
Abstract
Background
Human adenovirus 14 (HAdV-14) is a recognized causative agent of epidemic febrile respiratory illness (FRI). Last reported in Eurasia in 1963, this virus has since been conspicuously absent in broad surveys, and was never isolated in North America despite inclusion of specific tests for this serotype in surveillance methods. In 2006 and 2007, this virus suddenly emerged in North America, causing high attack rate epidemics of FRI and, in some cases, severe pneumonias and occasional fatalities. Some outbreaks have been relatively mild, with low rates of progression beyond uncomplicated FRI, while other outbreaks have involved high rates of more serious outcomes.
Methodology and Findings
In this paper we present the complete genomic sequence of this emerging pathogen, and compare genomic sequences of isolates from both mild and severe outbreaks. We also compare the genome sequences of the recent isolates with those of the prototype HAdV-14 that circulated in Eurasia 30 years ago and the closely related sequence of HAdV-11a, which has been circulating in southeast Asia.
Conclusions
The data suggest that the currently circulating strain of HAdV-14 is closely related to the historically recognized prototype throughout its genome, though it does display a couple of potentially functional mutations in the fiber knob and E1A genes. There are no polymorphisms that suggest an obvious explanation for the divergence in severity between outbreak events, suggesting that differences in outcome are more likely environmental or host determined rather than viral genetics.
Similar content being viewed by others

Introduction
Adenoviruses are double-stranded DNA viruses. The 52 recognized serotypes of human adenovirus (HAdV) cause a broad range of symptoms: community-acquired gastrointestinal, conjunctival, and febrile respiratory illness (FRI; both upper and lower respiratory tract), hemorrhagic cystitis associated with bone marrow transplant, hepatic and urinary tract infections, and perhaps even obesity [[1–4], http://www.ncbi.nlm.nih.gov/ICTVdb/Ictv/index.htm].
The 10 serotypes of HAdV associated with FRI and pneumonia are grouped into 3 species, B (including subspecies B1 and B2), C and E, on the basis of hemagglutination and phylogenetic criteria [5–9]. HAdV-1, 2, 5, and 6, belonging to species C, cause generally endemic patterns of FRI in children and young adults [8, 10, 11]. In contrast, HAdV-4 (the sole serotype of species E) and the remaining respiratory species B serotypes (HAdV-3, 7, 11, 14, 16, and 21), often cause distinctive outbreaks of FRI, conjunctivitis, and pneumonia in crowded civilian populations such as dorms, public swimming pools, and boarding schools [7, 8]. In the absence of vaccines, these viruses also cause almost continuous outbreaks of FRI among recruits in military training throughout the world [8, 7, 12, 13].
Four of these seven adult human respiratory adenoviruses, HAdV-3, 7 and 21 (subspecies B1) and HAdV-4 (species E) are common, intraserotypically diverse, and inevitably represented in broad surveys [7, 8, 10, 14]. In different populations and at different times one serotype may completely dominate this niche, several serotypes may intermingle, or multiple serotypes can appear in series through distinct replacement events [13, 15–19]. The remaining three serotypes that cause FRI in healthy adults, HAdV-16 of subspecies B1 [11] and HAdV-11 and 14 of subspecies B2 [13], have only infrequently been associated with FRI. These rare associations often appear to involve more severe symptoms, outcomes, and outbreak characteristics than do those of the more common species E and subspecies B1 serotypes [11, 20–24]. HAdV-14 was reported only four times in the twentieth century, always in transient, concentrated, and generally nonlethal but severely incapacitating FRI outbreaks in healthy (though crowded) adult and adolescent populations [25–28]. These outbreaks occurred between 1955 and 1963, all in Eurasia, and HAdV-14 was not reported again even in broad geographical and temporal surveys until 2001 when it was reported in 10% of FRI specimens in a retrospective analysis of clinic samples in Taiwan [29]. (Author's note: upon whole-genome analysis, this strain was identified as the very closely-related HAdV-11a; HSH, AK, data not shown). HAdV-11a has recently been seen in increased numbers of FRI cases in Asia, including some significant outbreaks [30]. Phenotypic intermediates of the closely related serotypes HAdV-11 and HAdV-14 were identified in a military camp in Spain in 1969 [31], and in Germany from a severe case of acute respiratory disease following a military training exercise (and apparently associated outbreak) in Turkey in 2004 [32].
HAdV-14 had never been identified in North America before its emergence in 2006. Following the recognized outbreaks in 2006 and 2007 [13, 22, 24], retrospective analysis of specific cases and collections uncovered isolated occurrences of the disease dating back a few years before the larger outbreaks (for example, see [23]). HAdV-14 was first seen in greater numbers and associated with significant outbreaks in March 2006, when it simultaneously emerged at four military recruit training centers throughout the United States, causing several hundred cases (estimated from partial surveillance) of FRI over the course of the calendar year [13]. The impact amounted to a partial replacement of the recently dominant HAdV-4, rather than an increase in overall adenoviral impact at these sites. These emergence events were not associated with symptoms or epidemiological patterns outside the normal range of those seen with the typical species E and subspecies B1 HAdVs seen in surveillance of recruit FRI and pneumonia [13].
Starting in March 2007, HAdV-14 was recognized as the cause of several severe civilian outbreaks, prompting attention from the Centers for Disease Control and Prevention (CDC) [22]. The same pathogen was recognized as the cause of prolonged outbreaks at three military installations where HAdV-14 either emerged against an adenovirus-free background ([22], and an outbreak at Coast Guard Training Center, Cape May; unpublished Naval Health Research Center [NHRC] data) or completely replaced the existing HAdV-4 strain (Marine Corps Recruit Depot [MCRD], Parris Island, NJ; unpublished NHRC data). Reported civilian HAdV-14 outbreaks were transient, lasting four months and involving nine casualties [22]. The two noted outbreaks in recruit facilities where there was no immediate history of ongoing adenovirus transmission were initially severe, involving greatly increased rates of disease among the recruits and also spreading to medical support personnel, training staff, and others. One death was reported among infected recruits as were many pneumonia hospitalizations, several requiring ventilation assistance [22].
Whole-genome restriction enzyme analysis (genome typing [33]) and partial gene sequence analysis (hexon, fiber, E1A) have shown that the currently emergent US strains of HAdV-14 (see result section for the definition of genome type HAdV-14p1), both civilian and military, are all similar at the genome type level and essentially identical (> 99.9%) at the sequence level in the hexon and fiber genes (AK, unpublished data). The circulating genome type, however, is significantly diverged from the prototype (HAdV-14p) de Wit strain (isolated from ill military recruits in the Netherlands [28]).
To further characterize the newly emergent US HAdV-14 strains, three recent HAdV-14p1 isolates were completely sequenced and compared with the prototype HAdV-14p de Wit genome sequence as well as other genetically related HAdV-11a isolates from southeast Asia. One isolate was collected in March 2006 at Marine Corps Recruit Depot, San Diego (MCRD-SD), when HAdV-14 was initially detected and identified in US recruits [13]. This isolate came from an emergence of HAdV-14p1 that did not exhibit uniquely severe outbreak dynamics or symptoms - in fact, it was observed during this outbreak, in which both HAdV-4 and HAdV-14 were present in approximately equal proportions, that HAdV-14 did not seem to cause as much pneumonia as did HAdV-4 (unpublished NHRC syndromic surveillance data). This outbreak did not involve increased rates, but rather a simple and temporary replacement of HAdV-4 with HAdV-14 [13], and the studied isolate was collected from a recruit with uncomplicated FRI. The other two sequenced HAdV-14 isolates were collected at Lackland Air Force Base during the severe and prolonged outbreak of HAdV-14 that started in February 2007 and drew the attention of the CDC a month later, as the outbreak spread [22]. One was from a fatal case of respiratory failure from viral pneumonia, which followed several weeks of intubation and life support. The other was from a mild case of FRI.
The primary goal of our study was to determine if there were any apparent genetic correlates that might distinguish viruses causing mild and severe outbreaks or mild and severe symptoms, or differences between the currently circulating strain of HAdV-14p1 and the prototype HAdV-14p that was seen in Eurasia in the 1950 s and 1960 s. A secondary goal was to identify unique signature sequences that might allow us to track individual strains of HAdV-14 of US origin for the purposes of epidemiological investigations.
Materials and methods
Sample Collection
The isolate from a mild FRI outbreak at MCRD San Diego was collected with consent under an institutional review board-approved research protocol (NHRC.1999.0002), identified, cultured, and analyzed as part of NHRC's ongoing population-based FRI surveillance program. The two isolates from Lackland Air Force Base included one NHRC surveillance isolate from a recruit with uncomplicated FRI and one fatal pneumonia isolate collected from a severely ill recruit at Wilford Hall Medical Center, initially collected for diagnostic viral culture and later provided to LRRI and WRAIR as a de-identified isolate.
NHRC samples were collected as oropharyngeal (throat) swabs in VTM (Remel, Lenexa, KS), immediately frozen in either -80°C freezers or on dry ice, and transported on dry ice to NHRC under College of American Pathologists (CAP)-accredited collection and transport protocols. Lackland Air Force Base samples were collected as throat swabs in VTM, cultured in A549 cells, and transported to NHRC as above. All samples were tested at NHRC for HAdV-14 [13] as raw specimens, then subsequently cultured in A549 cells (Diagnostic Hybrids Inc., Athens, OH), and stored frozen as infected tissue culture fluid (isolated virus) at Lovelace Respiratory Research Institute (LRRI). Sequencing work on these samples was performed at Walter Reed Army Institute of Research (WRAIR) on the resulting isolates.
Sanger Sequencing of HAdV-14
PCR and sequencing were accomplished at the virology facility at Walter Reed Army Institute of Research (WRAIR), Silver Spring, Maryland, USA. PCR primer pairs were designed from the prototype HAdV-14p de Wit sequence (GenBank accession number AY803294) and used to generate overlapping 1-2 kilobase amplicons covering the entire genome. All PCR products were sequenced in both directions by using forward and reverse PCR primers corresponding to each individual PCR product. All clean and verified readable sequences were used to assemble full HAdV-14 genome sequences using the Sequencher software (Gene Codes Corporation, Ann Arbor, MI).
Two hundred microliter aliquots of each isolate were extracted using the Invitrogen ChargeSwitch DNA extraction kit (Invitrogen Corporation, Carlsbad, CA) per the manufacturer's instructions, and resuspended in 200 μl elution buffer. One hundred microliter PCR amplification reactions consisted of 2 mM MgCl2, 0.6 mM dNTP (1.5 mM each A, C, T, and G), 200 μM each primer, 2.5 units Platinum Taq Polymerase (Invitrogen), and 1 ul of extracted isolate in 1X ABI Buffer II (Applied Biosystems Inc., Foster City, CA). Thermal cycling was carried out on an ABI9700 platform (Applied Biosystems) using the following parameters: initial activation for 2 min at 94°C, then 35 cycles of: 20 s at 94°C, 20 s at 53°C, and 2 min at 72°C. Final extension was for 7 min at 72°C. PCR cleanup was performed using the Qiagen PCR cleanup kit (Qiagen, Valencia, CA) per the manufacturer's instructions. Sequencing reactions were set up per the manufacturer's instructions using the ABI BigDye Terminator kit (manual version 3.2, Applied Biosystems), and run on an ABI9700 platform. Reaction products were analyzed on an ABI3130XL automated sequencer (Applied Biosystems) per the manufacturer's instructions. Resulting data were then edited and aligned using Sequencher software (Gene Codes).
Results and Discussions
HAdV-14 Genomes from 2006-7 US Outbreaks
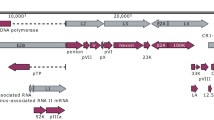
Genomic sequences from three different recent US HAdV-14 isolates, including two from Lackland Air Force Base (303600 and 1986T, associated with mild FRI and fatal pneumonia, respectively, both from a severe outbreak) and one from Marine Corps Recruit Depot, San Diego (NHRC22039, associated with a mild infection during a mild outbreak) were fully sequenced, assembled and submitted to the NCBI GenBank database (GenBank Accession #s FJ822614, EU827616 and EU833993, respectively). The genome size of Lackland strains 303600 and 1986T is identical to each other, 34,764 base pairs (bp) that is also identical in size with the HAdV-14 prototype deWit strain, 34,764 base pairs (bp). The San Diego strain NHRC22039 has a genome size of 34,768 bp. All three recent US HAdV-14 strains are highly homologous with each other. The two Lackland strains are 100% identical to each other, while the San Diego isolate differed only by a 4 bp extension of the polyadenylation signal (a poly-T on the coding strand) at the 5' end of the terminal binding protein (TBP) gene, and a single noncoding (synonymous) base substitution in the fiber gene. The poly-T repeat is 13 bp long [T(13)] in the Lackland strains and T(17) in the San Diego strain. HAdV-14p strain deWit contains a corresponding T(11) repeat. The genomes of all three recent US HAdV-14 isolates share identical coding regions for all genes. As recently described, and after detailed characterization by restriction enzyme analysis, all North American isolates of HAdV-14 correspond to genome type 14p1 [34]. Table 1 shows the summary of alignment results. HAdV-14 deWit and the recent US HAdV-14p1 isolates differ by 0.3%, scattered quite evenly through the genome. All 4 HAdV-14 s examined in this study have the same base composition of 51.2% A/T, 48.8% G/C. The GC content and the number of open reading frames (ORFs) were identical to the HAdV-14p de Wit strain. A map of the organization of predicted ORFs within the genome of the emerging HAdV-14p1 strain is shown in Figure 1, and is identical to that of the prototype HAdV-14 de Wit strain.

Map of apparent open reading frames and their identities in the genome of the emerging North American HAdV-14p1.
All HAdV genomes are bounded by inverted terminal repeats (ITR) ranging from 100 to 200 bp in size, which serve as viral replication origins. The representative ITRs of various HAdV species, such as species A (HAdV-12, 18, 31), B (HAdV-3, 7, 11), and C (HAdV-1, 2, 5) are available in GenBank. Among these HAdVs, ITRs are highly conserved within species but diverse between species. For example, the ITRs of HAdV-2 and HAdV-5 (species C) are identical 103 bp sequences. Similarly conserved ITR patterns are observed for the 137 bp ITRs of species B (HAdV-3, 7 and 11). However, all three recent US HAdV-14 s share identical inverted terminal repeat (ITR) sequence of 133 bp, in contrast to other species B HAdVs. The HAdV-14 prototype deWit also contains a 133 bp ITR, but this differs from recent HAdV-14 s of US origin by one substitution at base pair 68 (T68C). This level of polymorphism in ITR sequences among closely related strains is unusual.
We compared HAdV-14p1 to selected HAdV-B prototype strains (HAdV-14, 3, 7, 11, and 21) using the mVISTA Limited Area Global Alignment of Nucleotides (LAGAN) tool (MontaVista Software, Inc., Santa Clara, CA) [35] (Figure 2). With the exception of the hexon gene of HAdV-11p, HAdV-14p1 showed strong homology with both HAdV-14p and -11p. This was expected, since HAdV-11 and HAdV-14 are both members of subspecies B2. Comparison of HAdV-14p1 to HAdV-3, 7, and 21, members of subspecies B1, revealed sequence divergence throughout the genome, especially the penton, hexon, and fiber genes (Figure 2). These data are consistent with serological identification of the new strain as HAdV-14, since the hexon in the primary antigenic determinant and the penton and fiber act as secondary antigenic determinants.

Global pairwise comparison of multiple species B HAdV genomes.
The nucleotide identity scores for HAdV-14p1 genes with less than 100% identity with HAdV-14p are shown in Table 2. There were 19 nucleotide polymorphisms observed. Only two of these affected amino acid coding sequences. The first was a 3-bp insertion in the HAdV-14p1 sequence, which resulted in an inserted serine at position #147 of the 25.7 K and 28 K protein sequences (Figure 3). The proteins encoded by E1A regulate the transcription of viral as well as cellular genes [36, 37]. The second was a 6-bp deletion in the fiber gene of the HAdV-14p1 sequence. This resulted in a two amino acid deletion in the FG loop of the fiber gene (Figure 4). The fiber gene is responsible for mediating attachment of the adenovirus to the host cell [38, 39].

E1A alignments. Alignment of selected E1A 28K amino acid sequences from HAdV-3, 7, 11, 14p, 14p1, and 21. Black arrow demarcates the S147 insertion, shared by HAdV-3, 7, and 21.

Fiber knob binding site alignment. Alignment of the amino acid sequences on the exposed regions of the fiber loops FG, HI, and IJ that are involved in binding to CD46. Black arrow demarcates the ΔK250-E251 deletion. All sequences were aligned using the ClustalX [40] alignment method. The following HAdV genomes (GenBank accession numbers) were used: HAdV-14p (AY803294), HAdV-11p (AY163756), HAdV-3 (NC_011203), HAdV-7 (AC_000018), and HAdV-21 (AY601633).
Although at the nucleotide level the genomes of HAdV-14p de Wit and HAdV-14p1 strains were highly homologous, we wanted to determine whether there was evidence of recombination. SimPlot bootscan analysis of HAdV-14p1 with respect to prototypical strains of HAdV-3, 4, 7, 11, 14, and 21 demonstrated that this virus is mostly closely related to the HAdV-14p prototype strain and is not a recombinant with respect to other recognized serotypic clades (data not shown). As noted previously, polymorphisms between these two strains were distributed evenly throughout the genome.
The three sequenced strains of HAdV-14p1 were almost identical. The two Lackland isolates were exactly the same, while the San Diego strain differed by the addition of four extra Ts to the TBP polyadenylation signal repeat, and by a single synonymous base substitution in the fiber gene.
Conclusions
HAdV-14p1 (strain 303600) appears to be a closely related direct drift variant of the HAdV-14p (strain de Wit) prototype seen in the past, differing primarily by an insertion in E1A, a small deletion in the fiber gene, and a few other coding single nucleotide polymorphisms (SNPs) in E3 and other genes. The deletion (ΔK250-E251) in the fiber gene is the most notable genetic difference between the HAdV-14p and HAdV-14p1 (Figure 4). Despite the observed ΔK250-E251 deletion, the fiber gene sequence of HAdV-14p1 shares greater overall homology with the fiber of HAdV-11a than that of HAdV-11p. Whereas, HAdV-11p causes mostly urinary tract infections and shares very low fiber homology with HAdV-11p1, HAdV-11a and HAdV-14p all causing respiratory infections [34]. This is consistent with the receptor-binding role of the fiber and the close relationship between receptor specificity and organ tropism. HAdV-14p1 and HAdV-11a both cause upper respiratory infections, while HAdV-11p causes mostly urinary tract and bone marrow infections in transplant patients. Species B viruses are unique in that they use CD46, a complement protein, as a receptor [38]. Many other human adenoviruses use the CAR protein [38]. The deleted amino acids could affect the exposed region of the FG loop by altering the overall affinity for CD46. A less likely alternative is that the fiber deletion influences an interaction with a receptor other than CD46, such as CAR. A third possibility is that this deletion has no affect at all on the fiber gene. Whether this mutation affects the pathogenicity of HAdV-14p1 compared with HAdV-14p will require further studies.
When HAdV-14p was first identified in Eurasia in the 1950 s and 1960 s, it generated localized, high attack rate epidemics of FRI similar to those seen with the current strain. After a decade of sporadic activity, it disappeared and remained almost completely undetected for 4 decades. As a recently emerged virus, HAdV-14p1 has an increased potential for high rates of transmission and high attack rates, simply because the vast majority of North Americans are likely to have never been exposed (essentially the entire population is susceptible). This is similar to the situation long recognized for HAdV-4, which, in the absence of vaccines, has always been the dominant serotype affecting military recruits. Serosurveys have generally indicated that a greater proportion of the young adult population is susceptible to HAdV-4 than to other common respiratory serotypes such as HAdV-7.
The two sequenced strains from Lackland Air Force Base, one from a severe (fatal) pneumonia and one from a mild case of acute respiratory disease, were identical. There were only two noncoding polymorphisms distinguishing the Lackland isolates from the San Diego isolate (another mild case). The poly-T length polymorphism was studied in a wide range of isolates from multiple sites, and found to be a hypervariable and useful source of geographically specific strain identity information [40]. Neither mutation suggested a significant genetic source of variation in clinical severity. The results supported previous observations of a high degree of conservation in hexon and fiber genes relative to the prototype HAdV-14p and to the closely related HAdV-11a.
References
Hierholzer JC, Stone YO, Broderson JR: Antigenic relationships among the 47 human adenoviruses determined in reference horse serum. Arch Virol 1991, 121:179–197.
Wadell G: Adenoviruses. In Encyclopedia of virology. Volume 1. New York: Academic Press; 1994:1–7.
Rogers PM, Mashtalir N, Rathod MA, Dubuisson O, Wang ZQ, et al.: Metabolically favorable remodeling of human adipose tissue by human adenovirus Ad-36. Diabetes 2008,57(9):2321–2331.
Jones MS II, Harrach B, Ganac RD, Gozum MMA, delaCruz WP, Riedel B, et al.: New adenovirus species found in a patient presenting with gastroenteritis. J Virol 2007,81(11):5978–5984.
Davison AJ, Benkő M, Harrach B: Genetic content and evolution of adenoviruses. J Gen Virol 2003, 84:2895–2908.
Sambrook J, Sleigh M, Engler JA, Broker TR: The evolution of the adenoviral genome. Ann NY Acad Sci 1980, 354:426–452.
Schmitz H, Wigand R, Heinrich W: Worldwide epidemiology of human adenovirus infections. Am J Epidemiol 1983, 117:455–466.
Rubin BA: Clinical picture and epidemiology of adenovirus infections. Acta Microbiol Hung 1993, 40:303–323.
Green M, Mackay JK, Wold WSM, Rigden P: Thirty-one human adenovirus serotypes (Ad1-Ad31) form five speciess (A-E) based upon DNA genome homologies. Virology 1979, 93:481–492.
Fox JP, Hall CE, Cooney MK: The Seattle virus watch. VII. Observations of adenovirus infections. Am J Epidemiol 1977, 105:362–386.
Metzgar D, Osuna M, Yingst S, Rakha M, Earhart K, et al.: PCR analysis of Egyptian respiratory adenovirus isolates, including identification of species, serotypes, and coinfections. J Clin Microbiol 2005, 43:5743–5752.
Hierholzer J: Adenoviruses. In Diagnostic procedures for viral, rickettsial and chlamydial infections. 7th edition. Edited by: Lennette EH, Lennette DA, Lennette ET. Washington, DC: American Public Health Association; 1995:169–188.
Metzgar D, Osuna M, Kajon AE, Hawksworth AW, Irvine M, et al.: Abrupt emergence of diverse species B adenoviruses in US military recruit training centers. J Infect Dis 2007, 196:1465–1473.
Brandt CD, Kim HW, Jeffries BC, Pyles G, Christmas EE, Reid JL, et al.: Infections in 18,000 infants and children in a controlled study of respiratory tract disease. II. Variation in adenovirus infections by year and season. Am J Epidemiol 1972, 95:218–227.
Ryan MA, Gray GC, Smith B, McKeehan JA, Hawksworth AW, et al.: Large epidemic of respiratory illness due to adenovirus types 7 and 3 in healthy young adults. Clin Infect Dis 2002, 34:577–582.
Kajon AE, Moseley JM, Metzgar D, Huong HS, Wadleigh A, et al.: Molecular epidemiology of adenovirus type 4 infections in US military recruits in the postvaccination era (1997–2003). J Infect Dis 2007, 196:67–75.
Kendall EJC, Riddle RW, Tuck HA, Rodan KS, Andrews BE, et al.: Pharyngo-conjunctival fever: school outbreaks in England during the summer of 1955 associated with adenovirus types 3, 7, and 14. BMJ 1957, 2:131–136.
Lin KH, Lin YC, Chen HL, Ke GM, Chiang CJ, et al.: A two-decade survey of respiratory adenovirus in Taiwan: the reemergence of adenovirus types 7 and 4. J Med Virol 2004, 73:274–279.
van der Veen J, Oei KG, Abarbanel MFW: Patterns of infections with adenovirus types 4, 7 and 21 in military recruits during a 9-year survey. J Hyg (Lond) 1969, 67:255–268.
Morgan PN, Moses EB, Fody EP, Barron AL: Association of adenovirus type 16 with Reye's-syndrome-like illness and pneumonia. South Med J 1984, 77:827–830.
Mufson MA, Belshe RB: A review of adenoviruses in the etiology of acute hemorrhagic cystitis. J Urol 1976, 115:191–194.
Centers for Disease Control and Prevention: Acute respiratory disease associated with adenovirus serotype 14 -- four states, 2006–2007. MMWR 2007, 56:1181–1184.
Allibhai TF, Spinella PC, Meyer MT, Hall BH, Kofos D, et al.: Survival after prolonged pediatric extracorporeal membrane oxygenation support for adenoviral pneumonia. J Pediatr Surg 2008, 43:E9-E11.
Louie JK, Kajon AE, Holodniy M, Guardia-LaBar L, Lee B, Petru AM, et al.: Severe pneumonia due to adenovirus serotype 14: a new respiratory threat? Clin Infect Dis 2008, 46:421–425.
Mevzos LM, Il'ina TS, Makhmudov OS, Zolotarskaia EE, Dreizin RS: An outbreak of acute respiratory infections among adults caused by adenovirus serotype 14. Vopr Virusol [Russian] 1966, 11:426–431.
Bruj J, Farnik J, Sedmidubsky V: Epidemic of acute respiratory disease due to adenovirus type 14. Cesk Epidemiol Mikrobiol Imunol [Czech] 1966, 15:165–171.
Cooper RJ, Hallett R, Tullo AB, Klapper PE: The epidemiology of adenovirus infections in Greater Manchester, UK 1982–96. Epidemiol Infect 2000, 125:333–345.
van der Veen J, Kok G: Isolation and typing of adenoviruses recovered from military recruits with acute respiratory disease in the Netherlands. Am J Hyg 1957, 65:119–129.
Chen HL, Chiou SS, Hsiao HP, Ke GM, Lin YC, et al.: Respiratory adenoviral infections in children: a study of hospitalized cases in southern Taiwan in 2001–2002. J Trop Pediatr 2004, 50:279–284.
Zhu Z, Zhang Y, Xu S, Yu P, Tian X, et al.: Outbreak of acute respiratory disease in China caused by B2 species of adenovirus type 11. J Clin Microbiol 2009,47(3):697–703.
Hierholzer JC, Pumarola A: Antigenic characterization of intermediate adenovirus 14–11 strains associated with upper respiratory illness in a military camp. Infect Immun 1976, 13:354–359.
Chmielewicz B, Benzler J, Pauli G, Krause G, Bergmann F, et al.: Respiratory disease caused by a species B2 adenovirus in a military camp in Turkey. J Med Virol 2005, 77:232–237.
Li QG, Wadell G: Analysis of 15 different genome types of adenovirus type 7 isolated on five continents. J Virol 1986, 60:331–335.
Kajon AE, Lu X, Erdman DD, Louie J, Schnurr D, George KS, Koopmans MP, Allibhai T, Metzgar D: Molecular epidemiology and brief history of emerging adenovirus 14-associated respiratory disease in the United States. J Infect Dis 2010, 202:93–103.
Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, et al.: LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Research 2003,13(4):721–731.
Frisch SM, Mymryk JS: Adenovirus-5 E1A: paradox and paradigm. Nat Rev Mol Cell Biol 2002, 3:441–452.
Gallimore PH, Turnell AS: Adenovirus E1A: remodelling the host cell, a life or death experience. Oncogene 2001, 20:7824–7835.
Gaggar A, Shayakhmetov DM, Liszewski MK, Atkinson JP, Lieber A: Localization of regions in CD46 that interact with adenovirus. J Virol 2005, 79:7503–7513.
Wang H, Tuve S, Erdman DD, Lieber A: Receptor usage of a newly emergent adenovirus type 14. Virology 2009, 387:436–441.
Houng HSH, Lott L, Gong H, Kuschner RA, Lynch JA, Metzgar D: Adenovirus microsatellite reveals dynamics of transmission during a recent HAdV-14 epidemic. J Clin Microbiol 2009, 47:2243–2248.
Acknowledgements
The authors acknowledge the Clinic Commanders and medical staff at Lackland Air Force Base and Wilford Hall Medical Center, San Antonio, TX (US Air Force) and Marine Corps Recruit Depot, San Diego, CA for the permissions, access, and assistance necessary to conduct these studies. The authors also acknowledge the administrative support of the Henry M. Jackson Foundation for Military Medicine and the efforts of the entire WRAIR, NHRC, LRRI, and DGMC teams, especially the technicians and collection personnel whose efforts are represented in this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
HG carried out the sequencing of Ad14 genomes. AEK, MSJ, RAK, AL, LL, KL and DM all participated in the samples collections, sequencing alignment and draft of manuscript. All authors read and approved the final manuscript submission.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Houng, HS.H., Gong, H., Kajon, A.E. et al. Genome sequences of Human Adenovirus 14 isolates from mild respiratory cases and a fatal pneumonia, isolated during 2006-2007 epidemics in North America. Respir Res 11, 116 (2010). https://doi.org/10.1186/1465-9921-11-116
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1465-9921-11-116