
Abstract
Adhesions are permanent fibrovascular bands between peritoneal surfaces, which develop following virtually all body cavity surgeries. The susceptibility to develop, and the severity, of adhesions following intra-abdominal surgery varies within and between individuals, suggesting that heritable factors influence adhesion development. In this manuscript, we discuss the pathophysiology of adhesion development from the perspective of genetic susceptibility. We restrict our discussion to genes and single-nucleotide polymorphisms (SNPs) that are specifically involved in, or that cause modification of, the adhesion development process. We performed a literature search using the PubMed database for all relevant English language articles up to March 2020 (n = 186). We identified and carefully reviewed all relevant articles addressing genetic mutations or single-nucleotide polymorphisms (SNPs) that impact the risk for adhesion development. We also reviewed references from these articles for additional information. We found several reported SNPs, genetic mutations, and upregulation of messenger RNAs that directly or indirectly increase the propensity for postoperative adhesion development, namely in genes for transforming growth factor beta, vascular endothelial growth factor, interferon-gamma, matrix metalloproteinase, plasminogen activator inhibitor-1, and the interleukins. An understanding of genetic variants could provide insight into the pathophysiology of adhesion development. The information presented in this review contributes to a greater understanding of adhesion development at the genetic level and may allow modification of these genetic risks, which may subsequently guide management in preventing and treating this challenging complication of abdominal surgery. In particular, the information could help identify patients at greater risk for adhesion development, which would make them candidates for anti-adhesion prophylaxis. Currently, agents to reduce postoperative adhesion development exist, and in the future, development of agents, which specifically target individual genetic profile, would be more specific in preventing intraperitoneal adhesion development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Postoperative adhesions are fibrovascular bands between peritoneal surfaces that follow abdominal and pelvic surgeries, and remain largely unavoidable despite new advancements in the field of minimally invasive surgery [1]. The presence of intraperitoneal adhesions is associated with immediate-, short-, and long-term sequelae, including but not limited to difficult reoperations, increased operating time secondary to lysis of adhesions, and the need to take time to repair injury to other organs such as bowel, bladder or the ureters [2], infertility [3], and chronic abdominal or pelvic pain [4]. It is therefore incumbent on scientists to elucidate the etiology of adhesion development.
One area of research that needs exploration is the genetics of adhesion development. From an epidemiological perspective, susceptibility to develop, and the severity, of adhesions following intra-abdominal surgery varies within and between individuals [5]. In addition, while some patients develop little to no scar tissue, others develop dense adhesions from seemingly equivalent procedures by the same surgeon despite comparable surgical techniques [5]. Furthermore, adhesions tend to recur in these same patients. These raise the question as to whether there is a genetic predisposition to the development of postoperative adhesions [6]. This possibility is also supported by epidemiological data that suggest that the pathogenesis of intraperitoneal adhesion development may be similar in several aspects to the pathogenesis of other benign fibro-proliferative disorders such as keloids [7] and retroperitoneal fibrosis [8]. In several studies related to intraperitoneal adhesion development, knockout gene experiments of growth and transcription factors in mice compared to wild types [9,10,11] raise the possibility that mutations in genes involved in the pathogenesis of adhesions [12] may predispose patients to adhesion development, even when such mutations are currently unrecognized in humans. These would suggest that heritable factors influence intraperitoneal adhesion development and are, likely, inherited as a complex genetic trait for which the phenotype reflects interactions between allelic variants of susceptibility genes and gene-environment interactions. Even where genetic mutation or single-nucleotide polymorphism (SNP) are established, it remains unclear whether the genotype is directly related to predisposition to adhesion development or whether they simply act as permissive factors in the pathogenesis of adhesion formation. This lack of understanding has limited our ability to develop a personalized approach to prevent adhesions. Currently, there are no available models to help predict an individual’s susceptibility to develop adhesions following an intra-abdominal surgery. Furthermore, there is a lack of reliable methods to prevent adhesion development in predisposed individuals. At present, we are not aware of any concise publication that summarizes the published literature on the genetic predisposition to postoperative adhesion development.
In this manuscript, we discuss the pathophysiology of adhesion development from the perspective of genetic susceptibility. We restrict our discussion to genes and single-nucleotide polymorphisms (SNPs) that are specifically involved in, or that cause modification of, the adhesion development process. A review of the molecular and genetic aberrations associated with adhesion development could direct future research directions and prevention efforts, which may subsequently guide the management of women who develop adhesions following intra-abdominal and pelvic surgery.
Methods
We performed a literature search using the PubMed database for all relevant English language articles up to March 2020 that addressed the genetics of adhesion development. We used medical subject headings (MeSH) terms (genetic mutations) AND (single nucleotide polymorphism) AND (individual known mutations and SNP previously described to be associated with intraperitoneal adhesion development) AND (adhesion development), which revealed 186 articles. Of these, we identified and reviewed 84 that were relevant and/or addressed genetic mutations or single-nucleotide polymorphisms (SNPs) that influence the risk for adhesion development. We also reviewed references from these articles for additional information.
Pathophysiology of Adhesion Formation
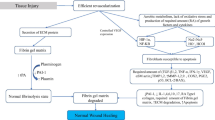
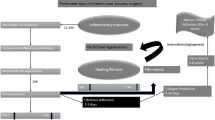
The surface covering of the peritoneum consists of a layer of the mesothelium, below which are fibroblasts, macrophages, and blood vessels. Under normal physiological conditions, eukaryotic cells are under aerobic conditions and have an innate defense system against reactive oxygen species (ROS), which contributes to the maintenance of the balance between pro-oxidants (free radical species) and the body’s scavenging ability (antioxidants). When the peritoneum is breached, it releases mesothelial cells, macrophages, fibroblasts, and blood containing cytokines and several cell types at the site of injury. Peritoneal injury and de-vascularization also result in tissue hypoxia at the injured site. Tissue hypoxia following peritoneal injury, if it persists, necessitates anaerobic metabolism [13], the production of superoxide (O2•−), and the formation of peroxynitrite (ONOO−) (Fig. 1). Collectively, these events result in increased oxidative stress [12], which maintains continuous signaling of hypoxia-induced factor (HIF)-1α [14] and nuclear factor-kB (NF-kB) family of proteins [15] produced by altered normal fibroblasts. These fibroblasts acquire the adhesion phenotype [16] and are designated adhesion fibroblasts [17] (Fig. 2). The adhesion fibroblast cells if they persist, together with mesothelial cells and macrophages [18], produce less nitrous oxide (NO) than normal fibroblasts [19] under the hypoxic environment at the injured site. These adhesion fibroblasts manifest elevated expression and increased production of adhesiogenic transcription factors and their proteins [12]. The production of HIF-1α [14], and NF-kB [15], stimulates the cascading pathways [12], which includes increased expression and cross activations of a host of transcription and growth factors and cytokines (Fig. 2). These include increased interferon regulatory factor-1 (IRF-1) [20], B cell leukemia/lymphoma-2 {(BCl-2)/BCl2-associated X (BAX)} [21], and cytokine production such as TGF-β1 and -β2 [22,23,24]. Also, increased are granulocyte macrophage colony-stimulating factor and interleukins-1, -4, -6, and -10 (IL-1, -4, -6, -10) [17, 25, 26], lipopolysacharide (LPS), and tumor necrotic factor (TNF)-α [27]. In addition, there are associated increase; angiogenesis associated with increased production of vascular endothelial growth factor (VEGF) [28], alpha-smooth muscle actin (α-SMA) [29] glycoprotein, components of the extracellular matrix (ECM), type I and type III collagen and fibronectin [18], and expression of cyclooxygenase (COX-2) [30]. Also, involved are reduced proteolytic enzyme productions such as decreased ratios of tissue plasminogen activator/plasminogen activator inhibitor ((tPA)/PAI-1) [31] and matrix metalloproteinase-1/tissue inhibitor of metalloproteinase (MMP-1/TIMP-1) [17, 32]. Adhesion tissue (AT) fibroblasts also exhibit lower apoptosis and higher protein nitration as compared to normal peritoneal (NP) fibroblasts, potentiating intraperitoneal adhesion development [32, 33] (Figs. 1 and 2). In addition, work by Detti and colleagues [34] showed that the inflammatory-like changes manifested by adhesion fibroblasts enhance the expression of receptors of anabolic hormones estrogen (ER-alpha, ER-beta) but not progesterone (PR), when exposed to estradiol. Oxidative stress, if it persists, leads to alteration in the innate immune system [35] and ultimately an increase in the expression of the genes responsible for the aforementioned transcription factors and cytokines. These genes and the proteins they produce are involved in vascular and inflammatory responses, fluid exudation, ECM deposition, cell growth and differentiation, angiogenesis, and eventual tissue remodeling [5, 25, 36] leading to adhesion bands that join adjacent and injured peritoneum (Figs. 1 and 2) [37,38,39,40].

Pathogenesis of adhesion development. ECM, extracellular matrix; HIF, hypoxia-induced factor; NADPH, nicotine adenine dinucleotide phosphate; PAI, plasminogen activator inhibitor; TGF-β, transforming growth factor beta; tPA, tissue plasminogen activator; VEGF, vascular endothelial growth factor

Proposed chain of events; growth factors, cytokines, associated gene mutations, and SNPs involved in adhesion development. ↑ denotes an increase and ↓ a decrease; BCl-2, B cell CLL/lymphoma 2; BAX, BCl2-associated X; COX-2, cyclooxygenase 2; ECM, extracellular matrix; HO, hydroxyl radical; H2O2, hydrogen peroxide; HIF, hypoxia-induced factor; HSP90, heat shock protein 90; IFN-γ, interferon-gamma; IL, interleukin; iNOS, inducible nitrous oxide synthase; IRF-1, interferon regulatory factor-1; MMP, matrix metalloproteinases; NADP, nicotine adenine dinucleotide phosphate; NF-kB, nuclear factor-kB; NO, nitric oxide; NOS, nitric oxide synthase; O2•−, superoxide; ONOO−, peroxynitrite; P53, tumor protein 53; PAI-1, plasminogen activator inhibitor; PGE2, prostaglandin E2; TGF-β1, transforming growth-beta1; TIMP, tissue inhibitor of matrix metalloproteinases; TNF-α, tumor necrosis factor; tPA, tissue plasminogen activator; VEGF, vascular endothelial growth factor
Inbuilt into the normal wound healing process without adhesion development is a balance between the fibrinolysis by plasmin (the active form of the proenzyme plasminogen, cleaved by tissue plasminogen activator (tPA) or urokinase-type plasminogen activator (uPA)) that degrades ECM, and tPA suppression by plasminogen activator inhibitor-1 (PAI-1), a serine protease inhibitor [39, 41,42,43] (Fig. 1). The induced hypoxemic conditions in intraperitoneal adhesion development [28, 44,45,46] are theorized to disrupt the balance of persistence of the fibrin gel matrix with fibroblast infiltration laying down collagen (a prerequisite to adhesion development) [47], lower apoptosis through S-nitrosylation of caspase-3 [33] and lower fibrinolysis [48], leading to the formation of disarrayed dense tissue. Similarly, hyperoxemia as with oxygen treatment can also cause oxidative stress via an increase in the NADH oxidase and P450 reductase pathway [49] as shown in Fig. 1. The resulting adhesions, once vascularized, are hypothesized to be a means of providing re-oxygenation to de-vascularized tissue [45].
Genetic Predisposition to Postoperative Adhesions Development
Advances in molecular biology, reverse transcription polymerase chain reaction (RT-PCR) [36], chromosomal microarray [50], and the development of transgenic mice [51] technologies have provided new insight into the function of many different genes involved in wound healing. These technologies allow the gain of function (overexpression of genes) as well as the loss of function experiments (gene knockout) and have made it possible to determine the functions of proteins necessary for wound healing. In addition, the availability of genetically modified mice have helped researchers to elucidate the role of the deleted [52], mutated, or overexpressed genes [53] in different types of repair processes. Genes that predispose to intraperitoneal adhesion development might include any of those that direct the molecular processes that control the development, proliferation, survival, and migration of the adhesion fibroblast into the injured site after surgery (Fig. 2). Indeed, there is mounting evidence that mutations or SNPs in some of these genes exhibit abnormal products that are relevant to the pathogenesis of the disease. This may help explain why some patients develop little to no scar tissue, while others develop dense adhesions from similar procedures by the same surgeon employing comparable surgical techniques [5]. Mutations and SNPs or manipulation of some of the genes responsible for the production of relevant transcription and growth factors may provide insight into their role in adhesion development.
In this manuscript, we summarize current evidence, when they exist, regarding the effects and manifestations of reported allelic variants in genes coding for individual transcription and growth factors, and cytokines involved in the adhesion cascade following intraperitoneal surgery (Table 1). A good understanding of adhesion genetics will not only assist pharmaceuticals to develop targeted therapy but may assist clinicians to institute measures to prevent this disorder and better manage patients with intraperitoneal adhesions. We also discuss the preliminary efforts—when they exist—by researchers to use this predisposing genetic susceptibility to design targeted pharmaco-therapeutics and preventive measures against adhesion development.
Hypoxia-Induced Factor and the Nuclear Factor-kB
Hypoxia-induced factor (HIF) [14] and (NF)-kB [15, 54] family of proteins are known to increase the production of hydrogen peroxide and TGF-β1 expression, which in turn augments the production of TIMP-1,2, COX-2, and prostaglandin E2 leading to the propensity for adhesion development as shown in Fig. 2. Hypoxia-induced factor-1α gene is assigned to human chromosome 14q21-q24 [54] and (NK)-kB located on chromosome 11q13.1 but no SNP in these regions to our knowledge has been associated with adhesion development. Cyclooxygenase-2 gene is located on chromosome 1q31.1 [55]. Wei and colleagues [56] found increased expression of hypoxia-induced COX-2 in peritoneal fibroblasts of postoperative intra-abdominal adhesions in a rat model. The authors also showed that inhibition of COX-2 attenuated the activating effect of hypoxia on NP fibroblasts in vitro. Their data suggest that selective COX-2 inhibition prevents in vivo intra-abdominal adhesion development by inhibition of basic fibroblast growth factor and TGF-β expression. In addition, Saed et al. [57] transfected normal and adhesion fibroblasts with an adenovirus COX-2 mRNA in sense or antisense orientation. The authors demonstrated that adhesion fibroblasts transfected with COX-2 antisense exhibited markedly reduced mRNA levels of type I and type III collagen, fibronectin, and TGF-β1, suggesting that inhibition of COX-2 may reduce the development of postoperative adhesions by preventing the formation of the adhesion phenotype. A search of the literature did not reveal any mutation or SNPs in the COX-2 gene in relation to adhesion development.
Transforming Growth Factor Beta
Transforming growth factor beta is a cytokine linked to adhesion formation as it regulates the expression of other growth factors and cytokines involved in the adhesion cascade such as interferon-gamma (IFN-1γ) and interleukin (IL)-17, PAI, and MMPs [46]. In mammals, TGF-β exists in three isoforms namely TGF-β1, TGF-β2, and TGF-β3 [5]. Transforming growth factor-β1 is the main isoform involved in regulating ECM deposition in the wound healing process and acts as a principal profibrotic cytokine [5]. The TGF-β1 gene is located on chromosome 19q13.2. In fact, recent evidence shows that the unregulated activity of TGF-β1 protein results in fibrotic diseases throughout the body [5]. As with other pathways already discussed, hypoxia is a key player in TGF-β-induced adhesion formation. Diamond et al. [46] demonstrated that TGF-β mRNA expression in AT fibroblasts was 96% higher than in NP fibroblasts, and these levels were further increased by 37% when exposed to hypoxia. In murine studies, Krause et al. [58] demonstrated that mice heterozygous for TGF-β1 had significantly lower amounts of adhesions compared to wild-type mice. Furthermore, neutralizing antibodies directed against TGF-β1 resulted in decreased adhesion formation [58].
Dichloroacetic acid (DCA) is known to stimulate the pyruvate dehydrogenase (PD) complex by inhibiting the PD kinase enzyme thereby trapping all PD in its active form. This way, DCA stimulates oxidative metabolism, allowing all pyruvate resulting from the 6-carbon sugar, glucose, and three carbon lactate, alanine, and glycerol to shunt into Kreb’s cycle converting anaerobic metabolism resulting from hypoxia at the injured site to aerobic metabolism. Diamond and colleagues [46] exploited this mechanism by exposing human fibroblasts cultures from normal peritoneum and adhesions to DCA using multiplex RT-PCR and reported that not only did DCA inhibit TGF-β1 mRNA expression under normoxic and hypoxic conditions; it markedly reduced ECM component (fibronectin and type III collagen) expression under hypoxic conditions in both fibroblasts. This is also in keeping with previous work from the same group [59] that showed that DCA reduces type 1 collagen mRNA levels in a dose-dependent manner in normal and adhesion fibroblasts cultured under hypoxic conditions with the adhesion fibroblast showing a greater reduction, including under hypoxic conditions. Thus, with the conversion to aerobic metabolism at the site of injury, cells may not sense the need to produce TGF-β1, and therefore, the cascades of molecular event downstream (Fig. 2) should lead to a decrease in adhesion development. As TGF-β1 also regulates the expression of PAI-1, these results indicate that modulation of the TGF-β1 pathway can result in significantly fewer adhesions postoperatively in at-risk individuals. To date, however, no report of polymorphism in the TGF-β1 gene exists in humans in relation to intraperitoneal adhesion development (IPAD).
Vascular Endothelial Growth Factor
The vascular endothelial growth factor (VEGF) gene is located on chromosome 6p12 [60], and its protein product induces and regulates angiogenesis by several mechanisms. It acts as an endothelial cell mitogen, increases vascular hyperpermeability, promotes the deposition of an ECM, and finally, it helps modulate ECM proteolysis, an essential component of the angiogenic process [61, 62]. The vascular endothelial growth factor is therefore involved in the development of intraperitoneal adhesions. Hypoxic cells show an increased expression of VEGF as a regulatory mechanism to increase oxygenation to de-vascularized tissue by activating HIF-1α [45]. Saltzman et al. [63] were the first to suggest a role for VEGF in the pathogenesis of adhesion formation. The authors used an antiserum to vascular permeability factor/VEGF to inhibit postoperative adhesion formation in a murine model in a randomized study. They performed intraperitoneal administration of either vascular permeability factor antiserum (n = 14) or pre-immune serum (n = 14) after a standardized peritoneal injury and found that mice treated with the study medication had significantly lower adhesion scores than did the control group. In addition, they reported that when the groups were analyzed for the presence of grade 2 or 3 adhesions, the group treated with vascular permeability factor antiserum had a significantly lower incidence of advanced adhesions (38% vs control 92%). This study demonstrates that the intraperitoneal administration of a neutralizing antiserum to vascular permeability factor/VEGF limits intraperitoneal adhesion development.
An increase in VEGF mRNA has been isolated in mouse studies with tissue injury, as well as from adhesions in human samples [28]. In fact, Diamond and colleagues [28] demonstrated that VEGF mRNA is 32% higher in fibroblasts isolated from adhesions compared to NP fibroblasts. Additionally, oxygen deprivation led to increased VEGF mRNA expression compared to normal conditions. Using this data, Diamond and collaborators [28] showed that aerobic conditions led to a statistically significant decrease in VEGF mRNA in both peritoneal and adhesion fibroblasts. Research involving modulation of the VEGF pathway led Molinas et al. [64] to demonstrate that VEGF knockout mice had decreased rates of adhesion formation compared to wild-type mice. This information sheds light on potential approaches for decreasing postoperative adhesions through regulating VEGF expression in hypoxic tissues. Currently, there is no literature on specific VEGF polymorphisms as they relate to IPAD in humans.
Plasminogen Activator Inhibitor-1
Plasminogen activator inhibitor-1 (PAI-1) is the main inhibitor of urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), the enzymes responsible for cleaving plasminogen into its active form plasmin [46]. Thus, PAI-1 serves as an inhibitor to the fibrinolysis pathway by preventing fibrin matrix breakdown (Fig. 1). The PAI-1 gene lies on chromosome 7q21.3-q22 and consists of nine exons and eight introns [65]. A well-known 4G/5G polymorphism at the − 675 position in the promoter region was implicated in intraperitoneal adhesion development [66]. Of the two alleles in the PAI-1 gene, the 4G allele has more transcriptional activity than the 5G allele as the latter contains an additional binding site for a repressor [67, 68]. Consequently, carriers of 4G/4G genotype have a higher concentration of PAI-1 than individual carrying 5G. This has been confirmed by one case-control study (Ramon et al.) [69] and one meta-analysis of five case-control studies (Zhao and colleagues) [70] that demonstrated that PAI-1 levels were significantly higher in women with the 4G/4G genotype compared to women with the 5G/5G genotype. A caveat to the study by Zhao et al. [70] was that after subgroup analysis by ethnicity, the significantly increased risks were found only among Asians and Brazilians but not in Caucasians. Nevertheless, the upregulation of PAI-1 may result in the impairment of the fibrinolytic system. Saed and Diamond [31] showed that fibroblasts isolated from adhesive tissue had higher levels of PAI-1 expression compared to normal fibroblasts, and this activity was further increased by hypoxia.
Efforts to identify high-risk individuals (i.e., those carrying the 4G/4G polymorphism) are underway with a view to intervening pharmacologically with PAI-1 inhibition to reduce postoperative adhesions [71]. In fact, Honjo et al. [71] have shown decreased adhesion formation in murine models using a novel PAI-1 inhibitor, named TM5275, a drug that blocks the binding site between tPA and PAI-1, thus rendering PAI-1 ineffective. Mice treated with this drug showed reduced intra-abdominal adhesions as well as decreased histologically detectable fibrotic changes, without increased evidence of hemorrhage. This study reveals a promising new avenue to explore for reducing adhesion formation, especially in those patients predisposed to higher PAI-1 levels.
Matrix Metalloproteinases
The MMP-1 gene is located on chromosome 11q22.2. Matrix metalloproteinase expressions are limited to areas undergoing extensive tissue remodeling given that they degrade ECM components as part of normal healing [45]. It is estimated that there are approximately 18 MMPs, with MMP-1 being the most important in ECM and collagen degradation [45]. Tissue inhibitors of matrix metalloproteinases (TIMPs) regulate matrix metalloproteinases activity and typically bind in a 1:1 ratio. Therefore, one would expect alterations in the expression of TIMP to affect adhesion formation. Diamond et al. [45] exposed human fibroblast cultures from normal peritoneum and adhesions to dichloroacetic acid (DCA) under normal and hypoxic conditions and found that under hypoxic conditions, DCA reduced TIMP-1 mRNA expression from both, which would lead to a decreased propensity for adhesion formation. Therefore, similar to TGF-β1 mRNA [46], increasing intracellular energy stores at the cellular level can be altered in such a way that cells at the site of peritoneal injury no longer senses hypoxic conditions, which could decrease adhesion formation [28]. Diamond et al. [45] proposed that this could be done by simulating aerobic metabolism, leading to alterations in the interaction between MMP/TIMP and thus decreased de novo adhesion formation in response to hypoxia. To our knowledge, no study has looked at polymorphisms in the MMP-1 gene as it relates to adhesion development.
HLA System
Recently, attention has turned to the field of immunogenetics and the potential role it plays in abnormal wound healing. Initial research on this topic focused on the major histocompatibility complex (MHC), a set of cell surface proteins that allow the immune system to recognize foreign material. In humans, the MHC is known as the human leukocyte antigen (HLA) system, and increasing attention has been given to the HLA class II region for its role in wound healing [6]. Erdogan et al. [72] focused on whether there was an association between different phenotypes of the HLA system and intestinal adhesion formation in children. The authors study showed a statistically significant association between the HLA subtypes A24 and DR11; however, the authors were unable to elucidate a clear mechanism linking the MHC system to adhesion formation.
Alpay and colleagues [35] through various experiments confirmed that natural immune response plays a role in the elimination of AT fibroblasts. The authors performed flow cytometry on primary cell cultures of NP and AT fibroblasts obtained from the same four patients and found that the average expression of CD54, CD40, and CD120b was significantly greater in AT compared with NP fibroblasts. In addition, they reported that the lymphokine-activated killer (LAK) cell-mediated fibroblast elimination correlated significantly with the increased expression of these three lymphocytes while the average LAK cell-mediated fibroblast killing was 1.8 ± 0.8-fold greater in AT fibroblast over NP fibroblast (P = 0.008). In another study, the same authors [73] demonstrated that hypoxia treatment enhances NP fibroblast elimination by LAK cells to the level of AT fibroblasts. These results demonstrate that AT fibroblasts are more susceptible to lymphocyte-mediated elimination than NP fibroblasts. Therefore, the development of adhesions despite the susceptibility of AT fibroblasts to immune elimination could be explained by either impaired or overwhelmed autologous natural immune response against reactive fibroblasts.
Interleukins
An initial step following tissue trauma is the release of cytokines during the inflammatory phase prior to wound healing. Key mediators of the inflammatory response are the interleukins (IL-1, -4, -6, -10, -17). These pro-inflammatory acute-phase reaction cytokines lead to systemic toxic effects [74], complement and clotting cascade activation, and increased expression of PAI-1 and TGF-β [38, 75]. An increase in these interleukins can lead to an increased propensity for adhesion development (Fig. 2).
Interleukin-1 is regulated by the IL-1 receptor antagonist (IL-1RN), both of which are located on chromosome 2q13 [38, 75, 76]. A mutant allele has been identified on the IL-1RN gene in Caucasian women, named IL-1RN*2 [75]. This mutation leads to decreased production of IL-1RN, hence, decreased fibrinolytic activity and increased risk for intraperitoneal adhesion development, a risk that is independent of previous abdominal surgery and the presence of endometriosis [75]. Furthermore, these authors [75] showed that rats treated with a monoclonal antibody against IL-1 prior to surgery developed significantly fewer postoperative adhesions compared to controls [75]. Stocks et al. [77] demonstrated a significant decrease in the amount of intraperitoneal adhesions in their murine studies when these mice were given a diet rich in fish oil. They postulated that the anti-inflammatory properties of omega-3 fatty acids decreased activation of large protein complexes called inflammasomes, which serve as immune system receptors and sensors to regulate the innate immune system [77]. Inflammasomes have sparked an area of interest in modulating many immune-mediated disease states. The most widely studied inflammasome complex is the NLR family pyrin domain containing 3 (NLRP3), which activates IL-1β. In this study, Stocks et al. [77] injected endometrial tissue from women with endometriosis and controls into the peritoneum of mice, and found that significantly greater amounts of adhesions were formed in the experimental group, indicating the activation of inflammasomes and therefore IL-1β leading to greater adhesion development [77]. Likewise, by blocking the action of IL-1β, the researchers showed a significantly decreased amount of adhesion formation with minimal collagen deposition [77].
Interleukin (IL)-6 and tumor necrosis factor (TNF)-α are also implicated in adhesion development. Studies by Ambler et al. [50] showed significantly higher IL-6 and TNF-α expression in AT fibroblasts compared to NP fibroblasts. This effect was more prominent in hypoxic conditions; however, there was a significant increase in adhesion formation even in a normoxic environment. Zhang et al. [78] were able to prevent postoperative adhesion formation in rats with ligustrazine, a substance routinely used to treat patients with coronary artery disease in China, by decreasing the serum levels of IL-1β, IL-6, connective tissue growth factor (CTGF), TNF-α, and the expression of TGF-β1 in the peritoneal fluid in a dose-dependent manner. To date, however, there has been no report of mutations or SNPs in IL-10 and -17 in association with the risk of intraperitoneal adhesion development.
Class I MHC-restricted T cell-associated molecule (CRTAM) is a member of the nectin-like adhesion molecule family. CRTAM plays a role in IFN-γ and IL-17 production and T cell proliferation. Interleukin-17 also plays a role in the development of adhesions by promoting the production of PAI-1 and inhibiting tPA [79]. Valle-Rios and colleagues [80] detected an activator protein (AP)-1 binding site, located at 1.4 kb from the ATG translation codon of CRTAM gene as an essential element for CRTAM expression in activated but not resting human CD8(+) T cells. CRTAM expression was reduced in activated CD8(+) T cells treated with the JNK inhibitor SP600125, indicating that CRTAM expression is driven by the JNK-AP-1 signaling pathway, which suggests that AP-1 is a positive transcriptional regulator of this gene. In addition, blocking IL-17 might have a therapeutic potential in preventing intraperitoneal adhesion development after surgery [81].
Interferon-Gamma
Another major cytokine of interest is IFN-γ. Interferon-gamma downregulates the expression of TGF-β in murine studies, likely by blocking its signal transduction pathways [82]. Similarly, Saed and Diamond [83] using primary cultured fibroblasts showed that IFN-γ can block the stimulating effects of hypoxia on type I collagen and fibronectin mRNA expression, supporting the antifibrogenic nature of this cytokine. The mechanism involved is unknown but may be due to the ability of IFN-γ to modulate the Smads 2–4 pathway of TGF-β1 signal transduction that prevents the upregulation of ECM components [84] albeit in subconjunctival fibroblasts. Therefore, IFN-γ could be a good candidate for consideration for intervention in the prevention of peritoneal adhesions. A caveat to these findings is work by Wang et al. [81] that showed that decreasing IFN-γ postoperatively significantly decreased adhesion formation. Given these relatively contradictory results, further work is required to determine whether IFN-γ is an anti-adhesion [83] or pro-adhesion-forming cytokine [81]. At this time, no study has investigated the role of IFN-γ polymorphism in relation to intraperitoneal adhesion development.
Conclusion
Postoperative adhesions form because of abnormal ECM protein deposition in response to tissue hypoxia. It remains problematic for surgeons, patients, and hospitals due to increased morbidity and utilization of health care resources from treating postoperative adhesion complications. Epidemiological data exist to support a genetic predisposition to the development of postoperative adhesions and murine studies have shed light on pathways involved in the adhesion-forming cascade. However, the interplay between the numerous growth factors and cytokines they interact with in the adhesion cascade remains a challenge for researchers to pinpoint targets for therapeutic advances. In addition, much of the research involving cell cultures renders difficulties in applying these data to in vivo events. These notwithstanding, our knowledge has been illuminated by the various studies enumerated in this manuscript and can help in formulating therapies going forward.
Although there are few known SNPs and mutations in relation to intraperitoneal adhesion development, the different genetic components that signal adhesion formation could help in identifying patients at risk of adhesion prior to surgery and such individuals targeted for anti-adhesion measures. In the same way, blocking changes in the expression or function of genes necessary for the transformation of NP fibroblasts to AT fibroblasts may curtail adhesion formation. Another novel approach to preventing postoperative adhesions could focus on recombinant therapy against the profibrotic transcription factor and cytokines involved in the adhesion development process.
Future studies should concentrate on the examination of SNPs in genes within the adhesion cascade that induce apoptosis at the site of injury or that have been associated with decreased propensity for other profibrotic or fibrotic diseases. Researchers should ensure that the identified SNPs are accurate and reproducible before introducing them into the clinical arena to help identify candidates that may be at risk of adhesion development. Scientists can also use such information to develop a pharmacologic intervention for those most at risk for developing postoperative adhesions.
References
Awonuga AO, Saed GM, Diamond MP. Laparoscopy in gynecologic surgery: adhesion development, prevention, and use of adjunctive therapies. Clin Obstet Gynecol. 2009;52(3):412–22.
Holmdahl L, Risberg B. Adhesions: prevention and complications in general surgery. Eur J Surg. 1997;163(3):169–74.
Vrijland WW, Jeekel J, van Geldorp HJ, Swank DJ, Bonjer HJ. Abdominal adhesions: intestinal obstruction, pain, and infertility. Surg Endosc. 2003;17(7):1017–22.
Duffy DM, di Zerega GS. Adhesion controversies: pelvic pain as a cause of adhesions, crystalloids in preventing them. J Reprod Med. 1996;41(1):19–26.
Chegini N. TGF-beta system: the principal profibrotic mediator of peritoneal adhesion formation. Semin Reprod Med. 2008;26(4):298–312.
Brown JJ, Bayat A. Genetic susceptibility to raised dermal scarring. Br J Dermatol. 2009;161(1):8–18.
Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med. 2011;17(1-2):113–25.
Corradi D, Maestri R, Palmisano A, Bosio S, Greco P, Manenti L, et al. Idiopathic retroperitoneal fibrosis: clinicopathologic features and differential diagnosis. Kidney Int. 2007;72(6):742–53.
Yang Z, Kyriakides TR, Bornstein P. Matricellular proteins as modulators of cell-matrix interactions: adhesive defect in thrombospondin 2-null fibroblasts is a consequence of increased levels of matrix metalloproteinase-2. Mol Biol Cell. 2000;11(10):3353–64.
Kosaka H, Yoshimoto T, Yoshimoto T, Fujimoto J, Nakanishi K. Interferon-gamma is a therapeutic target molecule for prevention of postoperative adhesion formation. Nat Med. 2008;14(4):437–41.
Agah A, Kyriakides TR, Bornstein P. Proteolysis of cell-surface tissue transglutaminase by matrix metalloproteinase-2 contributes to the adhesive defect and matrix abnormalities in thrombospondin-2-null fibroblasts and mice. Am J Pathol. 2005;167(1):81–8.
Awonuga AO, Belotte J, Abuanzeh S, Fletcher NM, Diamond MP, Saed GM. Advances in the pathogenesis of adhesion development: the role of oxidative stress. Reprod Sci. 2014;21(7):823–36.
Fletcher NM, Awonuga AO, Abusamaan MS, Saed MG, Diamond MP, Saed GM. Adhesion phenotype manifests an altered metabolic profile favoring glycolysis. Fertil Steril. 2016;105(6):1628–1637.e1621.
Shavell VI, Saed GM, Diamond MP. Review: cellular metabolism: contribution to postoperative adhesion development. Reprod Sci. 2009;16(7):627–34.
Cookson VJ, Chapman NR. NF-kappaB function in the human myometrium during pregnancy and parturition. Histol Histopathol. 2010;25(7):945–56.
Fletcher NM, Jiang ZL, Diamond MP, Abu-Soud HM, Saed GM. Hypoxia-generated superoxide induces the development of the adhesion phenotype. Free Radic Biol Med. 2008;45(4):530–6.
Saed GM, Zhang W, Diamond MP. Molecular characterization of fibroblasts isolated from human peritoneum and adhesions. Fertil Steril. 2001;75(4):763–8.
Saed GM, Zhang W, Chegini N, Holmdahl L, Diamond MP. Alteration of type I and III collagen expression in human peritoneal mesothelial cells in response to hypoxia and transforming growth factor-beta1. Wound Repair Regen. 1999;7(6):504–10.
Saed GM, Abu-Soud HM, Diamond MP. Role of nitric oxide in apoptosis of human peritoneal and adhesion fibroblasts after hypoxia. Fertil Steril. 2004;82(Suppl 3):1198–205.
Hecker M, Preiss C, Klemm P, Busse R. Inhibition by antioxidants of nitric oxide synthase expression in murine macrophages: role of nuclear factor kappa B and interferon regulatory factor 1. Br J Pharmacol. 1996;118(8):2178–84.
Saed GM, Diamond MP. Apoptosis and proliferation of human peritoneal fibroblasts in response to hypoxia. Fertil Steril. 2002;78(1):137–43.
Saed GM, Diamond MP. Hypoxia-induced irreversible up-regulation of type I collagen and transforming growth factor-beta1 in human peritoneal fibroblasts. Fertil Steril. 2002;78(1):144–7.
Chegini N. The role of growth factors in peritoneal healing: transforming growth factor beta (TGF-beta). Eur J Surg Suppl. 1997;577:17–23.
Idell S, Zwieb C, Boggaram J, Holiday D, Johnson AR, Raghu G. Mechanisms of fibrin formation and lysis by human lung fibroblasts: influence of TGF-beta and TNF-alpha. Am J Phys. 1992;263(4 Pt 1):L487–94.
diZerega GS. Biochemical events in peritoneal tissue repair. Eur J Surg Suppl. 1997;(577):10–6.
Holmdahl L, Ivarsson ML. The role of cytokines, coagulation, and fibrinolysis in peritoneal tissue repair. Eur J Surg. 1999;165(11):1012–9.
Ivarsson ML, Holmdahl L, Falk P, Molne J, Risberg B. Characterization and fibrinolytic properties of mesothelial cells isolated from peritoneal lavage. Scand J Clin Lab Invest. 1998;58(3):195–203.
Diamond MP, El-Hammady E, Munkarah A, Bieber EJ, Saed G. Modulation of the expression of vascular endothelial growth factor in human fibroblasts. Fertil Steril. 2005;83(2):405–9.
Saed GM, Diamond MP. Differential expression of alpha smooth muscle cell actin in human fibroblasts isolated from intraperitoneal adhesions and normal peritoneal tissues. Fertil Steril. 2004;82(Suppl 3):1188–92.
Saed GM, Munkarah AR, Abu-Soud HM, Diamond MP. Hypoxia upregulates cyclooxygenase-2 and prostaglandin E(2) levels in human peritoneal fibroblasts. Fertil Steril. 2005;83(Suppl 1):1216–9.
Saed GM, Diamond MP. Modulation of the expression of tissue plasminogen activator and its inhibitor by hypoxia in human peritoneal and adhesion fibroblasts. Fertil Steril. 2003;79(1):164–8.
Saed GM, Diamond MP. Molecular characterization of postoperative adhesions: the adhesion phenotype. J Am Assoc Gynecol Laparosc. 2004;11(3):307–14.
Jiang ZL, Fletcher NM, Diamond MP, Abu-Soud HM, Saed GM. S-nitrosylation of caspase-3 is the mechanism by which adhesion fibroblasts manifest lower apoptosis. Wound Repair Regen. 2009;17(2):224–9.
Detti L, Saed GM, Jiang ZL, Kruger ML, Diamond MP. The effect of estradiol on the expression of estrogen, progesterone, androgen, and prolactin receptors in human peritoneal fibroblasts. J Assist Reprod Genet. 2008;25(6):245–50.
Alpay Z, Ozgonenel MS, Savasan S, Buck S, Saed GM, Diamond MP. Possible role of natural immune response against altered fibroblasts in the development of post-operative adhesions. Am J Reprod Immunol. 2006;55(6):420–7.
Rout UK, Saed GM, Diamond MP. Expression pattern and regulation of genes differ between fibroblasts of adhesion and normal human peritoneum. Reprod Biol Endocrinol. 2005;3:1.
Dunn RC, Buttram VC Jr. Tissue-type plasminogen activator as an adjuvant for post surgical adhesions. Prog Clin Biol Res. 1990;358:113–8.
Fortin CN, Saed GM, Diamond MP. Predisposing factors to post-operative adhesion development. Hum Reprod Update. 2015;21(4):536–51.
Cheong YC, Laird SM, Li TC, Shelton JB, Ledger WL, Cooke ID. Peritoneal healing and adhesion formation/reformation. Hum Reprod Update. 2001;7(6):556–66.
Roy SG, Nozaki Y, Phan SH. Regulation of alpha-smooth muscle actin gene expression in myofibroblast differentiation from rat lung fibroblasts. Int J Biochem Cell Biol. 2001;33(7):723–34.
Ulisse S, Baldini E, Sorrenti S, D’Armiento M. The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr Cancer Drug Targets. 2009;9(1):32–71.
Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21(7):1104–17.
Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342(24):1792–801.
Diamond MP. Incidence of postsurgical adhesions. In: diZerega GS, editor. Peritoneal Surg. New York: Springer. 2000. https://doi.org/10.1007/978-1-4612-1194-5_17.
Diamond MP, El-Hammady E, Wang R, Saed G. Regulation of matrix metalloproteinase-1 and tissue inhibitor of matrix metalloproteinase-1 by dichloroacetic acid in human fibroblasts from normal peritoneum and adhesions. Fertil Steril. 2004;81(1):185–90.
Diamond MP, El-Hammady E, Wang R, Saed G. Regulation of transforming growth factor-beta, type III collagen, and fibronectin by dichloroacetic acid in human fibroblasts from normal peritoneum and adhesions. Fertil Steril. 2003;79(5):1161–7.
Reed KL, Heydrick SJ, Aarons CB, Prushik S, Gower AC, Stucchi AF, et al. A neurokinin-1 receptor antagonist that reduces intra-abdominal adhesion formation decreases oxidative stress in the peritoneum. Am J Physiol Gastrointest Liver Physiol. 2007;293(3):G544–51.
Heydrick SJ, Reed KL, Cohen PA, Aarons CB, Gower AC, Becker JM, et al. Intraperitoneal administration of methylene blue attenuates oxidative stress, increases peritoneal fibrinolysis, and inhibits intraabdominal adhesion formation. J Surg Res. 2007;143(2):311–9.
Jiang W, Maturu P, Liang YW, Wang L, Lingappan K, Couroucli X. Hyperoxia-mediated transcriptional activation of cytochrome P4501A1 (CYP1A1) and decreased susceptibility to oxygen-mediated lung injury in newborn mice. Biochem Biophys Res Commun. 2018;495(1):408–13.
Ambler DR, Fletcher NM, Diamond MP, Saed GM. Effects of hypoxia on the expression of inflammatory markers IL-6 and TNF-a in human normal peritoneal and adhesion fibroblasts. Syst Biol Reprod Med. 2012;58(6):324–9.
Cassidy MR, Sheldon HK, Gainsbury ML, Gillespie E, Kosaka H, Heydrick S, et al. The neurokinin 1 receptor regulates peritoneal fibrinolytic activity and postoperative adhesion formation. J Surg Res. 2014;191(1):12–8.
Guo SW, Ding D, Geng JG, Wang L, Liu X. P-selectin as a potential therapeutic target for endometriosis. Fertil Steril. 2015;103(4):990–1000.e1008.
Haertel E, Joshi N, Hiebert P, Kopf M, Werner S. Regulatory T cells are required for normal and activin-promoted wound repair in mice. Eur J Immunol. 2018;48(6):1001–13.
Semenza GL, Rue EA, Iyer NV, Pang MG, Kearns WG. Assignment of the hypoxia-inducible factor 1alpha gene to a region of conserved synteny on mouse chromosome 12 and human chromosome 14q. Genomics. 1996;34(3):437–9.
Sharma V, Nandan A, Sharma AK, Singh H, Bharadwaj M, Sinha DN, et al. Signature of genetic associations in oral cancer. Tumour Biol. 2017;39(10):1010428317725923.
Wei G, Chen X, Wang G, et al. Inhibition of cyclooxygenase-2 prevents intra-abdominal adhesions by decreasing activity of peritoneal fibroblasts. Drug Des Devel Ther. 2015;9:3083–98.
Saed GM, Al-Hendy A, Salama SA, Diamond MP. Adenovirus-mediated expression of cyclooxygenase-2 antisense reverse abnormal genetic profile of human adhesion fibroblasts. Fertil Steril. 2008;89(5 Suppl):1455–60.
Krause TJ, Katz D, Wheeler CJ, Ebner S, McKinnon RD. Increased levels of surgical adhesions in TGFbeta1 heterozygous mice. J Invest Surg. 1999;12(1):31–8.
Diamond MP, El-Hammady E, Wang R, Saed G. Metabolic regulation of collagen I in fibroblasts isolated from normal peritoneum and adhesions by dichloroacetic acid. Am J Obstet Gynecol. 2002;187(6):1456–60 discussion 1460-1451.
Wei MH, Popescu NC, Lerman MI, Merrill MJ, Zimonjic DB. Localization of the human vascular endothelial growth factor gene, VEGF, at chromosome 6p12. Hum Genet. 1996;97(6):794–7.
Chau CH, Clavijo CA, Deng HT, Zhang Q, Kim KJ, Qiu Y, et al. Etk/Bmx mediates expression of stress-induced adaptive genes VEGF, PAI-1, and iNOS via multiple signaling cascades in different cell systems. Am J Physiol Cell Physiol. 2005;289(2):C444–54.
Giugliano G, Pasquali D, Notaro A, Brongo S, Nicoletti G, D’Andrea F, et al. Verapamil inhibits interleukin-6 and vascular endothelial growth factor production in primary cultures of keloid fibroblasts. Br J Plast Surg. 2003;56(8):804–9.
Saltzman AK, Olson TA, Mohanraj D, Carson LF, Ramakrishnan S. Prevention of postoperative adhesions by an antibody to vascular permeability factor/vascular endothelial growth factor in a murine model. Am J Obstet Gynecol. 1996;174(5):1502–6.
Molinas CR, Campo R, Dewerchin M, Eriksson U, Carmeliet P, Koninckx PR. Role of vascular endothelial growth factor and placental growth factor in basal adhesion formation and in carbon dioxide pneumoperitoneum-enhanced adhesion formation after laparoscopic surgery in transgenic mice. Fertil Steril. 2003;80(Suppl 2):803–11.
Bosma PJ, van den Berg EA, Kooistra T, Siemieniak DR, Slightom JL. Human plasminogen activator inhibitor-1 gene. Promoter and structural gene nucleotide sequences. J Biol Chem. 1988;263(19):9129–41.
Ramon LA, Gilabert-Estelles J, Cosin R, et al. Plasminogen activator inhibitor-1 (PAI-1) 4G/5G polymorphism and endometriosis. Influence of PAI-1 polymorphism on PAI-1 antigen and mRNA expression. Thromb Res. 2008;122(6):854–60.
Iacoviello L, Burzotta F, Di Castelnuovo A, Zito F, Marchioli R, Donati MB. The 4G/5G polymorphism of PAI-1 promoter gene and the risk of myocardial infarction: a meta-analysis. Thromb Haemost. 1998;80(6):1029–30.
Canbaz MA, Ustun C, Kocak I, Yanik FF. The comparison of gonadotropin-releasing hormone agonist therapy and intraperitoneal Ringer’s lactate solution in prevention of postoperative adhesion formation in rat models. Eur J Obstet Gynecol Reprod Biol. 1999;82(2):219–22.
Hulboy DL, Rudolph LA, Matrisian LM. Matrix metalloproteinases as mediators of reproductive function. Mol Hum Reprod. 1997;3(1):27–45.
Zhao LG, Chenglei, Meng Y. Meta-analysis of the association between endometriosis and polymorphisms in ACE and PAI-1. Int J Clin Exp Med. 2016;9(6):10602–14.
Honjo K, Munakata S, Tashiro Y, Salama Y, Shimazu H, Eiamboonsert S, et al. Plasminogen activator inhibitor-1 regulates macrophage-dependent postoperative adhesion by enhancing EGF-HER1 signaling in mice. FASEB J. 2017;31(6):2625–37.
Erdogan E, Celayir S, Eroglu E, Yilmaz E. The relation between human leukocyte antigen (HLA) distribution and intestinal obstruction and adhesions in childhood: preliminary report. Pediatr Surg Int. 2000;16(5–6):374–6.
Alpay Z, Ozgonenel M, Savasan S, Buck S, Saed GM, Diamond MP. Altered in vitro immune response to hypoxia-treated normal peritoneal fibroblasts. Fertil Steril. 2007;87(2):426–9.
Okusawa S, Gelfand JA, Ikejima T, Connolly RJ, Dinarello CA. Interleukin 1 induces a shock-like state in rabbits. Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J Clin Invest. 1988;81(4):1162–72.
Wieser F, Tempfer C, Schneeberger C, van Trotsenburg M, Huber J, Wenzl R. Interleukin-1 receptor antagonist polymorphism in women with peritoneal adhesions. BJOG. 2002;109(11):1298–300.
Gajbhiye R, McKinnon B, Mortlock S, Mueller M, Montgomery G. Genetic variation at chromosome 2q13 and its potential influence on endometriosis susceptibility through effects on the IL-1 family. Reprod Sci. 2018;25(9):1307–17.
Stocks MM, Crispens MA, Ding T, Mokshagundam S, Bruner-Tran KL, Osteen KG. Therapeutically targeting the Inflammasome product in a chimeric model of endometriosis-related surgical adhesions. Reprod Sci. 2017;24(8):1121–8.
Zhang H, Song Y, Li Z, et al. Evaluation of ligustrazine on the prevention of experimentally induced abdominal adhesions in rats. Int J Surg. 2015;21:115–21.
Chung DR, Chitnis T, Panzo RJ, Kasper DL, Sayegh MH, Tzianabos AO. CD4+ T cells regulate surgical and postinfectious adhesion formation. J Exp Med. 2002;195(11):1471–8.
Valle-Rios R, Patino-Lopez G, Medina-Contreras O, et al. Characterization of CRTAM gene promoter: AP-1 transcription factor control its expression in human T CD8 lymphocytes. Mol Immunol. 2009;46(16):3379–87.
Wang G, Wu K, Li W, Zhao E, Shi L, Wang J, et al. Role of IL-17 and TGF-beta in peritoneal adhesion formation after surgical trauma. Wound Repair Regen. 2014;22(5):631–9.
Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397(6721):710–3.
Saed GM, Diamond MP. Effects of interferon-gamma reverse hypoxia-stimulated extracellular matrix expression in human peritoneal and adhesion fibroblasts. Fertil Steril. 2006;85(Suppl 1):1300–5.
Yamanaka O, Saika S, Okada Y, Ooshima A, Ohnishi Y. Effects of interferon-gamma on human subconjunctival fibroblasts in the presence of TGFbeta1: reversal of TGFbeta-stimulated collagen production. Graefes Arch Clin Exp Ophthalmol. 2003;241(2):116–24.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Thakur, M., Rambhatla, A., Qadri, F. et al. Is There a Genetic Predisposition to Postoperative Adhesion Development?. Reprod. Sci. 28, 2076–2086 (2021). https://doi.org/10.1007/s43032-020-00356-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-020-00356-7