
Abstract
Atherosclerosis is commonly appreciated to represent a chronic inflammatory response of the vascular wall, and its complications cause high mortality in patients. Angioplasty with stent replacement is commonly performed in patients with atherosclerotic disease. However, the restenosis usually has a high incidence rate in angioplasty patients. Although the pathophysiological mechanisms underlying atherosclerosis and restenosis have been well established, new signaling molecules that control the progress of these pathologies have continuously been discovered. MicroRNAs (miRs) have recently emerged as a novel class of gene regulators that work via transcriptional degradation and translational inhibition or activation. Over 30% of genes in the cell can be directly regulated by miRs. Thus, miRs are recognized as crucial regulators in normal development, physiology and pathogenesis. AIterations of miR expression profiles have been revealed in diverse vascular diseases. A variety of functions of vascular cells, such as cell differentiation, contraction, migration, proliferation and inflammation that are involved in angiogenesis, neointimal formation and lipid metabolism underlying various vascular diseases, have been found to be regulated by miRs. This review summarizes current research progress and knowledge on the roles of miRs in regulating vascular cell function in atherosclerosis and restenosis. These discoveries are expected to present opportunities for clinical diagnostic and therapeutic approaches in vascular diseases resulting from atherosclerosis and restenosis.
Similar content being viewed by others

Review
Introduction
Atherosclerosis is a chronic and progressive pathology characterized by the accumulation of lipid and fibrous elements in the large arteries, which causes a number of cardiovascular-related diseases. Atherosclerosis has a tremendous impact in developing and developed countries, representing the underlying cause of approximately 50% of deaths. Our knowledge of the pathophysiology for this important malady has evolved over the past century. Extensive evidence reveals that the pathogenic feature of atherosclerosis is an inflammatory process, in which vascular endothelial cells (ECs) become dysfunctional due to influences by chemical substances, such as cytokines and growth factors [1], and hemodynamic forces [2]. Activated ECs with high expression levels of various leukocyte adhesion molecules recruit leukocytes and monocytes to bind to the endothelium and migrate into the vessel wall. The lesion then experiences the following steps: foam cell formation, fatty streak accumulation, migration and proliferation of vascular smooth muscle cells (VSMCs), and fibrous cap formation. Finally, the rupture of the unstable fibrous cap causes thrombosis in complications of advanced lesions that lead to unstable coronary syndromes, myocardial infarction and stroke. The knowledge that atherosclerosis is a vascular pathology resulting from inflammatory response enables new approaches to treatment and prevention. Immunosuppressant and anti-inflammatory agents could potentially be applied in clinical trials. However, surgical treatments remain the prevalent method of treating in the patients with atherosclerosis, including percutaneous transluminal coronary angioplasty (PTCA) and stent placement.
Angioplasty and stent placement remove the occlusion to augment the inner diameter of the artery at various vascular locations. These treatments exclusively improve the hemodynamic flow rate and lead to normal blood flow. While these treatments have been used in many patients with atherosclerotic disease over the past decades, restenosis is an ongoing complication with an incidence of 30–40% within 3–6 months after treatment. Although restenosis and atherosclerosis are recognized as inflammatory processes in response to injury [3], restenosis is in fact a vascular injury caused by balloon dilation and stent replacement during angioplasty [4]. The development of restenosis is pathophysiologically distinct from atherosclerosis. These differences have been observed during the proliferation and migration of VSMCs, extracellular matrix remodeling and neointimal hyperplasia. Anatomic and procedural clinical variables are associated with an increased incidence of restenosis following angioplasty [5].
MicroRNAs (miRs) are recently emerging endogenous, noncoding, single-stranded RNAs of 18–22 nucleotides that constitute a novel class of gene regulators. The first miR, lin-4, was discovered during the development of Caenorhabditis elegans in 1993 [6]. Bentwich et al. [7] developed an integrative approach combining bioinformatic prediction with microarray analysis and sequence-directed cloning to reveal that more than 800 miRs exist in humans. Currently, over 15,000 miR gene loci have been identified in over 140 species, and more than 17,000 distinct mature miR sequences are present in the miRBase16 [8]. MiRs bind to their target genes in 3’-untranslated regions (3’-UTRs), leading to the direct degradation of messenger RNA (mRNA) or translational repression by a perfect or imperfect complement. This implies that miRs are able to regulate the expression of hundreds or thousands genes. Thus, it is not surprising that miRs are involved in the regulation of all major cellular functions [9].
The pathophysiological mechanisms of vascular pathologies such as atherosclerosis, hypertension, coronary artery disease and restenosis after angioplasty have been well established over the past decades. Vascular properties including angiogenesis, re-endothelialization and neointima formation contribute to these vascular pathologies/diseases. Inflammatory responses to injury, differentiation, proliferation, migration and apoptosis of VSMCs or ECs are critical cellular events for the development of these vascular diseases. Blood cell recruitment, infiltration, activation and differentiation are also involved in these complicated diseases. Vascular diseases have been widely explored, and many new molecules have been studied as potential clinical therapies. In recent years, the roles of miRs have gradually received increasing attention in the biology of vascular diseases. Altered miR expression profiles have been related to cardiovascular diseases in over 400 studies. Although several review articles have described the regulation of miRs in vascular remodeling, inflammation and diseases [10–12], the specific role of miRs in the regulation of atherosclerosis and restenosis is barely described. Hence, this review focuses on the roles of miRs in different types of vascular cells in relation to atherosclerosis and restenosis.
The biogenesis of microRNA
Primary miR
Most miR genes are located in intronic regions, which may be transcribed as part of the mRNA genes. As for general mRNA, miR genes are commonly transcribed by RNA polymerase II (pol II) [13] in the nucleus (Figure 1). The primary miR transcripts (pri-miRs) contain capped structures and polyadenylated (poly A) tails, the hallmark properties of class II gene transcripts [14]. Besides pol II, Borchert et al. [15] found that C19MC miRs, including miR-515-1, miR-517a, miR-517c and miR-519a-1, are expressed by RNA polymerase III (pol III). Some miRs contain primary transcripts to produce a single miR, whereas other transcripts encode proteins in their exons and miRs. The cluster miRs such as the miR-17~92 family are grouped together in one cluster on a single unprocessed transcript and expressed together.

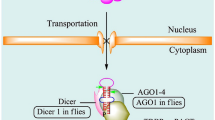
The canonical pathway of miR processing. The primary miR (pri-miR) are transcripted by either RNA polymerase II or III from independent gene in the nucleus. In the following processing, the microprocessor complex (Drosha-DGCR8) processes pri-miR into a ~60-100-nucleotide precursor hairpin (pre-miR). The resulting pre-miR is exported to the cytoplasm by Exportin-5-Ran-GTP. In the cytoplasm, the RNase III Dicer and TRBP cleave the pre-miR into ~22-nucleotide miR/miR* duplex. One strand termed as guide strand, further representing a mature miR, the miR* termed as passenger strand, which undergoes degradation rapidly. Mature miR is incorporated into a miRISC and base-paired to its target mRNAs for mRNA degradation or translational repression.
Precursor miR
Following transcription by pol II or pol III, the pri-miR received is endonucleolytically cleaved to an ~60–100 nucleotide hairpin structure with an ~2 nucleotide 3’ overhang termed the precursor-miR (pre-miR) by the nuclear microprocessor complex. This microprocessor complex is formed by the RNase III enzyme Drosha (RNASEN) and its partner DGCR8 (DiGeorge critical region 8), also known as Pasha (Partner of Drosha) in D. melanogaster and C. elegans[16–18]. Several molecules were identified to be involved in the post-transcriptional modulation of miR processing [19]. For example, RNA helicases p68 and p72, the cofactors of the microprocessor complex, promote the Drosha cleavage of a subset of miRs. p53, an important tumor suppressor protein, is present in the complex with p68 and Drosha to enhance the Drosha processing of a subset of miRs. Smad, transforming growth factor (TGF)-β and bone morphogenetic protein (BMP)-specific signaling transducer proteins are recruited to a consensus sequence (R-SBE) within the stem region of the primary transcripts of TGF-β/BMP-miRs in the Drosha and p68 complex. Thus, this Smad-Drosha-p68 complex promotes the processing of TGF-β/BMP-miRs [20]. After nuclear processing, the pre-miR is exported into the cytoplasm by Exportin-5 (XPO5) in complex with Ran-GTP cofactor [21].
Mature miR
The pre-miR is further processed in the cytoplasm by another RNase III Dicer, which forms the RISC complex with Argonaute 2 (Ago2) and TRBP (Tar RNA binding protein), which cleaves off the hairpin loop of the pre-miR to generate an ~22-nucleotide miR duplex [22–24]. This miR duplex contains mature miR referred to as the guide strand and a complementary strand referred to as the passenger strand (miR*). Following processing, one strand of the miR/miR* duplex (usually the guide strand) is preferentially incorporated into a miR-induced silencing complex (miRISC) that contains Dicer and other associated proteins [25], whereas the miR* is released and rapidly degraded. As part of the miRISC, the miR is base-paired to its target mRNA to induce translational repression or direct degradation [26, 27].
Atherosclerosis
A growing body of studies reveals that the pathogenic feature of atherosclerosis is an inflammatory process involving ECs in response to injury. These dysfunctional ECs lead to a sequence of inflammatory responses, blood-cell accumulation, foam cell formation, fibrous formation, advanced plaque formation and rupture [1, 28, 29]. These complicated processes are contributed by various blood cells such as monocytes, macrophages, and lymphocytes, and vascular cells such as ECs and VSMCs. Moreover, these cells influence each other and secrete various cytokines and growth factors to promote the formation of atherosclerosis.
Initiation step
The endothelium consists of a single layer of vascular ECs and serves as a selective barrier between blood and tissues. Atherosclerotic plaques preferentially occur in specific arterial sites such as branches, bifurcations and curvatures in which the flow pattern is disturbed, with a lower velocity and no particular orientation. ECs tend to turn over in these regions and show increased permeability to macromolecules such as low-density lipoprotein (LDL). As a result, LDL diffuses passively through EC junctions and accumulates in the subendothelial matrix. Subsequently, the LDL undergoes modification and oxidation, contributing to inflammation and further foam cell formation.
Inflammation
In the initial lesion, ECs have an activated and pro-inflammatory phenotype that leads to expression of various EC adhesion molecules (such as intercellular adhesion molecule-1 [ICAM-1], vascular cell adhesion molecule-1 [VCAM-1] and E-selectin), growth factors such as macrophage colony stimulating factor (M-CSF), and chemokines including monocyte chemotactic factor-1 (MCP-1) [30]. E-selectin is a member of the selectin family of adhesion molecules that plays a crucial role in the initial interaction between circulating leukocytes and ECs. E-selectin binds to carbohydrate ligands on the leukocytes and facilitates the rolling of leukocytes along the endothelial surface. Under the cooperation of adhesion molecules and chemotactic factors, the rolling leukocytes enter the vessel wall. In addition, the circulating monocytes and lymphocytes are recruited by MCP-1 and M-CSF into the vessel wall. M-CSF promotes macrophage proliferation and differentiation and the expression of scavenger receptors (SR), which increases the production of cytokines and growth factors by these cells. LDL has to be modified and oxidized before it can be taken up by macrophages. The reactive oxygen species (ROS) produced by vascular cells, including sphingomyelinase, secretory phospholipase-2 (sPLA2) and myeloperoxidase, are involved in the initiation of oxidation of the LDL (oxLDL) [31]. The oxLDL particles are recognized by macrophage scavenger receptors such as scavenger receptor-A (SR-A), CD36 antigen (CD36) and macrophage antigen CD68. Consequently, the oxLDL is rapidly taken up by macrophages that then become enlarged and full of lipids. These cells accumulate in the subendothelial matrix and transform into foam cells, characteristic of the early atherosclerotic lesion (atheroma).
Fibrous plaques
Arteries generally consist of three layers, the intima, media and adventitia. The normal media layer contains mostly contractile VSMCs and a few fibroblasts surrounded by their own basement membrane. The major components of the medial extracellular matrix are fibrillar collagen type I and III. In atherosclerosis, the inflammatory response triggers the activated macrophages and T-cells to secrete a number of cytokines and growth factors that promote the change of VSMCs from the quiescent contractile state (differentiation) to the active synthetic state (de-differentiation) [32], the migration from the media to the intima and the production of collagen (fragments of collagen type I, III, and collagen type VIII [33]), elastin and proteoglycan to form a fibrous matrix.
Advanced lesions and plaque disruption
The fibrous cap gradually covers the lipids, leading to the death of foam cells and other cell debris that form a necrotic core. The inflammatory response and continuous recruitment of leukocytes and macrophages lead to these lesions and the expansion of their area. The necrotic core represents the secretion of various growth factors (e.g., platelet-derived growth factor [PDGF] and TGF-β), cytokines (e.g., interleukin [IL]-1 and tissue necrotic factor-α [TNF-α]), osteopontin and matrix metalloproteinases (MMPs). Activated T-cells stimulate the production of MMPs, which promote the instability of the lesion and further complicate the inflammatory response. Thinning of the fibrous cap may result from MMPs such as collagenases, elastases, and stromelysins. These MMPs cause the degradation of the matrix, which could lead to hemorrhage from the vasa vasorum or from the lumen of the artery and cause thrombus formation and arterial occlusion.
Restenosis
Restenosis occurs in patients with atherosclerotic disease undergoing coronary angioplasty with stent replacement. Even with the best medical techniques, restenosis occurs in approximately 30% of patients [34]. Although restenosis and atherosclerosis are recognized as inflammatory processes in response to injury, restenosis has a different pathophysiological appearance than atherosclerosis and has already been considered a developmentally different process [4]. The swelling of diseased vessels by either angioplasty or stent insertion causes endothelial disruption, fragment of the internal elastic lamina, and dissection of the media, often extending into the adventitia. Thus, restenosis after angioplasty or stent insertion is a combination of biological processes, each of which contributes to the final luminal narrowing. Processes observed in animal models and patients include elastic recoil, thrombus, neointima formation and remodeling [35].
Elastic recoil
The human coronary artery is highly elastic, with elastin fibers comprising the internal elastin lamina (IEL) and external elastin lamina (EEL). In an eccentric atherosclerotic lesion, the balloon dilatation overstretches the segments of the artery. The elastic recoil occurs within seconds to minutes following balloon dilatation. Over the next days to weeks, the stretched segments become gradually relaxed, leading to a reduction of the luminal diameter. Vasoconstrictors such as serotonin and thromboxane are released by the aggregating platelets that promote vasoconstriction at the site of angioplasty [5].
Thrombus
Successful angioplasty usually causes endothelial denudation and induces medial dissection. The consequent exposure of subintimal components, such as collagen, von Willebrand factor, fibronectin, and laminin, causes platelet adherence and aggregation. Many platelets can then become cross-linked by fibrinogen, promoting more platelet aggregation. Platelet aggregation triggers the release of thromboxane A2 and serotonin, which also promotes further adhesion and aggregation. Platelets also secrete a number of mitogens and chemotactic factors for VSMCs, including PDGF and TGF-β, which lead to neointima formation at the site of angioplasty [5].
Neointima formation
Neointima formation, known as intimal hyperplasia, is caused by the proliferation and migration of VSMCs and the accumulation of fibroblasts at the site of injury. Based on the observation of specimens from patients, the migration and proliferation of VSMCs and fibroblasts in the neointimal layer occurs in the weeks to months after angioplasty. Angioplasty induces EC denudation and mechanical stretching of vessels, which lead to the release of various cytokines and growth factors by ECs, inflammatory cells and platelets promoting VSMC proliferation and migration and increasing the synthesis of the collagen, elastin and proteoglycan matrix [36].
Remodeling
Remodeling is described as a gradual process of relative changes in vessel size. Remodeling can be classified into positive remodeling (also termed outward/expansive remodeling) and negative remodeling (termed inward/constrictive remodeling). Restenosis may be caused by the negative remodeling of a dilated artery with less neointima formation. In contrast, the positive remodeling of a dilated artery may accumulate large amounts of neointimal tissue. Mintz et al. [34] further documented negative remodeling in a series of 209 angioplasty patients and observed that a significant portion of lumen loss was due to vessel constriction rather than neointimal thickening. However, the mechanisms by which negative remodeling can be involved in restenosis remain unclear. The extracellular matrix may be involved in the remodeling of dilated arteries after angioplasty. Angioplasty causes an acute alteration of extracellular matrix synthesis and degradation, resulting in an increase of collagen synthesis and a reduction of MMP activity, reducing matrix degradation.
Roles of microRNA in vascular cells
Endothelial cells
Inflammation
The pathogenic feature of atherosclerosis is an inflammatory process by which blood vessels respond to injury. Recent studies have reported that miRs are involved in these processes (Table 1). Vessels from swine exhibited decreased expression of miR-10a at athero-susceptible regions of the inner aortic arch and aorta-renal branches. To further demonstrate the role of miR-10a knockdown, the effects of miR-10a knockdown on the endothelial transcriptome were determined in cultured ECs by whole-genome microarray analyses. Bioinformatic analysis identified IκB/NF-κB–mediated inflammation as the main biological processes taking place in miR-10a knockdown cells. Downregulation of miR-10a enhances IκB/NF-κB activation and leads to significant upregulation of inflammatory biomarkers such as MCP-1, VCAM-1, E-selectin, IL-6 and IL-8. This evidence suggests that miR-10a suppresses pro-inflammatory molecules in endothelial phenotypes of the athero-susceptible region in vivo [37]. Through in silico analysis, Harris et al. [38] and Wang et al. [39] suggested that miR-126 may be a negative regulator of VCAM-1 expression. Overexpression of miR-126 by oligonucleotide transfection led to repression in TNF-α-induced protein expression of VCAM-1 and leukocyte adhesion. Moreover, miR-126 was identified to be involved in the regulation of VCAM-1 at the translational rather than transcriptional level. This result enhances the importance of miR-126 in posttranscriptional gene regulation in ECs. MiR-155 was demonstrated to play an anti-inflammatory role in ECs [40]. Overexpression of miR-155 decreased the adhesion of Jurkat T cells to angiotensin II (Ang II)-stimulated ECs. Endothelin-1 (ET-1) is a potent vasoconstrictive peptide and mitogen that plays multiple roles in the progression of atherosclerosis, vascular inflammation and remodeling. MiR-125a and miR-125b-5p were found to be highly expressed in ECs and are able to suppress the expression of oxLDL-induced ET-1 [41]. Furthermore, miR-132 [42] was also shown to be involved in the inflammatory response of ECs.
Angiogenesis
Angiogenesis is characterized by the formation of new blood vessels from the existing vascular network. Angiogenesis is required in various physiological and pathophysiological conditions such as embryonic development, tissue regeneration, wound healing, tumor growth and atherosclerosis [59]. Cell proliferation and mobility are critical steps for angiogenesis and are strictly controlled by various intracellular signals. MiR profiling of embryonic stem (ES) cell-derived ECs revealed a group of endothelia-enriched miRs, including miR-126, −146, −197 and −625. MiR-126 is most highly enriched in ECs and has been well-characterized as a pro-angiogenic miR. MiR-126 and miR-126* are encoded by intron 7 of the EGF-like domain 7 (Egfl7) gene, which encodes an EC-specific secreted peptide that has been reported to act as a chemoattractant and an inhibitor of smooth muscle cell migration [45]. Knockdown of miR-126 in zebrafish led to a loss of vascular integrity and induced hemorrhage during embryonic development [46]. Targeted deletion of miR-126 in mice led approximately 40% of the miR-126−/−mice to die embryonically or perinatally. Analysis of embryos obtained from timed matings revealed that miR-126−/− embryos were dead or dying, with severe systemic edema, multifocal hemorrhages and ruptured blood vessels throughout embryogenesis [39]. Analysis of gene expression profiles in ECs isolated from miR-126−/− and zebrafish morphants demonstrated that miR-126 promoted angiogenesis through VEGF/FGF signaling by targeting its negative regulators Sprouty-related protein-1 (Spred-1) and phosphoinositide-3 kinase regulatory subunit 2 (PIK3R2/p85-b) via the MAPK and PI3K pathways, respectively. The role of hemodynamic forces during embryonic development in the patterning and remodeling of the embryonic circulatory system has been investigated. Nicoli et al. [47] further demonstrated that the angiogenic sprouting of blood vessels required the blood flow-induced transcription factor KLF-2, which induced the expression of miR-126 to activate VEGF signaling. This study provided new insights into how ECs respond to flow and integrate developmental signals with miR-126 to promote angiogenesis. Anand et al. [48] identified that miR-132 was highly upregulated in human ES during vasculogenesis. Interestingly, miR-132 is also highly expressed in the endothelium of human tumors and hemangiomas, but it is undetectable in the normal endothelium. Overexpression of miR-132 leads to pro-angiogenic signals, proliferation and Ras activity via suppression of p120RasGAP in ECs. Furthermore, selective delivery of anti–miR-132 through αvβ3 integrin-targeted nanoparticles to the tumor endothelium of mice reduced the tumor burden and angiogenesis.
Dicer is an important RNase III enzyme for miR maturation. Suarez et al. [60] clarified that the knockdown of Dicer in ECs altered the expression of angiogenic regulators such as Tie-2, endothelial nitric oxide synthase (eNOS) and IL-8. Knockdown of Dicer in ECs results in a reduction of proliferation via cell cycle delay from the G1 into the S phase, along with impairment of cord formation. The miR-17~92 cluster (encoding miR-17, -18a, -19a/b-1, -20a, and -92a) is overexpressed in several tumor cells and in the regulation of angiogenesis. Bonauer et al. [43] demonstrated that miR-92a was highly expressed in ECs and exhibited anti-angiogenic activity by targeting several endothelial functional genes, including integrin subunit α5 and αv, sphingosine-1-phosphate receptor-1 (SIP-1), and mitogen-activated protein kinase (MAPK) kinase-4 (MKK-4). These endothelial functional genes mediate the cell-matrix interaction, cell migration and angiogenesis. Furthermore, the mouse hind limb ischemia model and myocardial infarction model demonstrated that antagomir-92a led to enhanced growth of blood vessels and functional recovery of damaged tissue. Moreover, miR-210 [49], miR-221 [50], miR-222 [51], miR-100 [44], miR-424 [52] and miR-503 [53] have also been shown to play critical roles in the modulation of angiogenesis (Table 1).
Migration
Endothelial migration is an important property of angiogenesis. This motile ability is regulated by growth factors, chemotactic factors and mechanical forces. These factors trigger several signaling networks that converge on cytoskeleton remodeling in migrating cells. In recent studies, several miRs were reported to be involved in the regulation of migration through impaired cytoskeleton remodeling related to transcription factors and signaling molecules (Table 1). An interesting article reported by Zhang et al. [55] demonstrated that secreted monocytic miR-150, which is packaged by the microvesicles (MVs), could enter and be delivered into human microvascular ECs (HMECs), thus enhancing cell migration and decreasing c-myc expression. These studies further revealed that blood cells and cultured THP-1 cells are able to selectively package other immune-related miRs such as miR-146a and miR-181a into MVs in response to various stimuli. In addition, miR-200a has been shown to promote EC migration via the repression of thrombrospodin-1 (THBS-1) [56]. The important miR-155 has multiple functions in ECs, not only in the regulation of inflammation, but also in the inhibition of EC migration in response to Ang II [40]. Ets-1 is an important endothelial transcription factor that robustly regulates endothelial inflammation, angiogenesis and vascular remodeling. Bioinformatic and luciferase assays demonstrate that Ets-1 can be directly targeted by miR-155 in two potential target sites of the 3’-UTR region. Slit-Robo signaling controls the angiogenesis and contributes to the development of the vascular network. Small et al. [57] demonstrated that miR-218 was expressed from the slit2 and slit3 genes, resulting in further direct repression of the expression of Robo1, Robo2, and glucuronyl C5-epimerase (GLCE), resulting in a reduction of EC migration. This intact miR-218-Slit-Robo regulatory network is necessary for the vascularization of the retina. MiRs have been reported to decrease EC migration, including miR-21 [54] and miR-320 [58] via repression of RhoB and insulin-like growth factor-1 (IGF-1), respectively.
Macrophages/monocytes
Monocytic differentiation and oxLDL uptake are critical processes in atherosclerosis. Wang et al. [61] integrated microarray data and a bioinformatic database to reveal the correlations between miR and target mRNA in the TPA-induced differentiation of U937 cells. Fontana et al. [62] demonstrated the role of miR-17-5p-20a-106a in one monocyte lineage from the cord blood CD34+ hematopoietic progenitor cells (HPCs). MiR-17-5p–20a–106a suppresses AML1 protein expression, leading to the downregulation of the M-CSF receptor (M-CSFR) and the inhibition of monocytopoiesis. In contrast, using the same model cell type, Rosa et al. [63] revealed that miR-424 promoted monocytic differentiation through the repression of NFI-A, the transcription factor used to regulate monocytic differentiation. An Agilent miR array revealed that miR-155, −222, -424, and −503 are involved in monocytic differentiation through cell cycle arrest and apoptosis [64]. In addition to these miRs, miR-155 is also implicated in the regulation of monocyte-derived dendritic cells [65], macrophage inflammatory responses [66] and uptake of oxLDL. Huang et al. [67] demonstrated the miR-155 could reduce the lipid uptake in oxLDL-stimulated and PMA-differentiated THP-1 cells. MiR-125a-5p was shown to decrease lipid uptake and the secretion of inflammatory cytokines, including IL-2, IL-6, TNF-α and TGF-β, in oxLDL-stimulated human primary monocytes via the repression of oxysteral binding protein like-9 (ORP9) [68]. MiR-33 has been reported to play a role in sterol transport [69, 70]. MiR-33 is an intronic miR that localizes within the gene encoding sterol-regulatory element–binding factor–2 (SREBF-2) and acts as a transcriptional regulator of cholesterol synthesis to modulate the expression of genes related to cholesterol transport. MiR target prediction algorithms and overexpression of miR-33 in mouse macrophages identified the adenosine triphosphate–binding cassette transporter (ABCA-1) as a miR-33 target gene. Antagonism of endogenous miR-33 increased ABCA1 protein and cholesterol efflux to apolipoprotein A1 in both murine and human macrophages (Table 2).
Smooth muscle cells
Neointima formation is commonly attributed to VSMC proliferation. Several reports have demonstrated the involvement of miRs in the mediation of VSMC proliferation and migration (Table 3). In rat balloon-injured carotid arteries and cultured rat VSMCs, miR-21 [71], miR-221 [72] and miR-222 were shown to play roles in the regulation of VSMC proliferation through phosphatase and tensin homology (PTEN), B-cell lymphoma 2 (Bcl-2) and p27(Kip1), p57(Kip2), respectively. PTEN and Bcl-2 have been reported to serve as important molecules associated with VSMC proliferation and apoptosis. p27(kip1) and p57(kip2) are critical molecules involved in cell cycle regulation and were demonstrated to be negative regulators in VSMC proliferation [73]. In general, miR-146a is known to serve an anti-inflammatory function in various cells (as mentioned above). Sun et al. [74] further substantiated that miR-146a directly targets Krupple-like factor-4 (KLF-4) and demonstrated its important role in promoting VSMC proliferation in cultured rat VSMCs and vascular neointimal hyperplasia. Interestingly, miR-146a and KLF-4 formed a feedback loop regulating each other’s expression. KLF-4 inhibited miR-146a at the transcriptional level, while miR-146a inhibited the expression of KLF-4 by targeting the 3’-UTR region of KLF-4. Another member of the KLF family, KLF-5, promoted the transcription of miR-146a and acted as a competitor with KLF-4. These molecules form a regulatory circuitry to precisely modulate the proliferation of VSMCs. Wu et al. [75] found that miR-130a correlated with vascular remodeling in spontaneously hypertensive rats (SHRs). MiR-130a was up-regulated in the thoracic aorta and mesenteric arteries of SHRs. In addition, the mRNA expression and protein level of growth arrest-specific homeobox (GAX) were downregulated by miR-130a. MiR-130a mimics at 25 or 50 nmol/l significantly promoted the proliferation of VSMCs.
Some miRs were found to take part in the repression of VSMC proliferation. The miR-143/145 cluster is abundantly expressed in the normal vessel walls. Interestingly, miR-143/145 is dramatically downregulated in injured carotid arteries after angioplasty [79, 80]. MiR-143 is highly conserved and lies within 1.7 kilobases (kb) of another miR145 on the mouse chromosome 18. Both miRs are downregulated in various cancer cell lines [81]. Cheng et al. [80] further demonstrated that miR-145 is a critical modulator for VSMC differentiation through its target gene KLF-5. The expression of VSMC differentiation marker genes such as SM α-actin, calponin and SM-MHC were increased at the gene and protein levels using a miR-145 mimic oligonucleotide. In contrast, overexpression of KLF-5 reduced the gene expression of SM α-actin. These results provide evidence of a correlation between miR-145 and KLF-5 in VSMC differentiation. MiR-26a was selected from growth-arrested human aortic SMCs by an miR array [77]. This profile revealed that miR-26a was significantly upregulated in differentiated VSMCs through a decrease of SMAD activity. In addition, miR-26a was dramatically downregulated in two murine AAA development models, abdominal aortic aneurysms (AAAs) and ApoE−/−/AngII aneurysm. MiR-133 is robustly expressed in VSMCs in vitro and in vivo [78]. In serum-starved synchronized adult rat carotid VSMCs, miR-133 was abundant and indirectly regulated VSMC marker genes and proteins through the Sp-1 transcription factor.
Roles of microRNA in atherosclerosis
Blood vessels are constantly subjected to various hemodynamic forces, including hydrostatic pressure, cyclic stretch and fluid shear stress. As the monolayer is in direct contact with flowing blood, vascular ECs are constantly exposed to blood flow-induced shear stress. Extensive evidence has shown that hemodynamic forces may play prominent roles in the development of vessel maturation, physiology and pathophysiology. Atherosclerosis occurs preferentially in arterial branches and curvatures where the shear stress is low and dynamic [2], and the initial step is attributed to EC dysfunction. Oscillatory shear stress (OSS) induces the expression of miR-21 at the transcriptional level in cultured ECs and eventually leads to an inflammatory response through peroxisome proliferators-activated receptor-α by 3'-UTR targeting [82]. Wu et al. [83] demonstrated that pulsatile shear stress (PSS) downregulated but OSS upregulated the expression of miR-92a in ECs. Previous studies have shown that KLF-2 was significantly upregulated by atheroprotective shear flow such as PSS and laminar shear stress. Bioinfomatic analysis demonstrated that KLF-2 serves as a target gene for miR-92a, and its gene and protein levels were downregulated by OSS-stimulated ECs. In addition, the KLF-2 regulated-genes such as eNOS and thrombomodulin (TM) were repressed by overexpression of miR-92a in ECs. This study provides a new concept for the regulatory circuitry of the responses of KLF-2 and miRs to atheroprotective shear flow. MiR-663 [84], miR-19a and miR-23b [85, 86] have also been studied and shown to be regulated by shear stress and involved in the modulation of EC inflammation and proliferation, respectively.
The functions of various miRs and their involvement in biological processes have been identified in various cultured cells or animal models. The expression profiles of circulating miRs [87] and peripheral blood mononuclear cells (PBMCs) [88–91] in patients with cardiovascular diseases have been extensively studied. Unfortunately, the involvement of miRs in human atherosclerotic plaques has received little attention. Raitoharju et al. [92] were the first to investigate the miR/mRNA expression profiles in human atherosclerotic plaques from peripheral arteries (carotid, femoral, and the aorta) in comparison to non-atherosclerotic left internal thoracic arteries (LITA), and they elucidated the relationship between miR/mRNA expression profiles and biological processes in atherosclerosis. They found that miR-21, -34a, -146a, -146b-5p, and −210 were expressed at significant levels, and numerous predicted targets of these miRs were downregulated in human atherosclerotic plaques. The combination of miR/mRNA profiles and bioinformatic analysis showed that nine KEGG pathways were enriched with predicted targets, including immunodeficiency, metabolism, p53 and cell proliferation signaling pathways. Interestingly, among these pathways, cancer-related pathways are significantly upregulated. In contrast, VSMC contraction and purine metabolism were downregulated in human atherosclerotic plaques compared to LITAs. MiR-34a was identified as a new target for atherosclerotic pathogenesis due to its function in apoptosis and cell cycle arrest, its modulation of the p53 signaling pathway, and its target genes related to VSMC proliferation and cholesterol metabolism. Taken together, these links strongly support the connection of miR-34a to cardiovascular diseases. MiR-146a is highly expressed in both human atherosclerotic plaques and PBMCs [88, 89] in patients with cardiovascular diseases It was previously shown that the miR-146 family (miR-146a/b) regulated downstream toll-like receptor 4 (TLR4) signaling, IL-1 receptor associated kinase-1 (IRAK1) and TNF-receptor associated factor-6 (TRAF6) through a negative-feedback regulation loop. IRAK and TRAF6 activated the downstream transcription factors NF-κB and AP-1 and then upregulated the TLR4-mediated immune response. Elevated miR-146 expression was shown to act in an NF-κB-dependent manner by utilizing an LPS (lipopolysaccharide)-stimulated human monocytic cell line [93].
Recent studies demonstrated that miRs can be transferred through the gap junction or secreted between cells [94–96]. Surprisingly, miRs are present in serum or plasma in a remarkably stable form that even resists repetitive freezing/thawing cycles and are protected against RNases. Fichtlscherer et al. [87] performed a miR profile using RNA isolated from 8 healthy volunteers and 8 patients with stable coronary artery disease. The circulating levels of angiogenesis-related miR-126 and miR-92a, the inflammation-associated miR-155, VSMC–enriched miR-145 and miR-17 are significantly reduced in patients with coronary artery disease compared with healthy controls. In contrast, cardiac muscle–enriched miRs, miR-133a and miR-208a levels were shown to be elevated in patients with coronary artery disease. The exact mechanisms of reduction of circulating miRs remain unclear. Supposedly, the activity of ECs may contribute to the lower levels of circulating miRs. Another implication may be that circulating miRs are taken up into atherosclerotic lesions, leading to a reduction of circulating miRs in blood. Overall, this article raises the potential role of circulating miRs as biomarkers for diagnosis of cardiovascular diseases.
Roles of microRNA in restenosis
Rat carotid artery balloon injury is a common animal model to study restenosis [36]. Ji et al. [71] were the first to determine the miR profile in the rat carotid artery after balloon injury utilizing an miR array. Aberrant overexpression of miR-21 was determined at a significant level in neointimal lesions. The miR-21 gene is located on the plus strand of chromosome 17q23.2 within the coding gene TMEM49 (also known as vacuole membrane protein). This gene was first described as an oncomir due to its abundant expression in various cancers [97]. MiR-21 is involved in the promotion of VSMC proliferation and anti-apoptosis by directly targeting PTEN and PDCD4 [98], respectively. In addition, Liu et al. [72] and Davis et al. [76] clarified the role of miR-221 and miR-222 in VSMC proliferation and neointimal hyperplasia. MiR-221 and miR-222 are encoded by a gene cluster on the X chromosome, they share the same seed and appear to have identical target genes and similar functions. Both miRs are significantly mediated by PDGF-BB and serum treatment in cultured VSMCs. Liu et al. [72] further demonstrated that expression of miR-221 and miR-222 were upregulated in balloon-injured rat carotid arteries and their target genes, p27(Kip1) and p57(Kip2), were downregulated. Downregulation of miR-221 and miR-222 reduced the proliferation of VSMCs and neointima formation in the rat carotid artery after angioplasty.
Recently, several studies demonstrated the role of the miR-143/miR-145 cluster in VSMC differentiation and vascular disease [79, 80, 99–101]. Cordes et al. [79] first revealed the distribution of miR-143/miR-145 during embryonic development. Postnatally, the transcript levels of the miR-143/miR-145 cluster are high in smooth muscle of the aorta, pulmonary artery and coronary vessels but undetectable in the ventricular myocardium. Furthermore, miR-143 and miR-145 cooperatively target a network of transcriptional factors including Elk-1 (ELK1 is a member of the ETS oncogene family), KLF-4 and myocardin to promote differentiation and repress the proliferation of VSMCs. MiR-143/miR-145 knockout (KO) mice were also established in advance to clarify the maintenance of the contractile phenotype of VSMCs [99–101]. Elia et al. [100] showed that the aorta of apolipoprotein E (ApoE) KO mice, in which vascular damage is enhanced by a hypercholesterol diet, exhibits markedly decreased constitutive levels of miR-143 and miR-145. Albinsson et al. [102] generated Dicer KO mice and found late embryonic lethality at embryonic day 16 to 17 associated with extensive internal hemorrhage. Expression of miRs, including miR−21, −221, -145 and VSMC-specific marker genes are significantly reduced in SMC-Dicer KO vessels. Interestingly, overexpression of miR-145 rescued SMC-specific mRNA and protein expression in dicer KO SMC by miR-145 mimics. This finding indicates that an additional miR-dependent mechanism is necessary during VSMC development rather than Dicer because the loss of Dicer in mice is lethal. These studies demonstrated the important role of miR-145 in VSMC differentiation and vascular disease.
Summary and conclusion
Atherosclerosis is a widespread condition with high morbidity and mortality in both developed and developing countries. Its complications, including unstable coronary syndromes, myocardial infarction and stroke, usually cause high mortality in patients. Several medications and surgical procedures have been used for clinical therapy. Patients with atherosclerotic disease are usually treated by angioplasty with stent replacement. However, restenosis is commonly observed in angioplasty patients. Both pathologies are underlined by complicated pathophysiological processes, and extensive studies on cellular mechanisms have been well established to seek opportunities for clinical therapy. MiRs are a novel class of gene regulators, and their important roles and functions in vascular biology have been demonstrated in over 400 reports. This review summarizes the current understanding of the roles of miRs in atherosclerosis and restenosis. ECs, VSMCs and blood cells contribute to both vascular pathologies. Each cell type has a specific role in these two conditions, with ECs exhibiting an inflammatory response, angiogenesis and migration; VSMCs undergoing differentiation and proliferation; and blood cells modulating oxLDL uptake and lipid metabolism. Hence, we focus on the different features of each type of cell to elucidate how miRs modulate these cellular functions. We discussed the significant changes in miR expression profiles that occur in human specimens with atherosclerosis and animal models with angioplasty. These profiles led to new insight into the potential clinical applications of miRs and emphasize the importance of miRs in the pathogenic processes of vascular diseases. Interestingly, some miRs are altered in vitro and in vivo studies, for example, miR−126, −17~92a, −145, −21 and −146a. Some miRs can only be expressed in specific tissues or cells with a special status. The EC-specific miR-126 and VSMC-specific miR-145 are usually enriched in blood vessels during embryonic development and in mature vessels. Supposedly, these miRs are involved in the maintenance of homeostasis or development of blood vessels. MiR-21 and miR-221/222 have been investigated as promoters of the proliferation of VSMCs via negative modulation of cell cycle regulation as well as of PTEN and p27. MiR-21 is also referred to as an oncomir due to its high expression levels in various cancer cell lines. This implies that these miRs contribute to vascular pathogenesis. Some miRs are expressed in multiple cells, such as miR-146a and miR-155, which are expressed in both ECs and blood cells to induce a cellular inflammatory response and protect blood vessels, respectively. This indicates that miRs may have great potential as therapeutics. Interestingly, recent advances have enabled the identification of miRs released into circulating blood from injured tissue or highly expressed in patients with cardiovascular diseases. This implies that circulating miRs and tissue/cell-specific miRs are potential biomarkers for clinical diagnosis in patients with cardiovascular diseases. The overall body of evidence shows that miRs have emerged as a new layer of complexity in vascular diseases and may represent novel biomarkers and new therapeutic targets for cardiovascular diseases.
Abbreviations
- 3’-UTRs:
-
3’-untranslated regions
- ABCA-1 transporter:
-
adenosine triphosphate–binding cassette
- Ang II:
-
angiotensin II
- ApoE:
-
apolipoprotein E
- Ago2:
-
argonaute 2
- Bcl-2:
-
B-cell lymphoma 2
- BMP:
-
bone morphogenetic protein
- CD36:
-
CD36 antigen
- 8DGCR:
-
DiGeorge critical region 8
- Egfl7:
-
EGF-like domain 7
- eNOS:
-
endothelial nitric oxide synthase
- ET-1:
-
endothelin-1
- XPO5:
-
exportin-5
- EEL:
-
external elastin lamina
- GLCE:
-
glucuronyl C5-epimerase
- GAX:
-
growth arrest-specific homeobox
- HPCs:
-
hematopoietic progenitor cells
- HMECs:
-
human microvascular ECs
- IRAK1:
-
IL-1 receptor associated kinase-1
- IGF-1:
-
insulin-like growth factor-1
- ICAM-1:
-
intercellular adhesion molecule-1
- IL-1:
-
interleukin
- IEL:
-
internal elastin lamina
- KO:
-
knockout
- KLF-4:
-
krupple-like factor-4
- LITA:
-
left internal thoracic arteries
- LPS:
-
lipopolysaccharide
- LDL:
-
low-density lipoprotein
- M-CSF:
-
macrophage colony stimulating factor
- MMPs:
-
matrix metalloproteinases
- M-CSFR:
-
M-CSF receptor
- mRNA:
-
messenger RNA
- miRs:
-
microRNAs
- MVs:
-
microvesicles
- miRISC:
-
miR-induced silencing complex
- MKK-4:
-
mitogen-activated protein kinase kinase-4
- MCP-1:
-
monocyte chemotactic factor-1
- OSS:
-
oscillatory shear stress
- oxLDL:
-
oxidation of the LDL
- ORP9:
-
oxysteral binding protein like-9
- Pasha:
-
partner of Drosha
- PTCA:
-
percutaneous transluminal coronary angioplasty
- PBMCs:
-
peripheral blood mononuclear cells
- PTEN:
-
phosphatase and tensin homology
- PIK3R2/p85-b:
-
phosphoinositide-3 kinase regulatory subunit 2
- PDGF:
-
platelet-derived growth factor
- pol II:
-
polymerase II
- pol III:
-
polymerase III
- pri-miRs:
-
primary miRs
- PSS:
-
pulsatile shear stress
- ROS:
-
reactive oxygen species
- RNASEN:
-
RNase III enzyme Drosha
- SR-A:
-
scavenger receptor-A
- SR:
-
scavenger receptors
- sPLA2:
-
secretory phospholipase-2
- SIP-1:
-
sphingosine-1-phosphate receptor-1
- Spred-1:
-
sprouty-related protein-1
- SREBF-2:
-
sterol-regulatory element–binding factor–2
- TRBP:
-
Tar RNA binding protein
- TM:
-
thrombomodulin
- THBS-1:
-
thrombrospodin-1
- TNF-α:
-
tissue necrotic factor-α
- TRAF6:
-
TNF-receptor associated factor-6
- TLR4:
-
toll-like receptor 4
- TGF-β:
-
transforming growth factor-β
- VCAM-1:
-
vascular cell adhesion molecule-1
- ECs:
-
vascular endothelial cells
- VSMCs:
-
vascular smooth muscle cells.
References
Libby P: Inflammation in atherosclerosis. Nature. 2002, 420 (6917): 868-874.
Chiu JJ, Chen LJ, Lee PL, Lee CI, Lo LW, Usami S, Chien S: Shear stress inhibits adhesion molecule expression in vascular endothelial cells induced by coculture with smooth muscle cells. Blood. 2003, 101 (7): 2667-2674.
Smith TP: Atherosclerosis and restenosis: an inflammatory issue. Radiology. 2002, 225 (1): 10-12.
Jukema JW, Verschuren JJ, Ahmed TA, Quax PH: Restenosis after PCI. Part 1: pathophysiology and risk factors. Nat Rev Cardiol. 2012, 9 (1): 53-62.
Lange RA, Flores ED, Hillis LD: Restenosis after coronary balloon angioplasty. Annu Rev Med. 1991, 42: 127-132.
Lee RC, Feinbaum RL, Ambros V: The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993, 75 (5): 843-854.
Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z: Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005, 37 (7): 766-770.
Kozomara A, Griffiths-Jones S: miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39: D152-D157. Database issue
Zhang C: MicroRNomics: a newly emerging approach for disease biology. Physiol Genomics. 2008, 33 (2): 139-147.
Weber C, Schober A, Zernecke A: MicroRNAs in arterial remodelling, inflammation and atherosclerosis. Curr Drug Targets. 2010, 11 (8): 950-956.
Nazari-Jahantigh M, Wei Y, Schober A: The role of microRNAs in arterial remodelling. Thromb Haemost. 2012, 107 (4): 611-618.
Qin S, Zhang C: MicroRNAs in vascular disease. J Cardiovasc Pharmacol. 2011, 57 (1): 8-12.
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN: MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23 (20): 4051-4060.
Cai X, Hagedorn CH, Cullen BR: Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004, 10 (12): 1957-1966.
Borchert GM, Lanier W, Davidson BL: RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006, 13 (12): 1097-1101.
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN: The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003, 425 (6956): 415-419.
Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R: The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004, 432 (7014): 235-240.
Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN: The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18 (24): 3016-3027.
Siomi H, Siomi MC: Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell. 2010, 38 (3): 323-332.
Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A: Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2010, 39 (3): 373-384.
Yi R, Qin Y, Macara IG, Cullen BR: Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17 (24): 3011-3016.
Knight SW, Bass BL: A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science. 2001, 293 (5538): 2269-2271.
Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD: A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001, 293 (5531): 834-838.
Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R: Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005, 123 (4): 631-640.
Diederichs S, Haber DA: Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007, 131 (6): 1097-1108.
Ghildiyal M, Xu J, Seitz H, Weng Z, Zamore PD: Sorting of Drosophila small silencing RNAs partitions microRNA* strands into the RNA interference pathway. RNA. 2010, 16 (1): 43-56.
Okamura K, Liu N, Lai EC: Distinct mechanisms for microRNA strand selection by Drosophila Argonautes. Mol Cell. 2009, 36 (3): 431-444.
Lusis AJ: Atherosclerosis. Nature. 2000, 407 (6801): 233-241.
Hansson GK: Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005, 352 (16): 1685-1695.
Burke-Gaffney A, Brooks AV, Bogle RG: Regulation of chemokine expression in atherosclerosis. Vascul Pharmacol. 2002, 38 (5): 283-292.
Steinberg D: Role of oxidized LDL and antioxidants in atherosclerosis. Adv Exp Med Biol. 1995, 369: 39-48.
Owens GK, Kumar MS, Wamhoff BR: Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004, 84 (3): 767-801.
Adiguzel E, Ahmad PJ, Franco C, Bendeck MP: Collagens in the progression and complications of atherosclerosis. Vasc Med. 2009, 14 (1): 73-89.
Mintz GS, Popma JJ, Pichard AD, Kent KM, Satler LF, Wong C, Hong MK, Kovach JA, Leon MB: Arterial remodeling after coronary angioplasty: a serial intravascular ultrasound study. Circulation. 1996, 94 (1): 35-43.
Bennett MR, O'Sullivan M: Mechanisms of angioplasty and stent restenosis: implications for design of rational therapy. Pharmacol Ther. 2001, 91 (2): 149-166.
Lafont A, Faxon D: Why do animal models of post-angioplasty restenosis sometimes poorly predict the outcome of clinical trials?. Cardiovasc Res. 1998, 39 (1): 50-59.
Fang Y, Shi C, Manduchi E, Civelek M, Davies PF: MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad Sci USA. 2010, 107 (30): 13450-13455.
Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ: MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci USA. 2008, 105 (5): 1516-1521.
Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN: The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008, 15 (2): 261-271.
Zhu N, Zhang D, Chen S, Liu X, Lin L, Huang X, Guo Z, Liu J, Wang Y, Yuan W, Qin Y: Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis. 2011, 215 (2): 286-293.
Li D, Yang P, Xiong Q, Song X, Yang X, Liu L, Yuan W, Rui YC: MicroRNA-125a/b-5p inhibits endothelin-1 expression in vascular endothelial cells. J Hypertens. 2010, 28 (8): 1646-1654.
Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, Soreq H: MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity. 2009, 31 (6): 965-973.
Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S: MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009, 324 (5935): 1710-1713.
Grundmann S, Hans FP, Kinniry S, Heinke J, Helbing T, Bluhm F, Sluijter JP, Hoefer I, Pasterkamp G, Bode C, Moser M: MicroRNA-100 regulates neovascularization by suppression of mammalian target of rapamycin in endothelial and vascular smooth muscle cells. Circulation. 2011, 123 (9): 999-1009.
Kuhnert F, Mancuso MR, Hampton J, Stankunas K, Asano T, Chen CZ, Kuo CJ: Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008, 135 (24): 3989-3993.
Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D: miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008, 15 (2): 272-284.
Nicoli S, Standley C, Walker P, Hurlstone A, Fogarty KE, Lawson ND: MicroRNA-mediated integration of haemodynamics and Vegf signalling during angiogenesis. Nature. 2010, 464 (7292): 1196-1200.
Anand S, Majeti BK, Acevedo LM, Murphy EA, Mukthavaram R, Scheppke L, Huang M, Shields DJ, Lindquist JN, Lapinski PE, King PD, Weis SM, Cheresh DA: MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med. 2010, 16 (8): 909-914.
Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC, Wu JC: MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation. 2010, 122 (11 Suppl): S124-S131.
Nicoli S, Knyphausen CP, Zhu LJ, Lakshmanan A, Lawson ND: miR-221 Is Required for Endothelial Tip Cell Behaviors during Vascular Development. Dev Cell. 2012, 22 (2): 418-429.
Dentelli P, Rosso A, Orso F, Olgasi C, Taverna D, Brizzi MF: microRNA-222 controls neovascularization by regulating signal transducer and activator of transcription 5A expression. Arterioscler Thromb Vasc Biol. 2010, 30 (8): 1562-1568.
Nakashima T, Jinnin M, Etoh T, Fukushima S, Masuguchi S, Maruo K, Inoue Y, Ishihara T, Ihn H: Down-regulation of mir-424 contributes to the abnormal angiogenesis via MEK1 and cyclin E1 in senile hemangioma: its implications to therapy. PLoS One. 2010, 5 (12): e14334
Caporali A, Meloni M, Vollenkle C, Bonci D, Sala-Newby GB, Addis R, Spinetti G, Losa S, Masson R, Baker AH, Agami R, le Sage C, Condorelli G, Madeddu P, Martelli F, Emanueli C: Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation. 2011, 123 (3): 282-291.
Sabatel C, Malvaux L, Bovy N, Deroanne C, Lambert V, Gonzalez ML, Colige A, Rakic JM, Noel A, Martial JA, Struman I: MicroRNA-21 exhibits antiangiogenic function by targeting RhoB expression in endothelial cells. PLoS One. 2011, 6 (2): e16979
Zhang Y, Liu D, Chen X, Li J, Li L, Bian Z, Sun F, Lu J, Yin Y, Cai X, Sun Q, Wang K, Ba Y, Wang Q, Wang D, Yang J, Liu P, Xu T, Yan Q, Zhang J, Zen K, Zhang CY: Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell. 2010, 39 (1): 133-144.
Li YX, Liu DQ, Zheng C, Zheng SQ, Liu M, Li X, Tang H: miR-200a modulate HUVECs viability and migration. IUBMB Life. 2011, 63 (7): 553-559.
Small EM, Sutherland LB, Rajagopalan KN, Wang S, Olson EN: MicroRNA-218 regulates vascular patterning by modulation of Slit-Robo signaling. Circ Res. 2010, 107 (11): 1336-1344.
Wang XH, Qian RZ, Zhang W, Chen SF, Jin HM, Hu RM: MicroRNA-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clin Exp Pharmacol Physiol. 2009, 36 (2): 181-188.
Slevin M, Krupinski J, Badimon L: Controlling the angiogenic switch in developing atherosclerotic plaques: possible targets for therapeutic intervention. J Angiogenes Res. 2009, 1: 4
Suarez Y, Fernandez-Hernando C, Pober JS, Sessa WC: Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007, 100 (8): 1164-1173.
Wang J, Xiang G, Mitchelson K, Zhou Y: Microarray profiling of monocytic differentiation reveals miRNA-mRNA intrinsic correlation. J Cell Biochem. 2011, 112 (9): 2443-2453.
Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, Croce CM, Brunetti E, Grignani F, Peschle C: MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol. 2007, 9 (7): 775-787.
Rosa A, Ballarino M, Sorrentino A, Sthandier O, De Angelis FG, Marchioni M, Masella B, Guarini A, Fatica A, Peschle C, Bozzoni I: The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc Natl Acad Sci USA. 2007, 104 (50): 19849-19854.
Forrest AR, Kanamori-Katayama M, Tomaru Y, Lassmann T, Ninomiya N, Takahashi Y, de Hoon MJ, Kubosaki A, Kaiho A, Suzuki M, Yasuda J, Kawai J, Hayashizaki Y, Hume DA, Suzuki H: Induction of microRNAs, mir-155, mir-222, mir-424 and mir-503, promotes monocytic differentiation through combinatorial regulation. Leukemia. 2010, 24 (2): 460-466.
Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, Pierre P: MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci USA. 2009, 106 (8): 2735-2740.
O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D: MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. 2007, 104 (5): 1604-1609.
Huang RS, Hu GQ, Lin B, Lin ZY, Sun CC: MicroRNA-155 silencing enhances inflammatory response and lipid uptake in oxidized low-density lipoprotein-stimulated human THP-1 macrophages. J Investig Med. 2010, 58 (8): 961-967.
Chen T, Huang Z, Wang L, Wang Y, Wu F, Meng S, Wang C: MicroRNA-125a-5p partly regulates the inflammatory response, lipid uptake, and ORP9 expression in oxLDL-stimulated monocyte/macrophages. Cardiovasc Res. 2009, 83 (1): 131-139.
Marquart TJ, Allen RM, Ory DS, Baldan A: miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci USA. 2010, 107 (27): 12228-12232.
Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C: MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010, 328 (5985): 1570-1573.
Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C: MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007, 100 (11): 1579-1588.
Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C: A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009, 104 (4): 476-487.
Dzau VJ, Braun-Dullaeus RC, Sedding DG: Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002, 8 (11): 1249-1256.
Sun SG, Zheng B, Han M, Fang XM, Li HX, Miao SB, Su M, Han Y, Shi HJ, Wen JK: miR-146a and Kruppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO Rep. 2011, 12 (1): 56-62.
Wu WH, Hu CP, Chen XP, Zhang WF, Li XW, Xiong XM, Li YJ: MicroRNA-130a mediates proliferation of vascular smooth muscle cells in hypertension. Am J Hypertens. 2011, 24 (10): 1087-1093.
Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A: Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009, 284 (6): 3728-3738.
Leeper NJ, Raiesdana A, Kojima Y, Chun HJ, Azuma J, Maegdefessel L, Kundu RK, Quertermous T, Tsao PS, Spin JM: MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J Cell Physiol. 2011, 226 (4): 1035-1043.
Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C: MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. 2011, 109 (8): 880-893.
Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D: miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009, 460 (7256): 705-710.
Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C: MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009, 105 (2): 158-166.
Calin GA, Croce CM: MicroRNA signatures in human cancers. Nat Rev Cancer. 2006, 6 (11): 857-866.
Zhou J, Wang KC, Wu W, Subramaniam S, Shyy JY, Chiu JJ, Li JY, Chien S: MicroRNA-21 targets peroxisome proliferators-activated receptor-alpha in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc Natl Acad Sci USA. 2011, 108 (25): 10355-10360.
Wu W, Xiao H, Laguna-Fernandez A, Villarreal G, Wang KC, Geary GG, Zhang Y, Wang WC, Huang HD, Zhou J, Li YS, Chien S, Garcia-Cardena G, Shyy JY: Flow-Dependent Regulation of Kruppel-Like Factor 2 Is Mediated by MicroRNA-92a. Circulation. 2011, 124 (5): 633-641.
Ni CW, Qiu H, Jo H: MicroRNA-663 upregulated by oscillatory shear stress plays a role in inflammatory response of endothelial cells. Am J Physiol Heart Circ Physiol. 2011, 300 (5): H1762-H1769.
Qin X, Wang X, Wang Y, Tang Z, Cui Q, Xi J, Li YS, Chien S, Wang N: MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc Natl Acad Sci USA. 2010, 107 (7): 3240-3244.
Wang KC, Garmire LX, Young A, Nguyen P, Trinh A, Subramaniam S, Wang N, Shyy JY, Li YS, Chien S: Role of microRNA-23b in flow-regulation of Rb phosphorylation and endothelial cell growth. Proc Natl Acad Sci USA. 2010, 107 (7): 3234-3239.
Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Roxe T, Muller-Ardogan M, Bonauer A, Zeiher AM, Dimmeler S: Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010, 107 (5): 677-684.
Guo M, Mao X, Ji Q, Lang M, Li S, Peng Y, Zhou W, Xiong B, Zeng Q: miR-146a in PBMCs modulates Th1 function in patients with acute coronary syndrome. Immunol Cell Biol. 2010, 88 (5): 555-564.
Takahashi Y, Satoh M, Minami Y, Tabuchi T, Itoh T, Nakamura M: Expression of miR-146a/b is associated with the Toll-like receptor 4 signal in coronary artery disease: effect of renin-angiotensin system blockade and statins on miRNA-146a/b and Toll-like receptor 4 levels. Clin Sci. 2010, 119 (9): 395-405.
Yao R, Ma Y, Du Y, Liao M, Li H, Liang W, Yuan J, Ma Z, Yu X, Xiao H, Liao Y: The altered expression of inflammation-related microRNAs with microRNA-155 expression correlates with Th17 differentiation in patients with acute coronary syndrome. Cell Mol Immuno. 2011, 8 (6): 486-495.
Li T, Cao H, Zhuang J, Wan J, Guan M, Yu B, Li X, Zhang W: Identification of miR-130a, miR-27b and miR-210 as serum biomarkers for atherosclerosis obliterans. Clin Chim Acta. 2011, 412 (1–2): 66-70.
Raitoharju E, Lyytikainen LP, Levula M, Oksala N, Mennander A, Tarkka M, Klopp N, Illig T, Kahonen M, Karhunen PJ, Laaksonen R, Lehtimaki T: miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis. 2011, 219 (1): 211-217.
Taganov KD, Boldin MP, Chang KJ, Baltimore D: NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006, 103 (33): 12481-12486.
Fichtlscherer S, Zeiher AM, Dimmeler S: Circulating microRNAs: biomarkers or mediators of cardiovascular diseases?. Arterioscler Thromb Vasc Biol. 2011, 31 (11): 2383-2390.
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO: Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007, 9 (6): 654-659.
Lim PK, Bliss SA, Patel SA, Taborga M, Dave MA, Gregory LA, Greco SJ, Bryan M, Patel PS, Rameshwar P: Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71 (5): 1550-1560.
Bonci D: MicroRNA-21 as therapeutic target in cancer and cardiovascular disease. Recent Pat Cardiovasc Drug Discov. 2010, 5 (3): 156-161.
Lin Y, Liu X, Cheng Y, Yang J, Huo Y, Zhang C: Involvement of MicroRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. J Biol Chem. 2009, 284 (12): 7903-7913.
Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T: Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009, 119 (9): 2634-2647.
Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G: The knockout of miR-143 and −145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009, 16 (12): 1590-1598.
Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN: MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009, 23 (18): 2166-2178.
Albinsson S, Suarez Y, Skoura A, Offermanns S, Miano JM, Sessa WC: MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler Thromb Vasc Biol. 2010, 30 (6): 1118-1126.
Acknowledgements
This work was supported by National Science Council (Taiwan) Grants 101-2321-B-400-001, 101-2325-B400-009 and National Health Research Institutes (Taiwan) Grant 100-MEPP06.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LJC and JJC wrote the manuscript. SHL, YTY, and SCL assisted with the revision of English grammar and style. All authors discussed the content and approved the final version of manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chen, LJ., Lim, S.H., Yeh, YT. et al. Roles of microRNAs in atherosclerosis and restenosis. J Biomed Sci 19, 79 (2012). https://doi.org/10.1186/1423-0127-19-79
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1423-0127-19-79