
Abstract
Epigenetic research focuses on heritable changes beyond the DNA sequence, which has led to a revolution in biological studies and benefits in many other fields. The well-known model ciliate, Tetrahymena thermophila offers a unique system for epigenetic studies due to its nuclear dimorphism and special mode of sexual reproduction (conjugation), as well as abundant genomic resources and genetic tools. In this paper, we summarize recent progress made by our research team and collaborators in understanding epigenetic mechanisms using Tetrahymena. This includes: (1) providing the first genome-wide base pair-resolution map of DNA N6-methyladenine (6mA) and revealed it as an integral part of the chromatin landscape; (2) dissecting the relative contribution of cis- and trans- elements to nucleosome distribution by exploring the unique nuclear dimorphism of Tetrahymena; (3) demonstrating the epigenetic controls of RNAi-dependent Polycomb repression pathways on transposable elements, and (4) identifying a new histone monomethyltransferase, TXR1 (Tetrahymena Trithorax 1), that facilitates replication elongation through its substrate histone H3 lysine 27 monomethylation (H3K27me1).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Eukaryotic genomes have multiple layers. Beyond genetic basis, DNA methylation, noncoding RNA and histone modifications/variants serve as a platform of epigenetic signals which regulate the gene expression leading to profound impact on cell development (Badeaux and Shi 2013; Bird 1992; Jeffares et al. 1998; Poole et al. 1998; Reisenauer et al. 1999; Zacharias 1993).
Tetrahymena thermophila is a eukaryotic unicellular model organism (see review in Orias et al. 2011). Having long been a genetic model system of choice, abundant genomic resources and tools have expanded Tetrahymena’s utility for epigenetic studies. Tetrahymena contains one diploid germline micronucleus (MIC) and one polyploid somatic macronucleus (MAC) within the same cell compartment (Fig. 1a) (Karrer 2012). MAC is transcriptionally active while MIC is inert during vegetative growth. During the sexual reproduction stage- (conjugation), the parental MAC degrades and a new MAC develops from the zygotic nucleus. This nuclear dimorphism features the distinct epigenetic landscapes established on the isogenic background, which makes Tetrahymena an ideal model for epigenetic studies. For example, histone variants and chromatin modifications were identified and were linked to the different transcription states by comparing the transcriptionally silent MIC with the active MAC during vegetative reproduction (Chalker et al. 2013). Among these, the histone variant H2A.Z was found to be associated with active gene transcription activity in the Tetrahymena MAC genome (Allis et al. 1980; Chalker et al. 2013; Hayashi et al. 1984; Henikoff and Smith 2015).

Generalized conceptual model diagrams. a–e are adapted from figures of (Chen et al. 2016; Wang et al. 2017a; Xiong et al. 2016; Gao et al. 2013; Zhao et al. 2019), respectively. a Photomicrographs of Tetrahymena. Note the oral apparatus (white left arrow) and the MIC (white right arrows). b Distribution of DNA 6mA methylation (6mA) is determined by both DNA sequence and the chromatin environment. c Coordinate contribution of cis- and trans-elements on nucleosome distribution. d Histone monomethytransferase TXR1 (Tetrahymena Trithorax-Related 1) regulates DNA replication elongation. e RNAi-dependent Polycomb repression controls transposable elements in Tetrahymena
Our group is dedicated to understanding epigenetic regulation mechanisms using Tetrahymena thermophila as a model system (Fig. 1b–e). A summary of our recent progress is presented here.
The genomic distribution and determinants of DNA N6-methyladenine (6mA) in Tetrahymena
N6-methyladenine (6mA) is reported to be involved in many biological processes in prokaryotes including the restriction modification system (Harrison and Karrer 1985). By contrast, 6mA in eukaryotes was largely neglected due to its low abundance in most genomes until recent studies that reported the existence of 6mA in multiple eukaryotes including Chlamydomonas reinhardtii (Fu et al. 2015), Caenorhabditis elegans (Greer et al. 2015), Drosophila melanogaster (Zhang et al. 2015), mouse embryonic stem cells (Schiffers et al. 2017; Wu et al. 2016), and humans (Xiao et al. 2018; Xie et al. 2018). Tetrahymena thermophila was among the first eukaryotes reported to contain 6mA (Gorovsky et al. 1973). 6mA is the only detectable DNA methylation in Tetrahymena and its 6mA level (0.6%–0.8%) is at least one order of magnitude higher than that in most of higher eukaryotes, thus rendering it an ideal system to study 6mA (Gorovsky et al. 1973; Wang et al. 2017a, b). Moreover, 6mA is present in the transcriptionally active MAC but absent in the transcriptionally silent MIC (Fig. 2a) (Gorovsky et al. 1973; Wang et al. 2017a), providing a near-perfect internal parallel control to more rigorously interrogate the impact of 6mA. However, lack of genome-wide distribution information drastically hinders further investigation of the functions and regulation mechanisms of 6mA.

Genomic distribution of 6mA in Tetrahymena. Adapted from figures of (Wang et al. 2017a). a 6mA is present in the transcriptionally active MAC but absent in the transcriptionally silent MIC). Note the absence of 6mA signal in the MIC (white arrowheads). b Genome-wide distribution of 6mA and its motif, as well as nucleosomes and H2A.Z signals on ten longest scaffolds. c SMRT sequencing identified 5′–AT–3′ as the most representative motif. d Distribution profiles of nucleosome (blue), H2A.Z (green) and 6mA (pink) around the transcription start site (TSS). e Linker DNA with 6mA is enriched with H2A.Z. f 6mA preferentially distributed on RNA polymerase II transcribed genes. g Relative distance of 6mA to nucleosome dyad. h Correlation matrix of 6mA and gene expression level
We generated the first genome-wide, base pair-resolution map of 6mA in Tetrahymena, by employing single-molecule real-time (SMRT) sequencing (Fig. 2b) (Wang et al. 2017a) 6mA preferentially located in the consensus sequence of 5′-AT-3′ (Fig. 2c), indicating that DNA sequence per se provides necessary information to locate 6mA. However, in a genome as AT rich as Tetrahymena, only a small percentage of adenines (0.66%) are methylated, suggesting that other factors beyond sequence also contribute to 6mA distribution. Further analysis revealed that 6mA accumulated in linker DNA regions between well-positioned nucleosomes (Fig. 2d, g), but the direction of the causal effect remains to be determined (Luo et al. 2018). Moreover, nucleosomes adjacent to 6mA were enriched with the conserved histone variant H2A.Z (Fig. 2e) which is dynamically regulated by ATP-dependent chromatin remodelers (Havas et al. 2001; Jaskelioff et al. 2000). Given that 6mA accumulated right downstream of the transcription start sites (TSS of RNA polymerase II (Pol II) transcribed genes) (Fig. 2f), we posit that 6mA was recruited to the promoter region by a similar mechanism to H2A.Z. It was previously reported that 6mA is either positively or negatively correlated with transcription (Fu et al. 2015; Liang et al. 2018; Wang et al. 2018; Wu et al. 2016; Zhou et al. 2018), and usually enriched in specific pathways in metazoa such as in mouse brain (Yao et al. 2017) and in human cancer cells (Xiao et al. 2018; Xie et al. 2018). In Tetrahymena, however, the correlation between gene expression level and the level/amount of 6mA was rather weak (Fig. 2h). Given that most Tetrahymena genes (> 90%) are decorated with 6mA, it is not surprising that 6mA in unicellular eukaryotes may have not evolved to regulate specific pathways. Collectively, the evidence suggests that distribution of 6mA in Tetrahymena is determined by both DNA sequence and the chromatin environment, and its special pattern is probably achieved by specific methyltransferase(s) deposition.
To better decipher the function and biological significance of 6mA, it is essential to perturb 6mA levels in vivo by manipulating its catalyzing enzymes. Our recent work identified a candidate 6mA methyltransferase (unpublished data), the deletion of which impaired cell growth and development. Systematic investigations are underway to find out how this methyltransferase regulates 6mA level and how the change in 6mA level affects cell fitness. It will also be interesting to look into other factors involved in the regulation of 6mA such as demethylase(s), reader(s), and chromatin modifications and how 6mA interacts with them to jointly shape the chromatin environment.
Epigenetic regulation of nucleosome distribution
How cis- and trans- determinants coordinate with each other and shape nucleosome distribution has long been debated. For example, an early study focusing on in vitro reconstituted nucleosomes showed that nucleosome distribution might be dependent largely on intrinsic DNA sequence rather than trans-acting factors (Sekinger et al. 2005). However, comprehensive in vivo mapping of nucleosomes in budding yeast, using high-throughput sequencing following micrococcal nuclease (MNase) digestion, showed that nucleosome distribution was affected by its chromatin contexts in which gene regulatory elements function on a genomic scale (Lee et al. 2007).
Tetrahymena offers an ideal system to resolve the relative contributions of cis- and trans- determinants as it benefits from nuclear dimorphism whereby the MAC and MIC share almost identical genetic sequences but possess different chromatin landscapes. A previous study, which reported the genome-wide nucleosome maps in the macronuclear genome of Tetrahymena thermophila, revealed the presence of stereotypical nucleosome pattern near TSSs, which is guided by DNA sequence and trans-acting factors (Beh et al. 2015). To acquire the nucleosome landscape in the MAC and MIC, we first optimized the nuclei isolation, MNase digestion and mono-nucleosome purification procedures in Tetrahymena (Fig. 3a). A detailed analysis of MNase-seq data revealed that the nucleosome positioning (e.g., translational positioning, the exact 147 bp of genomic DNA occupied by a subset of nucleosomes) differ significantly between Tetrahymena MAC and MIC (Xiong et al. 2016). Well-positioned and precisely phased nucleosomes are abundant in the transcriptionally active MAC, but are diminished in the transcriptionally inert MIC (Fig. 3b–c), suggesting that transcription-associated trans-determinants promote nucleosome positioning. Nonetheless, the nucleosome occupancy (the probability of a DNA sequence being associated with any nucleosome in a population) is similar between MAC and MIC, as well as with in vitro reconstitution and predictions based upon DNA sequence features (Fig. 3d), revealing strong contributions from cis-determinants (DNA sequence features). It should be noted that well-positioned nucleosomes in the MAC are often matched with GC content oscillations while stereotypical nucleosome arrays in the MIC, although diminished, are still detectable (Fig. 3e), suggesting the coordinate contribution of cis- and trans-determinants.

Nucleosome distribution in Tetrahymena. a is adapted from a figure of (Chen et al. 2016); b–e are adapted from figures of (Xiong et al. 2016); f is adapted from a figure of (Wang et al. 2017a). a Hierarchical organization of chromatin. b, c Strongly positioned nucleosomes are prevalent in the MAC, but not in the MIC. d Composite analysis of nucleosome positioning aligned to TSS. e In phase oscillation of nucleosome occupancy and GC content downstream of TSS over genes constrained +1 nucleosomes. f Nucleosomes exhibit a more consistent phase relative to TSS in genes marked with 6mA
Recently, N6-methyladenine (6mA) DNA methylation, as one of the potential cis-determinants, has been reported to play a role in nucleosome positioning (Fu et al. 2015). The result from our work showed that, in Tetrahymena, linker DNA regions with 6mA are usually flanked by well-positioned nucleosomes (Fig. 2e, 3f) (Wang et al. 2017a). By in vitro nucleosome assembly in Tetrahymena, a recent study revealed that the distribution pattern of nucleosomes could be recapitulated in native genomic DNA but not in DNA without 6mA (Luo et al. 2018). A study of another ciliate, Oxytricha trifallax, demonstrated that nucleosome fuzziness increased after loss of 6mA in the linker DNA (Beh et al. 2019). Overall, our studies and those of others have revealed the intrinsic repulsion between 6mA and nucleosomes which contributes to determining the nucleosome position. However, the causal functions of related factors and the precise mechanisms of how they coordinate with each other remain to be elucidated.
RNAi-dependent Polycomb repression controls transposable elements in Tetrahymena
Transposable elements (TEs) are DNA sequences which can induce mutations and affect the organism’s genome structure by changing their positions within a genome (Bourque et al. 2018). They are drivers of host genome evolution as well as threats to host genome integrity (Bennetzen and Wang 2014; Freeling et al. 2015; Lynch 2007). Therefore, different hosts have developed a wide variety of defense mechanisms to control TE expression, including small RNA, sequence-specific repressors, DNA, and chromatin modification pathways (Berrens et al. 2017; Ecco et al. 2017; Goodier 2016; Liu et al. 2017; Molaro and Malik 2016). In flies and mammals, Piwi-interacting RNAs (piRNAs), which are produced from piRNA clusters genomic loci, mediate TE silencing (Guzzardo et al. 2013; Siomi et al. 2011). In nematodes, Argonaute–small RNA complexes are used as transgenerational binary signals and program the ON/OFF expression state for TEs (Seth et al. 2013). Plants and yeasts identify and repress TEs by recognizing their intrinsic features (Lee et al. 2013; Slotkin et al. 2009). Yet little is known about how TEs are controlled in the single cell organism Tetrahymena thermophila.
Under unfavorable environmental conditions, Tetrahymena cells of two different mating types pair with each other and initiate conjugation. The zygotic MIC differentiates into the new MAC, accompanied by a large quantity of programmed genome rearrangement events (Chalker et al. 2013; Noto et al. 2015; Xu et al. 2019). Thousands of MIC-specific internally eliminated sequences (IES), where most TEs and their remnants are nested, are removed, leaving behind MAC-destined sequences (MDS) (Fig. 4a) (Coyne et al. 2012; Fillingham et al. 2004; Wuitschick et al. 2002). Previous studies have revealed an RNAi and Polycomb group (PcG) proteins-related pathway (Fig. 4b) (Noto and Mochizuki 2017). First, this is initiated by the RNA polymerase II (Pol II)-catalyzed bi-directional transcription of long noncoding RNA (ncRNA) in the meiotic MIC (Chalker and Yao 2001; Mochizuki and Gorovsky 2004). Second, Tetrahymena scnRNAs (~ 26–32 nt siRNAs), produced from ncRNA in the MIC by the Dicer protein Dcl1p (Malone et al. 2005; Mochizuki and Gorovsky 2005), are bound to the Argonaute protein Twi1p (Noto et al. 2010; Woehrer et al. 2015). Third, H3K27 and H3K9 methylation are subsequently deposited by the RNAi machinery and EZL1 (Polycomb group protein, an E(z) homologue in Tetrahymena) complex (Liu et al. 2004, 2007). Lastly, these conserved histone modifications are recognized by PDD1 (chromodomain-containing effector, analogous to HP1), which plays an important role in forming heterochromatic structures containing the IES sequences (Coyne et al. 1999; Madireddi et al. 1996; Schwope and Chalker 2014). It was previously reported that many Tetrahymena IES sequences are from TEs (Fillingham et al. 2004), but the mechanism of how TEs are propagated and controlled in Tetrahymena remains elusive.

RNAi-dependent Polycomb repression controls transposable elements in Tetrahymena. Adapted from figures of (Zhao et al. 2019). a A schematic for DNA elimination in Tetrahymena. b A schematic for DNA elimination pathway. c Distribution of polyadenylated transcripts in IES. d Radar chart of codon usage pattern. e Mobilization of the Tc1 element in wild-type (WT) and mutants. f IES-specific polyadenylated transcripts are only produced during the developing MAC formation. g Localization of the key components involved in transcriptional and co-transcriptional machineries. Note the parental MAC (green arrowheads), the developing MAC (green arrows), the new MIC (white arrowheads) and the degrading parental MAC (white arrows). hPolycomb repression as an ancient alternative for RNAi-dependent TE silencing
In our recent work, we and our collaborators analyzed the polyadenylated RNA transcripts from ~ 10,000 IESs in knockout strains of three key players (DCL1, EZL1 and PDD1) in the RNAi-dependent Polycomb repression pathway (Zhao et al. 2019). We found a significantly higher level of polyadenylated RNA in the mutants than that in the wild type (Fig. 4c). These IES-specific polyadenylated transcripts (many containing TE-related sequences) have mRNA features, such as poly-A tailing, strand specificity, splice sites and a similar codon usage pattern with that of MDS (Fig. 4d), demonstrating the capacity of protein coding, and probably TE coding. As evidence, we detected a dramatic increase in germline mobilization of a recently active TE in the mutants, this mobilization being achieved via a “cut-and-paste” manner, i.e., TE is excised from the genomic locus, leaving an AT footprint in the original locus (Fig. 4e). Both mRNA and ncRNA (scnRNA) were detected in the same IES locus, but mRNA was only produced in the developing MAC where key components in Pol II-driven mRNA biogenesis are present. More intriguingly, mRNA levels of TE-containing IESs are positively correlated with late-scnRNA, while are negatively correlated with early-scnRNA (Fig. 4f, g) (Noto et al. 2015). Together, these results indicate that the balance between ncRNA and mRNA production was probably mediated by RNAi-dependent Polycomb repression and co-transcriptional processing, which is essential for TE control. Considering the conservation of key components in this RNAi-dependent Polycomb repression pathway and the wide distribution of similar pathways throughout eukaryotes (Fig. 4h), we posit that interplay between RNAi and Polycomb repression may be a universal way for TE silencing and transcriptional repression of developmental genes.
Previous studies have revealed the recent transposition of TEs in Tetrahymena based on TE insertion polymorphisms in some IES (Huvos 2004) and purifying selection in predicted coding sequences of some potentially active TEs (Fillingham et al. 2004; Gershan and Karrer 2000; Hamilton et al. 2006, 2016). Our results demonstrate that Polycomb repression defects result in not only the activation of TE transcription but also the germline mobilization of TE. Future studies will focus on the mechanism underlying the molecular nature of TEs, such as where the mobilized TEs reassemble into the genome and which factors determine their insertion sites.
Histone methylation H3K27me1 is involved in the regulation of DNA replication
Residues of histones, especially those in N-terminal tails, can be post-translationally modified, which has been proved to play important roles in many chromatin-templated processes such as DNA replication, RNA transcription, DNA repair, chromatin condensation, and segregation (Allis and Jenuwein 2016; Jenuwein and Allis 2001; Lantermann et al. 2010; Lee et al. 2007; Strahl and Allis 2000). Histone 3 lysine 27 (H3K27) methylation is one of the most well-studied histone post-translational modifications (PTMs) (Cyrus and Yi 2005; Gao et al. 2013; Jacob et al. 2009; Jamieson et al. 2017; Klose and Yi 2007; Liu et al. 2007; Ru and Yi 2004; Zhang et al. 2018). In Drosophila, enhancer of zeste (E(z)) is the histone methyltransferase (HMT) for H3K27 methylation (Cao et al. 2002; Czermin et al. 2002; Jürg et al. 2002). In Arabidopsis, however, besides three homologs to E(z), MEDEA (Langmead and Salzberg 2012), CURLY LEAF (CLF), and SWINGER (SWN), two SET domain-containing proteins, i.e., ARABIDOPSIS TRITHORAX-RELATED PROTEIN5 (ATXR5) and ATXR6, which are phylogenetically divergent from E(z), possess the ability to specifically add one methyl group to H3K27 (Baumbusch et al. 2001; Jacob et al. 2009). The hypomorphic mutant, atxr5/atxr6 showed a moderate yet incomplete reduction of H3K27me1 level (Raynaud et al. 2006), and, more dramatically, re-replication in heterochromatin regions enriched with transposons and repetitive elements, indicating that this clade of HMTs is involved in replication regulation (Jacob et al. 2009, 2010; Yannick et al. 2010).
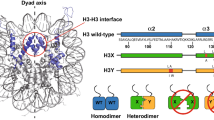
We and our collaborators identified the sole homolog to Arabidopsis ATXR5 and ATXR6 in Tetrahymena, i.e., Tetrahymena Trithorax-Related 1 (TXR1) (Fig. 5a) (Gao et al. 2013). TXR1 is present in the transcriptionally active MAC but is absent in the silent MIC and can specifically deposit one methyl group to H3K27 (Fig. 5b) (Zhang et al. 2013). Knockout of TXR1 resulted in severe replication stress, manifested by slower growth with a prolonged S-phase (Fig. 5c), excessive accumulation of single-stranded DNA (ssDNA) near replication origins (Fig. 5d, e), production of aberrant replication intermediates (RI) such as single-strand breaks (SSBs) and double-strand breaks (DSBs) (Fig. 5f), and the consequent activation of robust DNA damage response (DDR) including key players involved in ssDNA binding/sensing and DNA repair (Fig. 5g) (Gao et al. 2013). It should be noted that all the above-mentioned phenotypes occurred only in ΔTXR1 cells, but not in ΔEZL2 cells, in which one of the Tetrahymena E(z) homologues responsible for H3K27me2 and H3K27me3 was deleted, emphasizing the specific role of TXR1-dependent H3K27me1. Furthermore, a point mutation at histone H3 lysine 27 could partially phenocopy TXR1 knockout, corroborating H3K27me1 as a key player in DNA replication. A more recent study discovered that K27Q mutation in the replacement variant H3.3 further aggravated the replication stress of K27Q mutation in the canonical H3, supporting the assertion that both H3 and H3.3 are physiologically relevant substrates of TXR1 (Zhao et al. 2016). This is, however, distinct from the strong canonical H3 preference in Arabidopsis homologues ATXR5 and ATXR6 (Jacob et al. 2014). Additionally, TXR1 could functionally interact with the replication key player PCNA (proliferating cell nuclear antigen) via its PIP (PCNA-interacting protein) motif, which is potentially important for its recruitment to active replication forks (Raynaud et al. 2006). These findings support the presence of a conserved pathway through which TXR1 and H3K27me1 facilitate replication elongation at newly initiated origins.

DNA replication is regulated by the histone monomethyltransferase TXR1 (Tetrahymena Trithorax-Related 1). Adapted from figures of (Gao et al. 2013). a Domain structure comparison of Tetrahymena TXR1 with Arabidopsis ATXR5 and ATXR6. b TXR1 is required for H3K27me1 and EZL2 for H3K27me2 and H3K27me3. c ΔTXR1 cells have significantly increased doubling time (t2). d Accumulation of single-strand DNA (ssDNA) in ΔTXR1 cells. Note the absence of ssDNA signal in the MIC (white arrowheads). e ssDNA is specifically accumulated in intergenic regions, including replication origins. f Higher level of single-strand breaks (SSB) and double-strand breaks (DSB) in ΔTXR1 cells. g The activation of DNA damage response (DDR) in ΔTXR1 cells
Our work revealed a new scenario that deletion of a histone methyltransferase could severely impair the replication elongation instead of replication license, but the regulatory mechanism of lysine monomethylation in DNA methylation has yet to be elucidated. Focusing on the modulating mechanism of TXR1, we recently discovered that, in addition to its important role in replication, TXR1 may also be involved in transcription regulation (unpublished data). This is consistent with the finding that H3K27me1 in Arabidopsis exhibits two distinct distribution patterns in constitutive heterochromatin and genic regions, suggesting its role in transcription regulation (Roudier et al. 2011). These bring us to one of the most basic questions in biology: with the same DNA template, how can cells solve the conflicts of replication and transcription? (García-Muse and Aguilera 2016; Hamperl et al. 2017; Lin and Pasero 2017). It will be interesting to dissect roles played by TXR1 in replication and transcription, and how it contributes to coordinating conflicts between these two essential processes.
Summary and perspectives
Epigenetic studies have kept yielding cutting-edge knowledge in different eukaryotic models including Tetrahymena. Benefiting from the unique biological properties such as nuclear dimorphism, Tetrahymena has become a powerful model for epigenetic studies. In this paper, we review recent progress made by our group using Tetrahymena to elucidate the distribution and mechanism of DNA 6mA methylation, nucleosome occupancy driven by both cis- and trans-determinants, transposable elements regulated by RNAi-dependent Polycomb repression and H3K27me1-related regulation of DNA replication.
To improve the understanding of epigenetics using Tetrahymena, future studies will focus on: (1) the regulation mechanisms of 6mA together with its co-factors; (2) the relationship of 6mA and nucleosomes in the chromatin environment; (3) the molecular nature of TEs; and (4) roles of TXR1 in replication and transcription.
References
Allis CD, Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat Rev Genet 17:487–500
Allis CD, Glover CV, Bowen JK, Gorovsky MA (1980) Histone variants specific to the transcriptionally active, amitotically dividing macronucleus of the unicellular eucaryote, Tetrahymena thermophila. Cell 20:609–617
Badeaux AI, Shi Y (2013) Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Bio 14:211–224
Baumbusch LO, Thorstensen T, Krauss V, Fischer A, Naumann K, Assalkhou R, Schulz I, Reuter G, Aalen RB (2001) The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res 29:4319–4333
Beh LY, Müller MM, Muir TW, Kaplan N, Landweber LF (2015) DNA-guided establishment of nucleosome patterns within coding regions of a eukaryotic genome. Genome Res 25:1727–1738
Beh LY, Debelouchina GT, Clay DM, Thompson RE, Lindblad KA, Hutton ER, Bracht JR, Sebra RP, Muir TW, Landweber LF (2019) Identification of a DNA N6-adenine methyltransferase complex and its impact on chromatin organization. Cell 177:1781–1796
Bennetzen JL, Wang H (2014) The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu Rev Plant Biol 65:505–530
Berrens RV, Andrews S, Spensberger D, Santos F, Dean W, Gould P, Sharif J, Olova N, Chandra T, Koseki H, von Meyenn F, Reik W (2017) An endosiRNA-based repression mechanism counteracts transposon activation during global DNA demethylation in embryonic stem cells. Cell Stem Cell 21:694–703
Bird A (1992) The essentials of DNA methylation. Cell 70:5–8
Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, Imbeault M, Izsvák Z, Levin HL, Macfarlan TS, Mager DL, Feschotte C (2018) Ten things you should know about transposable elements. Genome Biol 19:199
Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y (2002) Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298:1039–1043
Chalker DL, Yao MC (2001) Nongenic, bidirectional transcription precedes and may promote developmental DNA deletion in Tetrahymena thermophila. Genes Dev 15:1287–1298
Chalker DL, Meyer E, Mochizuki K (2013) Epigenetics of ciliates. Cold Spring Harb Perspect Biol 5:a017764
Chen X, Gao S, Liu YF, Wang YY, Wang YR, Song WB (2016) Enzymatic and chemical mapping of nucleosome distribution in purified micro- and macronuclei of the ciliated model organism, Tetrahymena thermophila. Sci China Life Sci 59:909–919
Coyne RS, Nikiforov MA, Smothers JF, Allis CD, Yao MC (1999) Parental expression of the chromodomain protein Pdd1p is required for completion of programmed DNA elimination and nuclear differentiation. Mol Cell 4:865–872
Coyne RS, Stover NA, Miao W (2012) Whole genome studies of Tetrahymena. Methods Cell Biol 109:53–81
Cyrus M, Yi Z (2005) The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6:838–849
Czermin B, Melfi R, Mccabe D, Seitz V, Imhof A, Pirrotta V (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111:185–196
Ecco G, Imbeault M, Trono D (2017) KRAB zinc finger proteins. Development 144:2719–2729
Fillingham JS, Thing TA, Vythilingum N, Keuroghlian A, Bruno D, Golding GB, Pearlman RE (2004) A non-long terminal repeat retrotransposon family is restricted to the germ line micronucleus of the ciliated protozoan Tetrahymena thermophila. Eukaryot Cell 3:157–169
Freeling M, Xu J, Woodhouse M, Lisch D (2015) A solution to the C-value paradox and the function of junk DNA: the genome balance hypothesis. Mol Plant 8:899–910
Fu Y, Luo GZ, Chen K, Deng X, Yu M, Han D, Hao Z, Liu J, Lu X, Doré LC (2015) N 6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 161:879–892
Gao S, Xiong J, Zhang CC, Berquist BR, Yang RD, Zhao M, Molascon AJ, Kwiatkowski SY, Yuan DX, Qin ZH, Wen JF, Kapler GM, Andrews PC, Miao W, Fan LY (2013) Impaired replication elongation in Tetrahymena mutants deficient in histone H3 Lys 27 monomethylation. Genes Dev 27:1662–1679
García-Muse T, Aguilera A (2016) Transcription–replication conflicts: how they occur and how they are resolved. Nat Rev Mol Cell Bio 17:553–563
Gershan JA, Karrer KM (2000) A family of developmentally excised DNA elements in Tetrahymena is under selective pressure to maintain an open reading frame encoding an integrase-like protein. Nucleic Acids Res 28:4105–4112
Goodier JL (2016) Restricting retrotransposons: a review. Mobile DNA 7:16
Gorovsky MA, Hattman S, Pleger GL (1973) [6N] methyl adenine in the nuclear DNA of a eucaryote, Tetrahymena pyriformis. J Cell Biol 56:697–701
Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizábal-Corrales D, Hsu CH, Aravind L, He C, Shi Y (2015) DNA methylation on N 6-adenine in C. elegans. Cell 161:868–878
Guzzardo PM, Muerdter F, Hannon GJ (2013) The piRNA pathway in flies: highlights and future directions. Curr Opin Genet Dev 23:44–52
Hamilton EP, Williamson S, Dunn S, Merriam V, Lin C, Vong L, Russell-Colantonio J, Orias E (2006) The highly conserved family of Tetrahymena thermophila chromosome breakage elements contains an invariant 10-base-pair core. Eukaryot Cell 5:771–780
Hamilton EP, Kapusta A, Huvos PE, Bidwell SL, Zafar N, Tang H, Hadjithomas M, Krishnakumar V, Badger JH, Caler EV, Russ C, Zeng Q, Fan L, Levin JZ, Shea T, Young SK, Hegarty R, Daza R, Gujja S, Wortman JR et al (2016) Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. eLife 5:e19090
Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA (2017) Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170:774–786
Harrison GS, Karrer KM (1985) DNA synthesis, methylation and degradation during conjugation in Tetrahymena thermophila. Nucleic Acids Res 13:73–87
Havas K, Whitehouse I, Owen-Hughes T (2001) ATP-dependent chromatin remodeling activities. Cell Mol Life Sci 58:673–682
Hayashi T, Hayashi H, Fusauchi Y, Iwai K (1984) Tetrahymena histone H3. Purification and two variant sequences. J Biochem 95:1741–1749
Henikoff S, Smith MM (2015) Histone variants and epigenetics. Cold Spring Harb Perspect Biol 7:a019364
Huvos P (2004) A member of a repeat family is the source of an insertion–deletion polymorphism inside a developmentally eliminated sequence of Tetrahymena thermophila. J Mol Biol 336:1061–1073
Jacob Y, Feng S, Leblanc C, Bernatavichute Y, Stroud H, Cokus S, Johnson L, Pellegrini M, Jacobsen S, Michaels S (2009) ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat Struct Mol Biol 16:763–768
Jacob Y, Stroud H, LeBlanc C, Feng S, Zhuo L, Caro E, Hassel C, Gutierrez C, Michaels SD, Jacobsen SE (2010) Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature 466:987–991
Jacob Y, Bergamin E, Donoghue MT, Mongeon V, LeBlanc C, Voigt P, Underwood CJ, Brunzelle JS, Michaels SD, Reinberg D (2014) Selective methylation of histone H3 variant H3.1 regulates heterochromatin replication. Science 343:1249–1253
Jamieson K, Mcnaught KJ, Ormsby T, Leggett NA, Honda S, Selker EU (2017) Telomere repeats induce domains of H3K27 methylation in Neurospora. eLife 7:e31216
Jaskelioff M, Gavin IM, Peterson CL, Logie C (2000) SWI-SNF-mediated nucleosome remodeling: role of histone octamer mobility in the persistence of the remodeled state. Mol Cell Biol 20:3058–3068
Jeffares DC, Poole AM, Penny D (1998) Relics from the RNA world. J Mol Evol 46:18–36
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293:1074–1080
Jürg M, Hart CM, Francis NJ, Vargas ML, Aditya S, Brigitte W, Miller EL, O’Connor MB, Kingston RE, Simon JA (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111:197–208
Karrer KM (2012) Nuclear dualism. Methods Cell Biol 109:29–52
Klose RJ, Yi Z (2007) Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Bio 8:307–318
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Lantermann AB, Straub T, Strålfors A, Yuan GC, Ekwall K, Korber P (2010) Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat Struct Mol Biol 17:251–257
Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, Dhakshnamoorthy J, Taneja N, Yamanaka S, Zhou M, Grewal SI (2013) Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155:1061–1074
Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C (2007) A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 39:1235–1244
Liang Z, Shen L, Cui X, Bao S, Geng Y, Yu G, Liang F, Xie S, Lu T, Gu X (2018) DNA N 6-adenine methylation in Arabidopsis thaliana. Dev Cell 45:406–416
Lin YL, Pasero P (2017) Transcription-replication conflicts: orientation matters. Cell 170:603
Liu YF, Mochizuki K, Gorovsky MA, Blackburn E (2004) Histone H3 lysine 9 methylation is required for DNA elimination in developing macronuclei in Tetrahymena. Proc Natl Acad Sci USA 101:1679–1684
Liu YF, Taverna SD, Muratore TL, Shabanowitz J, Hunt DF, Allis CD (2007) RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev 21:1530–1545
Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, Wysocka J (2017) Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553:228–232
Luo GZ, Hao Z, Luo L, Shen M, Sparvoli D, Zheng Y, Zhang Z, Weng X, Chen K, Cui Q (2018) N 6-methyldeoxyadenosine directs nucleosome positioning in Tetrahymena DNA. Genome Biol 19:200
Lynch M (2007) The origins of genome architecture. Sinauer Associates, Sunderland
Madireddi MT, Coyne RS, Smothers JF, Mickey KM, Yao MC, Allis CD (1996) Pdd1p, a novel chromodomain-containing protein, links heterochromatin assembly and DNA elimination in Tetrahymena. Cell 87:75–84
Malone CD, Anderson AM, Motl JA, Rexer CH, Chalker DL (2005) Germ line transcripts are processed by a dicer-like protein that is essential for developmentally programmed genome rearrangements of Tetrahymena thermophila. Mol Cell Biol 25:9151–9164
Mochizuki K, Gorovsky MA (2004) RNA polymerase II localizes in Tetrahymena thermophila meiotic micronuclei when micronuclear transcription associated with genome rearrangement occurs. Eukaryot Cell 3:1233–1240
Mochizuki K, Gorovsky MA (2005) A Dicer-like protein in Tetrahymena has distinct functions in genome rearrangement, chromosome segregation, and meiotic prophase. Genes Dev 19:77–89
Molaro A, Malik HS (2016) Hide and seek: how chromatin-based pathways silence retroelements in the mammalian germline. Curr Opin Genet Dev 37:51–58
Noto T, Mochizuki K (2017) Whats, hows and whys of programmed DNA elimination in Tetrahymena. Open Biol 7:170172
Noto T, Kurth HM, Kataoka K, Aronica L, DeSouza LV, Siu KWM, Pearlman RE, Gorovsky MA, Mochizuki K (2010) The Tetrahymena argonaute-binding protein Giw1p directs a mature argonaute–siRNA complex to the nucleus. Cell 140:692–703
Noto T, Kataoka K, Suhren JH, Hayashi A, Woolcock KJ, Gorovsky MA, Mochizuki K (2015) Small-RNA-mediated genome-wide trans-recognition network in Tetrahymena DNA elimination. Mol Cell 59:229–242
Orias E, Cervantes MD, Hamilton EP (2011) Tetrahymena thermophila, a unicellular eukaryote with separate germline and somatic genomes. Res Microbiol 162:578–586
Poole AM, Jeffares DC, Penny D (1998) The path from the RNA world. J Mol Evol 46:1–17
Raynaud C, Sozzani R, Glab N, Domenichini S, Perennes C, Cella R, Kondorosi E, Bergounioux C (2006) Two cell-cycle regulated SET-domain proteins interact with proliferating cell nuclear antigen (PCNA) in Arabidopsis. Plant J 47:395–407
Reisenauer A, Kahng LS, McCollum S, Shapiro L (1999) Bacterial DNA methylation: a cell cycle regulator? J Bacteriol 181:5135–5139
Roudier F, Ahmed I, Bérard C, Sarazin A, Mary-Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois-Berthet E, Al-Shikhley L, Giraut L, Després B, Drevensek S, Barneche F, Dèrozier S, Brunaud V, Aubourg S, Schnittger A, Bowler C, Martin-Magniette M et al (2011) Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J 30:1928–1938
Ru C, Yi Z (2004) The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev 14:155–164
Schiffers S, Ebert C, Rahimoff R, Kosmatchev O, Steinbacher J, Bohne AV, Spada F, Michalakis S, Nickelsen J, Müller M, Carell T (2017) Quantitative LC–MS provides no evidence for m6dA or m4dC in the genome of mouse embryonic stem cells and tissues. Angew Chem Int Ed 56:11268–11271
Schwope RM, Chalker DL (2014) Mutations in Pdd1 reveal distinct requirements for its chromodomain and chromoshadow domain in directing histone methylation and heterochromatin elimination. Eukaryot Cell 13:190–201
Sekinger EA, Moqtaderi Z, Struhl K (2005) Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast. Mol Cell 18:735–748
Seth M, Shirayama M, Gu W, Ishidate T, Conte D, Mello CC (2013) The C. elegans CSR-1 argonaute pathway counteracts epigenetic silencing to promote germline gene expression. Dev Cell 27:656–663
Siomi MC, Sato K, Pezic D, Aravin AA (2011) PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Bio 12:246–258
Slotkin RK, Vaughn M, Borges F, Tanurdžić M, Becker JD, Feijó JA, Martienssen RA (2009) Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136:461–472
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41–45
Wang YY, Chen X, Sheng YL, Liu YF, Gao S (2017a) N6-adenine DNA methylation is associated with the linker DNA of H2A. Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena. Nucleic Acids Res 45:11594–11606
Wang YY, Sheng YL, Liu YQ, Pan B, Huang J, Warren A, Gao S (2017b) N6-methyladenine DNA modification in the unicellular eukaryotic organism Tetrahymena thermophila. Eur J Protistol 58:94–102
Wang X, Li Z, Zhang Q, Li B, Lu C, Li W, Cheng T, Xia Q, Zhao P (2018) DNA methylation on N 6-adenine in lepidopteran Bombyx mori. Biochim Biophys Acta- Gene Regul Mech 1861:815–825
Woehrer SL, Aronica L, Suhren JH, Busch CJL, Noto T, Mochizuki K (2015) A Tetrahymena Hsp90 co-chaperone promotes siRNA loading by ATP-dependent and ATP-independent mechanisms. EMBO J 34:559–577
Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, Liu Y, Byrum SD, Mackintosh SG, Zhong M (2016) DNA methylation on N 6-adenine in mammalian embryonic stem cells. Nature 532:329–333
Wuitschick JD, Gershan JA, Lochowicz AJ, Li S, Karrer KM (2002) A novel family of mobile genetic elements is limited to the germline genome in Tetrahymena thermophila. Nucleic Acids Res 30:2524–2537
Xiao CL, Zhu S, He M, Chen D, Zhang Q, Chen Y, Yu G, Liu J, Xie SQ, Luo F, Liang Z, Wang DP, Bo XC, Gu XF, Wang K, Yan GR (2018) N6-methyladenine DNA modification in the human genome. Mol Cell 71:306–318
Xie Q, Wu TP, Gimple RC, Li Z, Prager BC, Wu Q, Yu Y, Wang P, Wang Y, Gorkin DU, Zhang C, Dowiak AV, Lin K, Zeng C, Sui Y, Kim LJY, Miller TE, Jiang L, Lee CH, Huang Z et al (2018) N6-methyladenine DNA modification in glioblastoma. Cell 175:1228–1243
Xiong J, Gao S, Dui W, Yang W, Chen X, Taverna SD, Pearlman RE, Ashlock W, Miao W, Liu Y (2016) Dissecting relative contributions of cis-and trans-determinants to nucleosome distribution by comparing Tetrahymena macronuclear and micronuclear chromatin. Nucleic Acids Res 44:10091–10105
Xu J, Li X, Song W, Wang W, Gao S (2019) Cyclin Cyc2p is required for micronuclear bouquet formation in Tetrahymena thermophila. Sci China Life Sci 62:668–680
Yannick J, Hume S, Chantal L, Suhua F, Luting Z, Elena C, Christiane H, Crisanto G, Michaels SD, Jacobsen SE (2010) Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature 466:987–991
Yao B, Cheng Y, Wang Z, Li Y, Chen L, Huang L, Zhang W, Chen D, Wu H, Tang B, Jin P (2017) DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat Commun 8:1122
Zacharias W (1993) Methylation of cytosine influences the DNA structure. EXS 64:27–38
Zhang CC, Molascon AJ, Gao S, Liu YF, Andrews PC (2013) Quantitative proteomics reveals that the specific methyltransferases Txr1p and Ezl2p differentially affect the mono-, di- and trimethylation states of histone H3 lysine 27 (H3K27). Mol Cell Proteomics 12:1678–1688
Zhang GQ, Huang H, Liu D, Cheng Y, Liu XL, Zhang WX, Yin RC, Zhang DP, Zhang P, Liu JZ, Li CY, Liu BD, Luo YW, Zhu YX, Zhang N, He SM, He C, Wang HL, Chen DH (2015) N 6-methyladenine DNA modification in Drosophila. Cell 161:893–906
Zhang Y, Chang JF, Sun J, Chen L, Yang XM, Tang HY, Jing YY, Kang X, He ZM, Wu JY, Wei HM, Wang DL, Xu RG, Zhu RB, Shen Y, Zeng SY, Wang C, Liu KN, Zhang Y, Mao ZY et al (2018) Histone H3K27 methylation is required for NHEJ and genome stability by modulating the dynamics of FANCD2 on chromatin. J Cell Sci 131:jcs215525
Zhao XL, Wang YY, Wang YR, Liu YF, Gao S (2016) Histone methyltransferase TXR1 is required for both H3 and H3.3 lysine27 methylation in the well-known ciliated protist Tetrahymena thermophila. Sci China Life Sci 60:264–270
Zhao XL, Xiong J, Mao FB, Sheng YL, Chen X, Feng LF, Dui W, Yang WT, Kapusta A, Feschotte C, Coyne RS, Miao W, Gao S, Liu YF (2019) RNAi-dependent Polycomb repression controls transposable elements in Tetrahymena. Genes Dev 33:348–364
Zhou C, Wang CS, Liu HB, Zhou QW, Liu Q, Guo Y, Peng T, Song JM, Zhang JW, Chen LL, Zhao Y, Zeng ZX, Zhou DX (2018) Identification and analysis of adenine N 6-methylation sites in the rice genome. Nat Plants 4:554–563
Acknowledgements
The authors would like to thank the following people for assistance with this study: Ms. Yalan Sheng, Mr. Bo Pan, Ms. Yuan Li, Ms. Lili Duan for their help in draft preparation. Our special thanks are due to our collaborators, in particular Dr. Yifan Liu (University of Michigan) and Dr. Wei Miao (Institute of Hydrobiology of Chinese Academy of Sciences).
Funding
This work was supported by Natural Science Foundation of Shandong Province (JQ201706), The Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (2018SDKJ0406-2), Fundamental Research Funds for the Central Universities (201841005), and the Blue Life Breakthrough Program of LMBB of Qingdao National Laboratory for Marine Science and Technology (MS2018NO04).
Author information
Authors and Affiliations
Contributions
All authors wrote the paper and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Animal and human rights statement
This article does not contain human participants or animals.
Additional information
Edited by Jiamei Li.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cheng, T., Wang, Y., Huang, J. et al. Our recent progress in epigenetic research using the model ciliate, Tetrahymena thermophila. Mar Life Sci Technol 1, 4–14 (2019). https://doi.org/10.1007/s42995-019-00015-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42995-019-00015-0