chemokine receptor expression by human Th1 and Th2 cells in vitro and in vivo. J Leukoc Biol. 1999, 65: 691-699.
Heijink IH, Van Oosterhout AJ: Strategies for targeting T-cells in allergic diseases and asthma. Pharmacol Ther. 2006, 112: 489-500. 10.1016/j.pharmthera.2006.05.005.
Kuhn CF, Bazin M, Philippe L, Zhang J, Tylaska L, Miret J, Bauer PH: Bipiperidinyl carboxylic acid amides as potent, selective, and functionally active CCR4 antagonists. Chem Biol Drug Des. 2007, 70: 268-272. 10.1111/j.1747-0285.2007.00551.x.
Vijayanand P, Durkin K, Hartmann G, Morjaria J, Seumois G, Staples KJ, Hall D, Bessant C, Bartholomew M, Howarth PH, et al: Chemokine receptor 4 plays a key role in T cell recruitment into the airways of asthmatic patients. J Immunol. 2010, 184: 4568-4574. 10.4049/jimmunol.0901342.
Woodfolk JA: T-cell responses to allergens. J Allergy Clin Immunol. 2007, 119: 280-294. 10.1016/j.jaci.2006.11.008. quiz 295-286
Panina-Bordignon P, Papi A, Mariani M, Di Lucia P, Casoni G, Bellettato C, Buonsanti C, Miotto D, Mapp C, Villa A, et al: The C-C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J Clin Invest. 2001, 107: 1357-1364. 10.1172/JCI12655.
Chung CD, Kuo F, Kumer J, Motani AS, Lawrence CE, Henderson WR, Venkataraman C: CCR8 is not essential for the development of inflammation in a mouse model of allergic airway disease. J Immunol. 2003, 170: 581-587.
Mikhak Z, Fukui M, Farsidjani A, Medoff BD, Tager AM, Luster AD: Contribution of CCR4 and CCR8 to antigen-specific T(H)2 cell trafficking in allergic pulmonary inflammation. J Allergy Clin Immunol. 2009, 123: 67-73. 10.1016/j.jaci.2008.09.049. e63
Fox JM, Najarro P, Smith GL, Struyf S, Proost P, Pease JE: Structure/function relationships of CCR8 agonists and antagonists. Amino-terminal extension of CCL1 by a single amino acid generates a partial agonist. J Biol Chem. 2006, 281: 36652-36661. 10.1074/jbc.M605584200.
Jin J, Wang Y, Wang F, Kerns JK, Vinader VM, Hancock AP, Lindon MJ, Stevenson GI, Morrow DM, Rao P, et al: Oxazolidinones as novel human CCR8 antagonists. Bioorg Med Chem Lett. 2007, 17: 1722-1725. 10.1016/j.bmcl.2006.12.076.
Gonzalo JA, Lloyd CM, Peled A, Delaney T, Coyle AJ, Gutierrez-Ramos JC: Critical involvement of the chemotactic axis CXCR4/stromal cell-derived factor-1 alpha in the inflammatory component of allergic airway disease. J Immunol. 2000, 165: 499-508.
Hu JS, Freeman CM, Stolberg VR, Chiu BC, Bridger GJ, Fricker SP, Lukacs NW, Chensue SW: AMD3465, a novel CXCR4 receptor antagonist, abrogates schistosomal antigen-elicited (type-2) pulmonary granuloma formation. Am J Pathol. 2006, 169: 424-432. 10.2353/ajpath.2006.051234.
Lukacs NW, Berlin A, Schols D, Skerlj RT, Bridger GJ: AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol. 2002, 160: 1353-1360. 10.1016/S0002-9440(10)62562-X.
Humbles AA, Lu B, Friend DS, Okinaga S, Lora J, Al-Garawi A, Martin TR, Gerard NP, Gerard C: The murine CCR3 receptor regulates both the role of eosinophils and mast cells in allergen-induced airway inflammation and hyperresponsiveness. Proc Natl Acad Sci USA. 2002, 99: 1479-1484. 10.1073/pnas.261462598.
Sallusto F, Mackay CR, Lanzavecchia A: Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997, 277: 2005-2007. 10.1126/science.277.5334.2005.
Mori A, Ogawa K, Someya K, Kunori Y, Nagakubo D, Yoshie O, Kitamura F, Hiroi T, Kaminuma O: Selective suppression of Th2-mediated airway eosinophil infiltration by low-molecular weight CCR3 antagonists. Int Immunol. 2007, 19: 913-921. 10.1093/intimm/dxm049.
Gauvreau GM, Boulet LP, Cockcroft DW, Baatjes A, Cote J, Deschesnes F, Davis B, Strinich T, Howie K, Duong M, et al: Antisense therapy against CCR3 and the common beta chain attenuates allergen-induced eosinophilic responses. Am J Respir Crit Care Med. 2008, 177: 952-958. 10.1164/rccm.200708-1251OC.
Kallinich T, Schmidt S, Hamelmann E, Fischer A, Qin S, Luttmann W, Virchow JC, Kroczek RA: Chemokine-receptor expression on T cells in lung compartments of challenged asthmatic patients. Clin Exp Allergy. 2005, 35: 26-33. 10.1111/j.1365-2222.2004.02132.x.
Thomas SY, Banerji A, Medoff BD, Lilly CM, Luster AD: Multiple chemokine receptors, including CCR6 and CXCR3, regulate antigen-induced T cell homing to the human asthmatic airway. J Immunol. 2007, 179: 1901-1912.
Mionnet C, Buatois V, Kanda A, Milcent V, Fleury S, Lair D, Langelot M, Lacoeuille Y, Hessel E, Coffman R, et al: CX3CR1 is required for airway inflammation by promoting T helper cell survival and maintenance in inflamed lung. Nat Med. 2010, 16: 1305-1312. 10.1038/nm.2253.
Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, et al: Phenotypic and functional features of human Th17 cells. J Exp Med. 2007, 204: 1849-1861. 10.1084/jem.20070663.
Pettipher R: The roles of the prostaglandin D(2) receptors DP(1) and CRTH2 in promoting allergic responses. Br J Pharmacol. 2008, 153 (Suppl 1): S191-199.
Royer JF, Schratl P, Carrillo JJ, Jupp R, Barker J, Weyman-Jones C, Beri R, Sargent C, Schmidt JA, Lang-Loidolt D, Heinemann A: A novel antagonist of prostaglandin D2 blocks the locomotion of eosinophils and basophils. Eur J Clin Invest. 2008, 38: 663-671. 10.1111/j.1365-2362.2008.01989.x.
Lukacs NW, Berlin AA, Franz-Bacon K, Sasik R, Sprague LJ, Ly TW, Hardiman G, Boehme SA, Bacon KB: CRTH2 antagonism significantly ameliorates airway hyperreactivity and downregulates inflammation-induced genes in a mouse model of airway inflammation. Am J Physiol Lung Cell Mol Physiol. 2008, 295: L767-779. 10.1152/ajplung.90351.2008.
Stebbins KJ, Broadhead AR, Correa LD, Scott JM, Truong YP, Stearns BA, Hutchinson JH, Prasit P, Evans JF, Lorrain DS: Therapeutic efficacy of AM156, a novel prostanoid DP2 receptor antagonist, in murine models of allergic rhinitis and house dust mite-induced pulmonary inflammation. Eur J Pharmacol. 2010, 638: 142-149. 10.1016/j.ejphar.2010.04.031.
Torres D, Paget C, Fontaine J, Mallevaey T, Matsuoka T, Maruyama T, Narumiya S, Capron M, Gosset P, Faveeuw C, Trottein F: Prostaglandin D2 inhibits the production of IFN-gamma by invariant NK T cells: consequences in the control of B16 melanoma. J Immunol. 2008, 180: 783-792.
Schuligoi R, Sedej M, Waldhoer M, Vukoja A, Sturm EM, Lippe IT, Peskar BA, Heinemann A: Prostaglandin H2 induces the migration of human eosinophils through the chemoattractant receptor homologous molecule of Th2 cells, CRTH2. J Leukoc Biol. 2009, 85: 136-145.
Tajima T, Murata T, Aritake K, Urade Y, Hirai H, Nakamura M, Ozaki H, Hori M: Lipopolysaccharide induces macrophage migration via prostaglandin D(2) and prostaglandin E(2). J Pharmacol Exp Ther. 2008, 326: 493-501. 10.1124/jpet.108.137992.
Porter JC, Hall A: Epithelial ICAM-1 and ICAM-2 regulate the egression of human T cells across the bronchial epithelium. Faseb J. 2009, 23: 492-502.
Furusho S, Myou S, Fujimura M, Kita T, Yasui M, Kasahara K, Nakao S, Takehara K, Sato S: Role of intercellular adhesion molecule-1 in a murine model of toluene diisocyanate-induced asthma. Clin Exp Allergy. 2006, 36: 1294-1302. 10.1111/j.1365-2222.2006.02568.x.
Ueki S, Kihara J, Kato H, Ito W, Takeda M, Kobayashi Y, Kayaba H, Chihara J: Soluble vascular cell adhesion molecule-1 induces human eosinophil migration. Allergy. 2009, 64: 718-724. 10.1111/j.1398-9995.2008.01871.x.
Parmley LA, Elkins ND, Fini MA, Liu YE, Repine JE, Wright RM: Alpha-4/beta-1 and alpha-L/beta-2 integrins mediate cytokine induced lung leukocyte-epithelial adhesion and injury. Br J Pharmacol. 2007, 152: 915-929. 10.1038/sj.bjp.0707443.
Kelly M, Hwang JM, Kubes P: Modulating leukocyte recruitment in inflammation. J Allergy Clin Immunol. 2007, 120: 3-10. 10.1016/j.jaci.2007.05.017.
Mackay CR: Moving targets: cell migration inhibitors as new anti-inflammatory therapies. Nat Immunol. 2008, 9: 988-998. 10.1038/ni.f.210.
Kenyon NJ, Liu R, O'Roark EM, Huang W, Peng L, Lam KS: An alpha4 beta1 integrin antagonist decreases airway inflammation in ovalbumin-exposed mice. Eur J Pharmacol. 2009, 603: 138-146. 10.1016/j.ejphar.2008.11.063.
Ravensberg AJ, Luijk B, Westers P, Hiemstra PS, Sterk PJ, Lammers JW, Rabe KF: The effect of a single inhaled dose of a VLA-4 antagonist on allergen-induced airway responses and airway inflammation in patients with asthma. Allergy. 2006, 61: 1097-1103. 10.1111/j.1398-9995.2006.01146.x.
Davenport RJ, Munday JR: Alpha4-integrin antagonism--an effective approach for the treatment of inflammatory diseases?. Drug Discov Today. 2007, 12: 569-576. 10.1016/j.drudis.2007.05.001.
Muro F, Iimura S, Yoneda Y, Chiba J, Watanabe T, Setoguchi M, Takayama G, Yokoyama M, Takashi T, Nakayama A, Machinaga N: A novel and potent VLA-4 antagonist based on trans-4-substituted cyclohexanecarboxylic acid. Bioorg Med Chem. 2009, 17: 1232-1243. 10.1016/j.bmc.2008.12.026.
Saku O, Ohta K, Arai E, Nomoto Y, Miura H, Nakamura H, Fuse E, Nakasato Y: Synthetic study of VLA-4/VCAM-1 inhibitors: synthesis and structure-activity relationship of piperazinylphenylalanine derivatives. Bioorg Med Chem Lett. 2008, 18: 1053-1057. 10.1016/j.bmcl.2007.12.014.
Li JP, Vlodavsky I: Heparin, heparan sulfate and heparanase in inflammatory reactions. Thromb Haemost. 2009, 102: 823-828.
Duong M, Cockcroft D, Boulet LP, Ahmed T, Iverson H, Atkinson DC, Stahl EG, Watson R, Davis B, Milot J, et al: The effect of IVX-0142, a heparin-derived hypersulfated disaccharide, on the allergic airway responses in asthma. Allergy. 2008, 63: 1195-1201. 10.1111/j.1398-9995.2008.01707.x.
Spinozzi F, de Benedictis D, de Benedictis FM: Apoptosis, airway inflammation and anti-asthma therapy: from immunobiology to clinical application. Pediatr Allergy Immunol. 2008, 19: 287-295. 10.1111/j.1399-3038.2007.00668.x.
Simon HU: Cell death in allergic diseases. Apoptosis. 2009, 14: 439-446. 10.1007/s10495-008-0299-1.
Ohkusu-Tsukada K, Tsukada T, Isobe K: Accelerated development and aging of the immune system in p53-deficient mice. J Immunol. 1999, 163: 1966-1972.
Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, Straus D, Samelson LE, Thompson CB, Bluestone JA: Molecular basis of T cell inactivation by CTLA-4. Science. 1998, 282: 2263-2266.
da Rocha Dias S, Rudd CE: CTLA-4 blockade of antigen-induced cell death. Blood. 2001, 97: 1134-1137. 10.1182/blood.V97.4.1134.
Ubaldi V, Gatta L, Pace L, Doria G, Pioli C: CTLA-4 engagement inhibits Th2 but not Th1 cell polarisation. Clin Dev Immunol. 2003, 10: 13-17. 10.1080/10446670310001598519.
Abdulamir AS, Hafidh RR, Abubakar F, Abbas KA: Changing survival, memory cell compartment, and T-helper balance of lymphocytes between severe and mild asthma. BMC Immunol. 2008, 9: 73-10.1186/1471-2172-9-73.
Ramsdell F, Seaman MS, Miller RE, Picha KS, Kennedy MK, Lynch DH: Differential ability of Th1 and Th2 T cells to express Fas ligand and to undergo activation-induced cell death. Int Immunol. 1994, 6: 1545-1553. 10.1093/intimm/6.10.1545.
Fang Y, Yu S, Ellis JS, Sharav T, Braley-Mullen H: Comparison of sensitivity of Th1, Th2, and Th17 cells to Fas-mediated apoptosis. J Leukoc Biol. 2010, 87: 1019-1028. 10.1189/jlb.0509352.
Tong J, Clay BS, Ferreira CM, Bandukwala HS, Moore TV, Blaine KM, Williams JW, Hoffman LM, Hamann KJ, Shilling RA, et al: Fas ligand expression on T Cells is sufficient to prevent prolonged airway inflammation in a murine model of asthma. Am J Respir Cell Mol Biol. 2010, 43: 342-348. 10.1165/rcmb.2008-0454OC.
Wang Y, Bi Y, Wu K, Wang C: Dendritic cell co-transfected with FasL and allergen genes induces T cell tolerance and decreases airway inflammation in allergen induced murine model. Mol Biol Rep. 2011, 38: 809-817. 10.1007/s11033-010-0170-7.
Devadas S, Das J, Liu C, Zhang L, Roberts AI, Pan Z, Moore PA, Das G, Shi Y: Granzyme B is critical for T cell receptor-induced cell death of type 2 helper T cells. Immunity. 2006, 25: 237-247. 10.1016/j.immuni.2006.06.011.
Hothersall E, McSharry C, Thomson NC: Potential therapeutic role for statins in respiratory disease. Thorax. 2006, 61: 729-734. 10.1136/thx.2005.057976.
Robinson AJ, Kashanin D, O'Dowd F, Fitzgerald K, Williams V, Walsh GM: Fluvastatin and lovastatin inhibit granulocyte macrophage-colony stimulating factor-stimulated human eosinophil adhesion to inter-cellular adhesion molecule-1 under flow conditions. Clin Exp Allergy. 2009, 39: 1866-1874. 10.1111/j.1365-2222.2009.03334.x.
Kim DY, Ryu SY, Lim JE, Lee YS, Ro JY: Anti-inflammatory mechanism of simvastatin in mouse allergic asthma model. Eur J Pharmacol. 2007, 557: 76-86. 10.1016/j.ejphar.2006.11.027.
Imamura M, Okunishi K, Ohtsu H, Nakagome K, Harada H, Tanaka R, Yamamoto K, Dohi M: Pravastatin attenuates allergic airway inflammation by suppressing antigen sensitisation, interleukin 17 production and antigen presentation in the lung. Thorax. 2009, 64: 44-49.
Chiba Y, Arima J, Sakai H, Misawa M: Lovastatin inhibits bronchial hyperresponsiveness by reducing RhoA signaling in rat allergic asthma. Am J Physiol Lung Cell Mol Physiol. 2008, 294: L705-713. 10.1152/ajplung.00531.2007.
Zhou Q, Liao JK: Rho kinase: an important mediator of atherosclerosis and vascular disease. Curr Pharm Des. 2009, 15: 3108-3115. 10.2174/138161209789057986.
Schaafsma D, Gosens R, Zaagsma J, Halayko AJ, Meurs H: Rho kinase inhibitors: a novel therapeutical intervention in asthma?. Eur J Pharmacol. 2008, 585: 398-406. 10.1016/j.ejphar.2008.01.056.
Aihara M, Dobashi K, Iizuka K, Nakazawa T, Mori M: Effect of Y-27632 on release of cytokines from peripheral T cells in asthmatic patients and normal subjects. Int Immunopharmacol. 2004, 4: 557-561. 10.1016/j.intimp.2003.12.014.
Lin J, Kane LP: Are TIM proteins involved in asthma development or pathology?. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2011, 41: 917-919.
Su EW, Lin JY, Kane LP: TIM-1 and TIM-3 proteins in immune regulation. Cytokine. 2008, 44: 9-13. 10.1016/j.cyto.2008.06.013.
Kuchroo VK, Dardalhon V, Xiao S, Anderson AC: New roles for TIM family members in immune regulation. Nat Rev Immunol. 2008, 8: 577-580. 10.1038/nri2366.
Grigorian A, Torossian S, Demetriou M: T-cell growth, cell surface organization, and the galectin-glycoprotein lattice. Immunol Rev. 2009, 230: 232-246. 10.1111/j.1600-065X.2009.00796.x.
Ge XN, Bahaie NS, Kang BN, Hosseinkhani MR, Ha SG, Frenzel EM, Liu FT, Rao SP, Sriramarao P: Allergen-induced airway remodeling is impaired in galectin-3-deficient mice. J Immunol. 2010, 185: 1205-1214. 10.4049/jimmunol.1000039.
Lopez E, del Pozo V, Miguel T, Sastre B, Seoane C, Civantos E, Llanes E, Baeza ML, Palomino P, Cardaba B, et al: Inhibition of chronic airway inflammation and remodeling by galectin-3 gene therapy in a murine model. J Immunol. 2006, 176: 1943-1950.
Katoh S, Ishii N, Nobumoto A, Takeshita K, Dai SY, Shinonaga R, Niki T, Nishi N, Tominaga A, Yamauchi A, Hirashima M: Galectin-9 inhibits CD44-hyaluronan interaction and suppresses a murine model of allergic asthma. Am J Respir Crit Care Med. 2007, 176: 27-35. 10.1164/rccm.200608-1243OC.
Varki A, Angata T: Siglecs--the major subfamily of I-type lectins. Glycobiology. 2006, 16: 1R-27R. 10.1093/glycob/cwj126.
Angata T, Margulies EH, Green ED, Varki A: Large-scale sequencing of the CD33-related Siglec gene cluster in five mammalian species reveals rapid evolution by multiple mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2004, 101: 13251-13256. 10.1073/pnas.0404833101.
Zhang M, Angata T, Cho JY, Miller M, Broide DH, Varki A: Defining the in vivo function of Siglec-F, a CD33-related Siglec expressed on mouse eosinophils. Blood. 2007, 109: 4280-4287. 10.1182/blood-2006-08-039255.
Song DJ, Cho JY, Lee SY, Miller M, Rosenthal P, Soroosh P, Croft M, Zhang M, Varki A, Broide DH: Anti-Siglec-F antibody reduces allergen-induced eosinophilic inflammation and airway remodeling. Journal of immunology. 2009, 183: 5333-5341. 10.4049/jimmunol.0801421.
Nguyen DH, Hurtado-Ziola N, Gagneux P, Varki A: Loss of Siglec expression on T lymphocytes during human evolution. Proceedings of the National Academy of Sciences of the United States of America. 2006, 103: 7765-7770. 10.1073/pnas.0510484103.
King NE, Rothenberg ME, Zimmermann N: Arginine in asthma and lung inflammation. J Nutr. 2004, 134: 2830S-2836S. discussion 2853S
Maarsingh H, Zaagsma J, Meurs H: Arginase: a key enzyme in the pathophysiology of allergic asthma opening novel therapeutic perspectives. Br J Pharmacol. 2009, 158: 652-664. 10.1111/j.1476-5381.2009.00374.x.
Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, Muntel EE, Witte DP, Pegg AA, Foster PS, et al: Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003, 111: 1863-1874.
Yang M, Rangasamy D, Matthaei KI, Frew AJ, Zimmmermann N, Mahalingam S, Webb DC, Tremethick DJ, Thompson PJ, Hogan SP, et al: Inhibition of arginase I activity by RNA interference attenuates IL-13-induced airways hyperresponsiveness. J Immunol. 2006, 177: 5595-5603.
Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, Thompson RW, Cheever AW, Murray PJ, Wynn TA: Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009, 5: e1000371-10.1371/journal.ppat.1000371.
Su RC, Becker AB, Kozyrskyj AL, Hayglass KT: Epigenetic regulation of established human type 1 versus type 2 cytokine responses. J Allergy Clin Immunol. 2008, 121: 57-63. 10.1016/j.jaci.2007.09.004. e53
Choi JH, Oh SW, Kang MS, Kwon HJ, Oh GT, Kim DY: Trichostatin A attenuates airway inflammation in mouse asthma model. Clin Exp Allergy. 2005, 35: 89-96. 10.1111/j.1365-2222.2004.02006.x.
Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, Zhang Y, Rembold M, Gaisberger M, Hartl A, et al: Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol. 2010, 185: 3489-3497. 10.4049/jimmunol.0903610.
Debarry J, Garn H, Hanuszkiewicz A, Dickgreber N, Blumer N, von Mutius E, Bufe A, Gatermann S, Renz H, Holst O, Heine H: Acinetobacter lwoffii and Lactococcus lactis strains isolated from farm cowsheds possess strong allergy-protective properties. J Allergy Clin Immunol. 2007, 119: 1514-1521. 10.1016/j.jaci.2007.03.023.
Smit JJ, Folkerts G, Nijkamp FP: Mycobacteria, genes and the 'hygiene hypothesis'. Curr Opin Allergy Clin Immunol. 2004, 4: 57-62. 10.1097/00130832-200402000-00012.
Christ AP, Rodriguez D, Bortolatto J, Borducchi E, Keller A, Mucida D, Silva JS, Leite LC, Russo M: Enhancement of Th1 Lung Immunity Induced by Recombinant Mycobacterium bovis BCG Attenuates Airway Allergic Disease. Am J Respir Cell Mol Biol. 2009
Yokoi T, Amakawa R, Tanijiri T, Sugimoto H, Torii Y, Amuro H, Son Y, Tajima K, Liu YJ, Ito T, Fukuhara S: Mycobacterium bovis Bacillus Calmette-Guerin suppresses inflammatory Th2 responses by inducing functional alteration of TSLP-activated dendritic cells. Int Immunol. 2008, 20: 1321-1329. 10.1093/intimm/dxn094.
Ou-Yang HF, Hu XB, Ti XY, Shi JR, Li SJ, Qi HW, Wu CG: Suppression of allergic airway inflammation in a mouse model by Der p2 recombined BCG. Immunology. 2009, 128: e343-352. 10.1111/j.1365-2567.2008.02970.x.
Wu Q, Martin RJ, Lafasto S, Chu HW: A low dose of Mycoplasma pneumoniae infection enhances an established allergic inflammation in mice: the role of the prostaglandin E2 pathway. Clin Exp Allergy. 2009, 39: 1754-1763. 10.1111/j.1365-2222.2009.03309.x.
Delayre-Orthez C, de Blay F, Frossard N, Pons F: Dose-dependent effects of endotoxins on allergen sensitization and challenge in the mouse. Clin Exp Allergy. 2004, 34: 1789-1795. 10.1111/j.1365-2222.2004.02082.x.
Ichikawa S, Takai T, Yashiki T, Takahashi S, Okumura K, Ogawa H, Kohda D, Hatanaka H: Lipopolysaccharide binding of the mite allergen Der f 2. Genes Cells. 2009, 14: 1055-1065. 10.1111/j.1365-2443.2009.01334.x.
Inohara N, Nunez G: ML -- a conserved domain involved in innate immunity and lipid metabolism. Trends Biochem Sci. 2002, 27: 219-221. 10.1016/S0968-0004(02)02084-4.
Klinman D, Shirota H, Tross D, Sato T, Klaschik S: Synthetic oligonucleotides as modulators of inflammation. J Leukoc Biol. 2008, 84: 958-964. 10.1189/jlb.1107775.
Fonseca DE, Kline JN: Use of CpG oligonucleotides in treatment of asthma and allergic disease. Adv Drug Deliv Rev. 2009, 61: 256-262. 10.1016/j.addr.2008.12.007.
Sur S, Wild JS, Choudhury BK, Sur N, Alam R, Klinman DM: Long term prevention of allergic lung inflammation in a mouse model of asthma by CpG oligodeoxynucleotides. J Immunol. 1999, 162: 6284-6293.
Constabel H, Stankov MV, Hartwig C, Tschernig T, Behrens GM: Impaired lung dendritic cell migration and T cell stimulation induced by immunostimulatory oligonucleotides contribute to reduced allergic airway inflammation. J Immunol. 2009, 183: 3443-3453. 10.4049/jimmunol.0804223.
Hessel EM, Chu M, Lizcano JO, Chang B, Herman N, Kell SA, Wills-Karp M, Coffman RL: Immunostimulatory oligonucleotides block allergic airway inflammation by inhibiting Th2 cell activation and IgE-mediated cytokine induction. J Exp Med. 2005, 202: 1563-1573. 10.1084/jem.20050631.
Senti G, Johansen P, Haug S, Bull C, Gottschaller C, Muller P, Pfister T, Maurer P, Bachmann MF, Graf N, Kundig TM: Use of A-type CpG oligodeoxynucleotides as an adjuvant in allergen-specific immunotherapy in humans: a phase I/IIa clinical trial. Clin Exp Allergy. 2009, 39: 562-570. 10.1111/j.1365-2222.2008.03191.x.
Angeli V, Hammad H, Staels B, Capron M, Lambrecht BN, Trottein F: Peroxisome proliferator-activated receptor gamma inhibits the migration of dendritic cells: consequences for the immune response. J Immunol. 2003, 170: 5295-5301.
Hammad H, de Heer HJ, Soullie T, Angeli V, Trottein F, Hoogsteden HC, Lambrecht BN: Activation of peroxisome proliferator-activated receptor-gamma in dendritic cells inhibits the development of eosinophilic airway inflammation in a mouse model of asthma. Am J Pathol. 2004, 164: 263-271. 10.1016/S0002-9440(10)63116-1.
Hutchison S, Choo-Kang BS, Gibson VB, Bundick RV, Leishman AJ, Brewer JM, McInnes IB, Garside P: An investigation of the impact of the location and timing of antigen-specific T cell division on airways inflammation. Clin Exp Immunol. 2009, 155: 107-116. 10.1111/j.1365-2249.2008.03800.x.
Idzko M, Hammad H, van Nimwegen M, Kool M, Muller T, Soullie T, Willart MA, Hijdra D, Hoogsteden HC, Lambrecht BN: Local application of FTY720 to the lung abrogates experimental asthma by altering dendritic cell function. J Clin Invest. 2006, 116: 2935-2944. 10.1172/JCI28295.
Nishiuma T, Nishimura Y, Okada T, Kuramoto E, Kotani Y, Jahangeer S, Nakamura S: Inhalation of sphingosine kinase inhibitor attenuates airway inflammation in asthmatic mouse model. Am J Physiol Lung Cell Mol Physiol. 2008, 294: L1085-1093. 10.1152/ajplung.00445.2007.
Sawicka E, Zuany-Amorim C, Manlius C, Trifilieff A, Brinkmann V, Kemeny DM, Walker C: Inhibition of Th1- and Th2-mediated airway inflammation by the sphingosine 1-phosphate receptor agonist FTY720. J Immunol. 2003, 171: 6206-6214.
Xia M, Sui Z: Recent developments in CCR2 antagonists. Expert Opin Ther Pat. 2009, 19: 295-303. 10.1517/13543770902755129.
Pichavant M, Charbonnier AS, Taront S, Brichet A, Wallaert B, Pestel J, Tonnel AB, Gosset P: Asthmatic bronchial epithelium activated by the proteolytic allergen Der p 1 increases selective dendritic cell recruitment. J Allergy Clin Immunol. 2005, 115: 771-778. 10.1016/j.jaci.2004.11.043.
Ip WK, Wong CK, Lam CW: Interleukin (IL)-4 and IL-13 up-regulate monocyte chemoattractant protein-1 expression in human bronchial epithelial cells: involvement of p38 mitogen-activated protein kinase, extracellular signal-regulated kinase 1/2 and Janus kinase-2 but not c-Jun NH2-terminal kinase 1/2 signalling pathways. Clin Exp Immunol. 2006, 145: 162-172. 10.1111/j.1365-2249.2006.03085.x.
Hammad H, Kool M, Soullie T, Narumiya S, Trottein F, Hoogsteden HC, Lambrecht BN: Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J Exp Med. 2007, 204: 357-367. 10.1084/jem.20061196.
Crosby JR, Guha M, Tung D, Miller DA, Bender B, Condon TP, York-DeFalco C, Geary RS, Monia BP, Karras JG, Gregory SA: Inhaled CD86 antisense oligonucleotide suppresses pulmonary inflammation and airway hyper-responsiveness in allergic mice. J Pharmacol Exp Ther. 2007, 321: 938-946. 10.1124/jpet.106.119214.
Yokomura K, Suda T, Matsuda H, Hashizume H, Asada K, Suzuki K, Chida K: Suplatast tosilate alters DC1/DC2 balance in peripheral blood in bronchial asthma. J Asthma. 2005, 42: 567-570. 10.1080/02770900500215913.
Chen YQ, Shi HZ: CD28/CTLA-4--CD80/CD86 and ICOS--B7RP-1 costimulatory pathway in bronchial asthma. Allergy. 2006, 61: 15-26.
Seshasayee D, Lee WP, Zhou M, Shu J, Suto E, Zhang J, Diehl L, Austin CD, Meng YG, Tan M, et al: In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J Clin Invest. 2007, 117: 3868-3878. 10.1172/JCI33559.
Singh AK, Stock P, Akbari O: Role of PD-L1 and PD-L2 in allergic diseases and asthma. Allergy. 2011, 66: 155-162. 10.1111/j.1398-9995.2010.02458.x.
Tilley SL, Boucher RC: A1 antagonism in asthma: better than coffee?. J Clin Invest. 2005, 115: 13-16.
Lopez-Castejon G, Baroja-Mazo A, Pelegrin P: Novel macrophage polarization model: from gene expression to identification of new anti-inflammatory molecules. Cell Mol Life Sci. 2010
Chen CL, Wang YM, Liu CF, Wang JY: The effect of water-soluble chitosan on macrophage activation and the attenuation of mite allergen-induced airway inflammation. Biomaterials. 2008, 29: 2173-2182. 10.1016/j.biomaterials.2008.01.023.
Korf JE, Pynaert G, Tournoy K, Boonefaes T, Van Oosterhout A, Ginneberge D, Haegeman A, Verschoor JA, De Baetselier P, Grooten J: Macrophage reprogramming by mycolic acid promotes a tolerogenic response in experimental asthma. Am J Respir Crit Care Med. 2006, 174: 152-160. 10.1164/rccm.200507-1175OC.
Perrigoue JG, Saenz SA, Siracusa MC, Allenspach EJ, Taylor BC, Giacomin PR, Nair MG, Du Y, Zaph C, van Rooijen N, et al: MHC class II-dependent basophil-CD4+ T cell interactions promote T(H)2 cytokine-dependent immunity. Nat Immunol. 2009, 10: 697-705. 10.1038/ni.1740.
Sokol CL, Barton GM, Farr AG, Medzhitov R: A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol. 2008, 9: 310-318. 10.1038/ni1558.
Sokol CL, Chu NQ, Yu S, Nish SA, Laufer TM, Medzhitov R: Basophils function as antigen-presenting cells for an allergen-induced T helper type 2 response. Nat Immunol. 2009, 10: 713-720. 10.1038/ni.1738.
Yoshimoto T, Yasuda K, Tanaka H, Nakahira M, Imai Y, Fujimori Y, Nakanishi K: Basophils contribute to T(H)2-IgE responses in vivo via IL-4 production and presentation of peptide-MHC class II complexes to CD4+ T cells. Nat Immunol. 2009, 10: 706-712. 10.1038/ni.1737.
Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MA, Kool M, Muskens F, Lambrecht BN: Inflammatory dendritic cells--not basophils--are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. 2010, 207: 2097-2111. 10.1084/jem.20101563.
Kim S, Prout M, Ramshaw H, Lopez AF, LeGros G, Min B: Cutting edge: basophils are transiently recruited into the draining lymph nodes during helminth infection via IL-3, but infection-induced Th2 immunity can develop without basophil lymph node recruitment or IL-3. Journal of immunology. 2010, 184: 1143-1147. 10.4049/jimmunol.0902447.
Ho IC, Tai TS, Pai SY: GATA3 and the T-cell lineage: essential functions before and after T-helper-2-cell differentiation. Nat Rev Immunol. 2009, 9: 125-135. 10.1038/nri2476.
Finotto S, De Sanctis GT, Lehr HA, Herz U, Buerke M, Schipp M, Bartsch B, Atreya R, Schmitt E, Galle PR, et al: Treatment of allergic airway inflammation and hyperresponsiveness by antisense-induced local blockade of GATA-3 expression. J Exp Med. 2001, 193: 1247-1260. 10.1084/jem.193.11.1247.
Lee CC, Huang HY, Chiang BL: Lentiviral-mediated GATA-3 RNAi decreases allergic airway inflammation and hyperresponsiveness. Mol Ther. 2008, 16: 60-65. 10.1038/sj.mt.6300309.
Stritesky GL, Muthukrishnan R, Sehra S, Goswami R, Pham D, Travers J, Nguyen ET, Levy DE, Kaplan MH: The transcription factor STAT3 is required for T helper 2 cell development. Immunity. 2011, 34: 39-49. 10.1016/j.immuni.2010.12.013.
Simeone-Penney MC, Severgnini M, Tu P, Homer RJ, Mariani TJ, Cohn L, Simon AR: Airway epithelial STAT3 is required for allergic inflammation in a murine model of asthma. J Immunol. 2007, 178: 6191-6199.
Kagami S, Nakajima H, Kumano K, Suzuki K, Suto A, Imada K, Davey HW, Saito Y, Takatsu K, Leonard WJ, Iwamoto I: Both stat5a and stat5b are required for antigen-induced eosinophil and T-cell recruitment into the tissue. Blood. 2000, 95: 1370-1377.
D'Cruz LM, Klein L: Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol. 2005, 6: 1152-1159. 10.1038/ni1264.
Ohga K, Kuromitsu S, Takezawa R, Numazaki M, Ishikawa J, Nagashima S, Shimizu Y: YM-341619 suppresses the differentiation of spleen T cells into Th2 cells in vitro, eosinophilia, and airway hyperresponsiveness in rat allergic models. Eur J Pharmacol. 2008, 590: 409-416. 10.1016/j.ejphar.2008.06.035.
Walker W, Healey GD, Hopkin JM: RNA interference of STAT6 rapidly attenuates ongoing interleukin-13-mediated events in lung epithelial cells. Immunology. 2009, 127: 256-266. 10.1111/j.1365-2567.2008.02951.x.
Amsen D, Antov A, Jankovic D, Sher A, Radtke F, Souabni A, Busslinger M, McCright B, Gridley T, Flavell RA: Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity. 2007, 27: 89-99. 10.1016/j.immuni.2007.05.021.
Fang TC, Yashiro-Ohtani Y, Del Bianco C, Knoblock DM, Blacklow SC, Pear WS: Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity. 2007, 27: 100-110. 10.1016/j.immuni.2007.04.018.
Kang JH, Kim BS, Uhm TG, Lee SH, Lee GR, Park CS, Chung IY: Gamma-secretase inhibitor reduces allergic pulmonary inflammation by modulating Th1 and Th2 responses. Am J Respir Crit Care Med. 2009, 179: 875-882. 10.1164/rccm.200806-893OC.
Tu L, Fang TC, Artis D, Shestova O, Pross SE, Maillard I, Pear WS: Notch signaling is an important regulator of type 2 immunity. J Exp Med. 2005, 202: 1037-1042. 10.1084/jem.20050923.
Kovall RA: More complicated than it looks: assembly of Notch pathway transcription complexes. Oncogene. 2008, 27: 5099-5109. 10.1038/onc.2008.223.
Fortini ME: Notch signaling: the core pathway and its posttranslational regulation. Dev Cell. 2009, 16: 633-647. 10.1016/j.devcel.2009.03.010.
Ho IC, Hodge MR, Rooney JW, Glimcher LH: The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 1996, 85: 973-983. 10.1016/S0092-8674(00)81299-4.
Ho IC, Lo D, Glimcher LH: c-maf promotes T helper cell type 2 (Th2) and attenuates Th1 differentiation by both interleukin 4-dependent and -independent mechanisms. J Exp Med. 1998, 188: 1859-1866. 10.1084/jem.188.10.1859.
Ko E, Rho S, Cho C, Choi H, Ko S, Lee Y, Hong MC, Shin MK, Jung SG, Bae H: So-Cheong-Ryong-Tang, tradititional Korean medicine, suppresses Th2 lineage development. Biol Pharm Bull. 2004, 27: 739-743. 10.1248/bpb.27.739.
Won HY, Min HJ, Ahn JH, Yoo SE, Bae MA, Hong JH, Hwang ES: Anti-allergic function and regulatory mechanisms of KR62980 in allergen-induced airway inflammation. Biochem Pharmacol. 2009
Liu Z, Li Z, Mao K, Zou J, Wang Y, Tao Z, Lin G, Tian L, Ji Y, Wu X, et al: Dec2 promotes Th2 cell differentiation by enhancing IL-2R signaling. J Immunol. 2009, 183: 6320-6329. 10.4049/jimmunol.0900975.
Yang XO, Angkasekwinai P, Zhu J, Peng J, Liu Z, Nurieva R, Liu X, Chung Y, Chang SH, Sun B, Dong C: Requirement for the basic helix-loop-helix transcription factor Dec2 in initial TH2 lineage commitment. Nat Immunol. 2009, 10: 1260-1266. 10.1038/ni.1821.
Shinnakasu R, Yamashita M, Kuwahara M, Hosokawa H, Hasegawa A, Motohashi S, Nakayama T: Gfi1-mediated stabilization of GATA3 protein is required for Th2 cell differentiation. J Biol Chem. 2008, 283: 28216-28225. 10.1074/jbc.M804174200.
Hirahara K, Yamashita M, Iwamura C, Shinoda K, Hasegawa A, Yoshizawa H, Koseki H, Gejyo F, Nakayama T: Repressor of GATA regulates TH2-driven allergic airway inflammation and airway hyperresponsiveness. J Allergy Clin Immunol. 2008, 122: 512-520. 10.1016/j.jaci.2008.06.004. e511
Kusam S, Toney LM, Sato H, Dent AL: Inhibition of Th2 differentiation and GATA-3 expression by BCL-6. J Immunol. 2003, 170: 2435-2441.
Yoshimura A, Naka T, Kubo M: SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007, 7: 454-465. 10.1038/nri2093.
Seki Y, Inoue H, Nagata N, Hayashi K, Fukuyama S, Matsumoto K, Komine O, Hamano S, Himeno K, Inagaki-Ohara K, et al: SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat Med. 2003, 9: 1047-1054. 10.1038/nm896.
Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, Esposito B, Perez N, Yasukawa H, Van Snick J, et al: Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med. 2009, 206: 2067-2077. 10.1084/jem.20090545.
Seki Y, Hayashi K, Matsumoto A, Seki N, Tsukada J, Ransom J, Naka T, Kishimoto T, Yoshimura A, Kubo M: Expression of the suppressor of cytokine signaling-5 (SOCS5) negatively regulates IL-4-dependent STAT6 activation and Th2 differentiation. Proc Natl Acad Sci USA. 2002, 99: 13003-13008. 10.1073/pnas.202477099.
Ohshima M, Yokoyama A, Ohnishi H, Hamada H, Kohno N, Higaki J, Naka T: Overexpression of suppressor of cytokine signalling-5 augments eosinophilic airway inflammation in mice. Clin Exp Allergy. 2007, 37: 735-742. 10.1111/j.1365-2222.2007.02707.x.
Lu TX, Munitz A, Rothenberg ME: MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J Immunol. 2009, 182: 4994-5002. 10.4049/jimmunol.0803560.
Mattes J, Collison A, Plank M, Phipps S, Foster PS: Antagonism of microRNA-126 suppresses the effector function of TH2 cells and the development of allergic airways disease. Proc Natl Acad Sci USA. 2009, 106: 18704-18709. 10.1073/pnas.0905063106.
Hur GY, Lee SY, Lee SH, Kim SJ, Lee KJ, Jung JY, Lee EJ, Kang EH, Jung KH, Kim JH, et al: Potential use of an anticancer drug gefinitib, an EGFR inhibitor, on allergic airway inflammation. Exp Mol Med. 2007, 39: 367-375.
Yamamoto N, Takeshita K, Shichijo M, Kokubo T, Sato M, Nakashima K, Ishimori M, Nagai H, Li YF, Yura T, Bacon KB: The orally available spleen tyrosine kinase inhibitor 2-[7-(3,4-dimethoxyphenyl)-imidazo[1,2-c]pyrimidin-5-ylamino]nicotinamide dihydrochloride (BAY 61-3606) blocks antigen-induced airway inflammation in rodents. J Pharmacol Exp Ther. 2003, 306: 1174-1181. 10.1124/jpet.103.052316.
Meltzer EO, Berkowitz RB, Grossbard EB: An intranasal Syk-kinase inhibitor (R112) improves the symptoms of seasonal allergic rhinitis in a park environment. J Allergy Clin Immunol. 2005, 115: 791-796. 10.1016/j.jaci.2005.01.040.
Kudlacz E, Conklyn M, Andresen C, Whitney-Pickett C, Changelian P: The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur J Pharmacol. 2008, 582: 154-161. 10.1016/j.ejphar.2007.12.024.
Malaviya R, Chen CL, Navara C, Liu XP, Keenan M, Waurzyniak B, Uckun FM: Treatment of allergic asthma by targeting janus kinase 3-dependent leukotriene synthesis in mast cells with 4-(3', 5'-dibromo-4'-hydroxyphenyl)amino-6,7-dimethoxyquinazoline (WHI-P97). J Pharmacol Exp Ther. 2000, 295: 912-926.
Malaviya R, Zhu D, Dibirdik I, Uckun FM: Targeting Janus kinase 3 in mast cells prevents immediate hypersensitivity reactions and anaphylaxis. J Biol Chem. 1999, 274: 27028-27038. 10.1074/jbc.274.38.27028.
Duan W, Chan JH, Wong CH, Leung BP, Wong WS: Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. J Immunol. 2004, 172: 7053-7059.
Duan W, Chan JH, McKay K, Crosby JR, Choo HH, Leung BP, Karras JG, Wong WS: Inhaled p38 alpha mitogen-activated protein kinase antisense oligonucleotide attenuates asthma in mice. Am J Respir Crit Care Med. 2005, 171: 571-578.
Adcock IM, Chung KF, Caramori G, Ito K: Kinase inhibitors and airway inflammation. Eur J Pharmacol. 2006, 533: 118-132. 10.1016/j.ejphar.2005.12.054.
Barnes PJ: Novel signal transduction modulators for the treatment of airway diseases. Pharmacol Ther. 2006, 109: 238-245. 10.1016/j.pharmthera.2005.08.001.
Gruenbaum LM, Schwartz R, Woska JR, DeLeon RP, Peet GW, Warren TC, Capolino A, Mara L, Morelock MM, Shrutkowski A, et al: Inhibition of pro-inflammatory cytokine production by the dual p38/JNK2 inhibitor BIRB796 correlates with the inhibition of p38 signaling. Biochem Pharmacol. 2009, 77: 422-432. 10.1016/j.bcp.2008.10.032.
Chialda L, Zhang M, Brune K, Pahl A: Inhibitors of mitogen-activated protein kinases differentially regulate costimulated T cell cytokine production and mouse airway eosinophilia. Respir Res. 2005, 6: 36-10.1186/1465-9921-6-36.
Nath P, Eynott P, Leung SY, Adcock IM, Bennett BL, Chung KF: Potential role of c-Jun NH2-terminal kinase in allergic airway inflammation and remodelling: effects of SP600125. Eur J Pharmacol. 2005, 506: 273-283. 10.1016/j.ejphar.2004.11.040.
Trifilieff A, Keller TH, Press NJ, Howe T, Gedeck P, Beer D, Walker C: CGH2466, a combined adenosine receptor antagonist, p38 mitogen-activated protein kinase and phosphodiesterase type 4 inhibitor with potent in vitro and in vivo anti-inflammatory activities. Br J Pharmacol. 2005, 144: 1002-1010. 10.1038/sj.bjp.0706132.
Lee KS, Park SJ, Kim SR, Min KH, Lee KY, Choe YH, Hong SH, Lee YR, Kim JS, Hong SJ, Lee YC: Inhibition of VEGF blocks TGF-beta1 production through a PI3K/Akt signalling pathway. Eur Respir J. 2008, 31: 523-531. 10.1183/09031936.00125007.
Park SJ, Min KH, Lee YC: Phosphoinositide 3-kinase delta inhibitor as a novel therapeutic agent in asthma. Respirology. 2008, 13: 764-771. 10.1111/j.1440-1843.2008.01369.x.
Birrell MA, Hardaker E, Wong S, McCluskie K, Catley M, De Alba J, Newton R, Haj-Yahia S, Pun KT, Watts CJ, et al: Ikappa-B kinase-2 inhibitor blocks inflammation in human airway smooth muscle and a rat model of asthma. Am J Respir Crit Care Med. 2005, 172: 962-971. 10.1164/rccm.200412-1647OC.
Ziegelbauer K, Gantner F, Lukacs NW, Berlin A, Fuchikami K, Niki T, Sakai K, Inbe H, Takeshita K, Ishimori M, et al: A selective novel low-molecular-weight inhibitor of IkappaB kinase-beta (IKK-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br J Pharmacol. 2005, 145: 178-192. 10.1038/sj.bjp.0706176.
Hirose K, Wakashin H, Oki M, Kagami S, Suto A, Ikeda K, Watanabe N, Iwamoto I, Furuichi Y, Nakajima H: GS143, an IkappaB ubiquitination inhibitor, inhibits allergic airway inflammation in mice. Biochem Biophys Res Commun. 2008, 374: 507-511. 10.1016/j.bbrc.2008.07.072.
Lu S, Liu N, Dass SB, Reiss TF, Knorr BA: Randomized, placebo-controlled study of a selective PDE4 inhibitor in the treatment of asthma. Respir Med. 2009, 103: 342-347. 10.1016/j.rmed.2008.10.024.
Singh D, Petavy F, Macdonald AJ, Lazaar AL, O'Connor BJ: The inhaled phosphodiesterase 4 inhibitor GSK256066 reduces allergen challenge responses in asthma. Respir Res. 2010, 11: 26-10.1186/1465-9921-11-26.
Boswell-Smith V, Spina D, Oxford AW, Comer MB, Seeds EA, Page CP: The pharmacology of two novel long-acting phosphodiesterase 3/4 inhibitors, RPL554 [9,10-dimethoxy-2(2,4,6-trimethylphenylimino)-3-(n-carbamoyl-2-aminoethyl) -3,4,6,7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one] and RPL565 [6,7-dihydro-2-(2,6-diisopropylphenoxy)-9,10-dimethoxy-4H-pyrimido[6,1-a]i soquinolin-4-one]. J Pharmacol Exp Ther. 2006, 318: 840-848. 10.1124/jpet.105.099192.
Hoyer KK, Dooms H, Barron L, Abbas AK: Interleukin-2 in the development and control of inflammatory disease. Immunol Rev. 2008, 226: 19-28. 10.1111/j.1600-065X.2008.00697.x.
Rothenberg ME, Owen WF, Silberstein DS, Woods J, Soberman RJ, Austen KF, Stevens RL: Human eosinophils have prolonged survival, enhanced functional properties, and become hypodense when exposed to human interleukin 3. The Journal of clinical investigation. 1988, 81: 1986-1992. 10.1172/JCI113547.
Valent P, Dahinden CA: Role of interleukins in the regulation of basophil development and secretion. Current opinion in hematology. 2010, 17: 60-66. 10.1097/MOH.0b013e328331fae9.
Asquith KL, Ramshaw HS, Hansbro PM, Beagley KW, Lopez AF, Foster PS: The IL-3/IL-5/GM-CSF common receptor plays a pivotal role in the regulation of Th2 immunity and allergic airway inflammation. J Immunol. 2008, 180: 1199-1206.
Webb DC, Cai Y, Matthaei KI, Foster PS: Comparative roles of IL-4, IL-13, and IL-4Ralpha in dendritic cell maturation and CD4+ Th2 cell function. J Immunol. 2007, 178: 219-227.
Corren J, Busse W, Meltzer EO, Mansfield L, Bensch G, Fahrenholz J, Wenzel SE, Chon Y, Dunn M, Weng HH, Lin SL: A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. American journal of respiratory and critical care medicine. 2010, 181: 788-796. 10.1164/rccm.200909-1448OC.
Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M: Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007, 370: 1422-1431. 10.1016/S0140-6736(07)61600-6.
Oh CK, Geba GP, Molfino N: Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. European respiratory review: an official journal of the European Respiratory Society. 2010, 19: 46-54.
Leckie MJ: Anti-interleukin-5 monoclonal antibodies: preclinical and clinical evidence in asthma models. Am J Respir Med. 2003, 2: 245-259.
Leckie MJ, ten Brinke A, Khan J, Diamant Z, O'Connor BJ, Walls CM, Mathur AK, Cowley HC, Chung KF, Djukanovic R, et al: Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000, 356: 2144-2148. 10.1016/S0140-6736(00)03496-6.
Busse WW, Katial R, Gossage D, Sari S, Wang B, Kolbeck R, Coyle AJ, Koike M, Spitalny GL, Kiener PA, et al: Safety profile, pharmacokinetics, and biologic activity of MEDI-563, an anti-IL-5 receptor alpha antibody, in a phase I study of subjects with mild asthma. J Allergy Clin Immunol. 2010, 125: 1237-1244. 10.1016/j.jaci.2010.04.005. e1232
Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, Marshall RP, Bradding P, Green RH, Wardlaw AJ, Pavord ID: Mepolizumab and exacerbations of refractory eosinophilic asthma. The New England journal of medicine. 2009, 360: 973-984. 10.1056/NEJMoa0808991.
Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, Hargreave FE, O'Byrne PM: Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009, 360: 985-993. 10.1056/NEJMoa0805435.
Rincon M, Anguita J, Nakamura T, Fikrig E, Flavell RA: Interleukin (IL)-6 directs the differentiation of IL-4-producing CD4+ T cells. J Exp Med. 1997, 185: 461-469. 10.1084/jem.185.3.461.
Mucida D, Salek-Ardakani S: Regulation of TH17 cells in the mucosal surfaces. J Allergy Clin Immunol. 2009, 123: 997-1003. 10.1016/j.jaci.2009.03.016.
Weissenbach M, Clahsen T, Weber C, Spitzer D, Wirth D, Vestweber D, Heinrich PC, Schaper F: Interleukin-6 is a direct mediator of T cell migration. Eur J Immunol. 2004, 34: 2895-2906. 10.1002/eji.200425237.
Renauld JC, Goethals A, Houssiau F, Merz H, Van Roost E, Van Snick J: Human P40/IL-9. Expression in activated CD4+ T cells, genomic organization, and comparison with the mouse gene. J Immunol. 1990, 144: 4235-4241.
McLane MP, Haczku A, van de Rijn M, Weiss C, Ferrante V, MacDonald D, Renauld JC, Nicolaides NC, Holroyd KJ, Levitt RC: Interleukin-9 promotes allergen-induced eosinophilic inflammation and airway hyperresponsiveness in transgenic mice. Am J Respir Cell Mol Biol. 1998, 19: 713-720.
Temann UA, Geba GP, Rankin JA, Flavell RA: Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med. 1998, 188: 1307-1320. 10.1084/jem.188.7.1307.
White B, Leon F, White W, Robbie G: Two first-in-human, open-label, phase I dose-escalation safety trials of MEDI-528, a monoclonal antibody against interleukin-9, in healthy adult volunteers. Clin Ther. 2009, 31: 728-740. 10.1016/j.clinthera.2009.04.019.
Parker JM, Oh CK, Laforce C, Miller SD, Pearlman DS, Le C, Robbie GJ, White WI, White B, Molfino NA: Safety profile and clinical activity of multiple subcutaneous doses of MEDI-528, a humanized anti-interleukin-9 monoclonal antibody, in two randomized phase 2a studies in subjects with asthma. BMC Pulm Med. 2011, 11: 14-10.1186/1471-2466-11-14.
O'Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C: Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev. 2008, 223: 114-131. 10.1111/j.1600-065X.2008.00635.x.
Trinchieri G: Proinflammatory and immunoregulatory functions of interleukin-12. Int Rev Immunol. 1998, 16: 365-396. 10.3109/08830189809043002.
Gavett SH, O'Hearn DJ, Li X, Huang SK, Finkelman FD, Wills-Karp M: Interleukin 12 inhibits antigen-induced airway hyperresponsiveness, inflammation, and Th2 cytokine expression in mice. J Exp Med. 1995, 182: 1527-1536. 10.1084/jem.182.5.1527.
Bryan SA, O'Connor BJ, Matti S, Leckie MJ, Kanabar V, Khan J, Warrington SJ, Renzetti L, Rames A, Bock JA, et al: Effects of recombinant human interleukin-12 on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000, 356: 2149-2153. 10.1016/S0140-6736(00)03497-8.
Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA: Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999, 103: 779-788. 10.1172/JCI5909.
Brightling CE, Saha S, Hollins F: Interleukin-13: prospects for new treatments. Clin Exp Allergy:journal of the British Society for Allergy and Clinical Immunology . 2010, 40: 42-49.
Kasaian MT, Miller DK: IL-13 as a therapeutic target for respiratory disease. Biochem Pharmacol. 2008, 76: 147-155. 10.1016/j.bcp.2008.04.002.
Ishimitsu R, Nishimura H, Yajima T, Watase T, Kawauchi H, Yoshikai Y: Overexpression of IL-15 in vivo enhances Tc1 response, which inhibits allergic inflammation in a murine model of asthma. J Immunol. 2001, 166: 1991-2001.
Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, Mathieu C, Ceuppens JL: Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003, 28: 42-50. 10.1165/rcmb.4832.
Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, Fossiez F, Ryffel B, Schnyder B: Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006, 203: 2715-2725. 10.1084/jem.20061401.
Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H: Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol. 2001, 19: 423-474. 10.1146/annurev.immunol.19.1.423.
Maecker HT, Hansen G, Walter DM, DeKruyff RH, Levy S, Umetsu DT: Vaccination with allergen-IL-18 fusion DNA protects against, and reverses established, airway hyperreactivity in a murine asthma model. J Immunol. 2001, 166: 959-965.
Huang F, Wachi S, Thai P, Loukoianov A, Tan KH, Forteza RM, Wu R: Potentiation of IL-19 expression in airway epithelia by IL-17A and IL-4/IL-13: important implications in asthma. J Allergy Clin Immunol. 2008, 121: 1415-1421. 10.1016/j.jaci.2008.04.016.
Liao SC, Cheng YC, Wang YC, Wang CW, Yang SM, Yu CK, Shieh CC, Cheng KC, Lee MF, Chiang SR, et al: IL-19 induced Th2 cytokines and was up-regulated in asthma patients. J Immunol. 2004, 173: 6712-6718.
Sivakumar PV, Foster DC, Clegg CH: Interleukin-21 is a T-helper cytokine that regulates humoral immunity and cell-mediated anti-tumour responses. Immunology. 2004, 112: 177-182. 10.1111/j.1365-2567.2004.01886.x.
Besnard AG, Sabat R, Dumoutier L, Renauld JC, Willart M, Lambrecht B, Teixeira MM, Charron S, Fick L, Erard F, et al: Dual Role of IL-22 in allergic airway inflammation and its cross-talk with IL-17A. Am J Respir Crit Care Med. 2011, 183: 1153-1163. 10.1164/rccm.201008-1383OC.
Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N, Saito Y, Hatano M, Tokuhisa T, Iwakura Y, et al: IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. 2008, 178: 1023-1032. 10.1164/rccm.200801-086OC.
Li Y, Sun M, Cheng H, Li S, Liu L, Qiao H, Hua S, Lu J: Silencing IL-23 expression by a small hairpin RNA protects against asthma in mice. Exp Mol Med. 2011, 43: 197-204. 10.3858/emm.2011.43.4.024.
Ballantyne SJ, Barlow JL, Jolin HE, Nath P, Williams AS, Chung KF, Sturton G, Wong SH, McKenzie AN: Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2007, 120: 1324-1331. 10.1016/j.jaci.2007.07.051.
Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, Hippe A, Corrigan CJ, Dong C, Homey B, et al: IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007, 204: 1837-1847. 10.1084/jem.20070406.
Yoshimoto T, Yasuda K, Mizuguchi J, Nakanishi K: IL-27 suppresses Th2 cell development and Th2 cytokines production from polarized Th2 cells: a novel therapeutic way for Th2-mediated allergic inflammation. J Immunol. 2007, 179: 4415-4423.
Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, Haugen HS, Maurer M, Harder B, Johnston J, et al: Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004, 5: 752-760.
Lei Z, Liu G, Huang Q, Lv M, Zu R, Zhang GM, Feng ZH, Huang B: SCF and IL-31 rather than IL-17 and BAFF are potential indicators in patients with allergic asthma. Allergy. 2008, 63: 327-332.
Lohning M, Stroehmann A, Coyle AJ, Grogan JL, Lin S, Gutierrez-Ramos JC, Levinson D, Radbruch A, Kamradt T: T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci USA. 1998, 95: 6930-6935. 10.1073/pnas.95.12.6930.
Xu D, Chan WL, Leung BP, Huang F, Wheeler R, Piedrafita D, Robinson JH, Liew FY: Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med. 1998, 187: 787-794. 10.1084/jem.187.5.787.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, et al: IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005, 23: 479-490. 10.1016/j.immuni.2005.09.015.
Shannon J, Ernst P, Yamauchi Y, Olivenstein R, Lemiere C, Foley S, Cicora L, Ludwig M, Hamid Q, Martin JG: Differences in airway cytokine profile in severe asthma compared to moderate asthma. Chest. 2008, 133: 420-426. 10.1378/chest.07-1881.
Yang M, Kumar RK, Foster PS: Pathogenesis of steroid-resistant airway hyperresponsiveness: interaction between IFN-gamma and TLR4/MyD88 pathways. J Immunol. 2009, 182: 5107-5115. 10.4049/jimmunol.0803468.
Boguniewicz M, Schneider LC, Milgrom H, Newell D, Kelly N, Tam P, Izu AE, Jaffe HS, Bucalo LR, Leung DY: Treatment of steroid-dependent asthma with recombinant interferon-gamma. Clin Exp Allergy. 1993, 23: 785-790. 10.1111/j.1365-2222.1993.tb00367.x.
Gorelik L, Fields PE, Flavell RA: Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol. 2000, 165: 4773-4777.
Fattouh R, Midence NG, Arias K, Johnson JR, Walker TD, Goncharova S, Souza KP, Gregory RC, Lonning S, Gauldie J, Jordana M: Transforming growth factor-beta regulates house dust mite-induced allergic airway inflammation but not airway remodeling. Am J Respir Crit Care Med. 2008, 177: 593-603. 10.1164/rccm.200706-958OC.
Alcorn JF, Rinaldi LM, Jaffe EF, van Loon M, Bates JH, Janssen-Heininger YM, Irvin CG: Transforming growth factor-beta1 suppresses airway hyperresponsiveness in allergic airway disease. Am J Respir Crit Care Med. 2007, 176: 974-982. 10.1164/rccm.200702-334OC.
Brightling C, Berry M, Amrani Y: Targeting TNF-alpha: a novel therapeutic approach for asthma. J Allergy Clin Immunol. 2008, 121: 5-10. 10.1016/j.jaci.2007.10.028. quiz 11-12
Berry MA, Pavord ID: Antagonism of tumour necrosis factor alpha in refractory asthma. Thorax. 2008, 63: 571-572. 10.1136/thx.2007.095042.
Wenzel SE, Barnes PJ, Bleecker ER, Bousquet J, Busse W, Dahlen SE, Holgate ST, Meyers DA, Rabe KF, Antczak A, et al: A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med. 2009, 179: 549-558. 10.1164/rccm.200809-1512OC.
Acknowledgements
We would like to thank Drs. P. Stuetz and O. Hoffmann for their critical reading of the manuscript, discussions and support. We acknowledge the support for Berislav Bosnjak from the P3AGI project funded by the European Commission through an FP7- IAPP Marie Curie Action - (GA 230739).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Berislav Bosnjak - was employee of GlaxoSmithKline Research Centre Zagreb Ltd. until December 2008. No other competing interests.
Barbara Stelzmüller - none
Klaus J. Erb - is an employee of BoerhingerIngelheim Pharma, Respiratory Diseases Research, Biberach an der Riss, Germany
Michelle M. Epstein - received funding from BoerhingerIngelheim Pharma, Respiratory Diseases Research, Biberach an der Riss, Germany for collaborative project
Authors' contributions
BB - was involved in drafting the manuscript, revising it critically for important intellectual content; and has given final approval of the version to be published.
BS - made substantial contributions to conception of the review, was involved in drafting the manuscript, revising it critically for important intellectual content; and has given final approval of the version to be published.
KE - made substantial contributions to conception of the review, was involved in drafting the manuscript, revising it critically for important intellectual content; and has given final approval of the version to be published.
ME - made substantial contributions to conception of the review, was involved in drafting the manuscript, revising it critically for important intellectual content; and has given final approval of the version to be published.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bosnjak, B., Stelzmueller, B., Erb, K.J. et al. Treatment of allergic asthma: Modulation of Th2 cells and their responses. Respir Res 12, 114 (2011). https://doi.org/10.1186/1465-9921-12-114
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1465-9921-12-114