
Abstract
In recent years, Bovine herpesvirus 4 (BoHV-4) has emerged as an attractive gene delivery viral vector, mainly for vaccination purposes in the veterinary field. In the present study, a new infectious clone of the BoHV-4 genome carrying a bacterial artificial chromosome vector (BoHV-4-BAC) was developed by homologous recombination in mammalian cell culture and bacterial systems, and exploited to express a truncated form of glycoprotein D (tgD) of Bovine herpesvirus 1 (BoHV-1) (BoHV-4-tgD∆TK) as a vaccine candidate. This construct’s immunogenicity was compared to a DNA vector expressing the same antigen (pC-tgD) in a BALB/c mouse model. After the mice were immunized, total and specific antibody responses, cytokine responses, total splenocyte cells proliferation/cytotoxicity, and virus neutralization assays were conducted to analyze the immune response elicited by both constructs. Mice from both vaccine groups developed significant humoral and cellular immune responses after a booster dose regime was conducted on day 28 post-injection. In almost all immunological assays, BoHV-4-tgDΔTK induced as high an immune response as pC-tgD. In both vaccine constructs, neutralizing antibodies were a significant determining factor in protection against BoHV-1, even after the first injection. We conclude that a BoHV-4-based viral vector offers an effective immunization strategy as an alternative to DNA-based immunization platforms, at least to combat BoHV-1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Viral vectors can be used to infect different target cells and deliver genes of interest into those cells. Experimental trials have used two main groups of viral vectors: integrated and non-integrated. Because each group has both advantages and disadvantages, researchers continue to explore new viral vectors for gene therapy. These newly developed viral vectors must be analyzed to determine their safety, identity, potency, purity, and quality before conducting further experiments, especially human trials [1, 2].
Herpesviruses are large, enveloped, spherical to pleomorphic, 150–200 nm in diameter, with T = 16 icosahedral symmetry, and with the potential to infect both animals and humans. Their capsid of 162 capsomers is surrounded by an amorphous tegument. The use of this family in the gene therapy field has an interesting background. The majority of studies have focused on herpes simplex 1 as an effective gene transfer platform for vaccine development and cancer targeting. However, other members of this large family deserve attention as potential gene expressing platforms [3].
The use of herpesviruses in gene therapy has an impressive background. The majority of studies have focused on herpes simplex 1 as an effective gene transfer platform for vaccine development and cancer targeting. However, other members of this large family deserve attention as potential gene expressing vehicles [3].
Bovine herpesvirus 4 (BoHV-4) is a member of the Herpesviridae family, gammaherpesvirinae subfamily, and rhadinovirus genus. Its genome is 144 kb ± 6 in length with a long unique central region (L-DNA) of 110 kb (with low CpG content) flanked by two poly-repetitive DNA (prDNA) regions of 1450–2850 bp [4]. Based on restriction enzyme mapping analyses, almost all American strains belong to the DN-599 group whereas European strains are grouped into the Movar 33/63 group, isolated in Hungary [5].
BoHV-4 has been isolated from cattle with endometritis, chronic metritis, vulvovaginitis, abortions, mastitis, and respiratory infections, as well as apparently healthy individuals [6,7,8,9].
BoHV-4 is a potential viral vector for numerous reasons, including its simple genomic structure, easy genome manipulation, lack of significant pathogenicity in humans and animals except rabbits (laboratory animal model), no potential of transformation in infected cells, easy propagation in cell culture systems, persistence in macrophages and monocytes (long-life expression of the gene of interest), and lack of vector-neutralizing antibodies in either humans or natural viral hosts [10,11,12,13,14].
The preferred method to manipulate herpesvirus genomes is to clone them into artificial bacterial chromosome (BAC) (F′ plasmid) vectors by homologous recombination (HR) [15]. For viral recombinant vaccine development, BACs combine the advantages of DNA vaccines and modified live viruses since the recombinant virus can be reconstituted in vivo after administration of infectious DNAs [16, 17]. The obtaining of herpesvirus genomes carrying a BAC vector has provided an efficient tool for studying viral molecular biology, both in vitro and in vivo [18,19,20]. In addition to other herpesviruses, recombinant BoHV-4-BACs, which express diverse immune-dominant antigens from different pathogens, can successfully immunize different animal models with satisfactory results [21,22,23].
Bovine herpesvirus 1 (BoHV-1) is a member of Herpesviridae family and Alphaherpesvirinae subfamily. Unlike BoHV-4, BoHV-1, which is one of the most important cattle pathogens, is distributed worldwide, except for a few European countries that have eradicated it. It causes significant economic losses to the cattle industry in endemic areas [24]. In Turkey, too, its prevalence and etiological role in specific clinical symptoms like respiratory tract infection, mastitis, and abortion have been previously reported in closed dairy herds [25].
Although licensed vaccines against BoHV-1 are based on inactivated and/or live attenuated glycoprotein E deleted (∆gE) marker constructs that are commercially available in endemic areas, their disadvantages make this virus an interesting candidate for new vaccine design studies [26, 27]. Previous researchers have demonstrated that glycoproteins B (gB), gC, and gD, being the most immune-dominant antigens, are effective targets for vaccines against BoHV-1. Numerous vaccines targeting gD or its truncated form (tgD) have promoted immune responses against BoHV-1 with significant outcomes [28, 29].
In the present study, we first constructed a BoHV-4-BAC viral vector using Movar 33/63 (European) strain by homologous recombination to deliver and express the truncated glycoprotein D of BoHV-1. Next, we analyzed its immunogenicity in the BALB/c mouse model in a homologous prime-boost regime. To evaluate the immunogenicity of this new construct, we developed a DNA vaccine and compared this with the data obtained from both constructs.
Material and method.
Cells and viruses
We used Bovine herpesvirus 4 (BoHV-4) Movar 33/63 (European) strain (Access Number: AB035516) as the template to create the BoHV-4-BAC viral vector. As the immuno-dominant antigen, truncated glycoprotein D (tgD) was amplified from Bovine herpesvirus 1 (BoHV-1) Cooper strain (Access Number: JX898220). All cell lines and both viruses were obtained from the cell culture and virus stocks of the Virology Department of the Veterinary Faculty, Ankara University, Turkey. The wild type and recombinant viruses used to perform homologous recombination were inoculated in Madin Darby bovine kidney (MDBK; (NBL-1) (ATCC® CCL-22™)) and bovine embryonic kidney (BEK; BS CL-94) cells, respectively [30]. For transfection assay and elimination of loxp-BAC-EGFP-Neo-loxp, we used, respectively, human embryonic kidney (HEK293FT; Thermo Fisher Scientific: Catalog number: R70007) and MDBK-cre cells [31].
All cell lines were maintained in Dulbecco’s modified essential medium (DMEM; Sigma, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS; Biological Industries, Kibbutz Beit-Haemek, Israel), 2 mM of L-glutamine (Sigma, St. Louis, MO, USA), 100 IU/ml penicillin (Biological Industries, Kibbutz Beit-Haemek, Israel), 100 µg/ml streptomycin (Biological Industries, Kibbutz Beit-Haemek, Israel), and 2.5 µg/ml Amphotericin B (Biological Industries, Kibbutz Beit-Haemek, Israel). For the immunological assays, splenocyte cells were collected from all immunized mice and red blood cells were lyzed by RBC lysis buffer (Biological Industries, Kibbutz Beit-Haemek, Israel). After 2 wash steps with 1 × phosphate-buffered saline (1 × PBS), the cell pellets were resuspended in RPMI-1640 medium (Sigma, St. Louis, MO, USA) and 250 × 103 cells were added to each well of 24- or 96-well plates.
BoHV-4-BAC (iBAC) viral vector development
The viral DNA was extracted from BoHV-4 Movar 33/63 infected MDBK cells, as previously described (Sambrook, 2001). All primer sets used in this study are shown in Table 1.
The gene area between ORF2 and ORF3 (Access Number: NC002665) of BoHV-4 was selected to insert the BAC. In the first step, these two genes were amplified by PCR using Phusion High Fidelity DNA polymerase enzyme (Thermo Fisher Scientific, Waltham, MA, USA), as previously described [32]. After digestion with the appropriate restriction enzymes listed in Table 1, they were inserted into the pSP72 vector (Promega, Madison, WI, USA) to create the pSP-TM23 vector. This plasmid was verified by restriction enzyme digestion and sequencing. In the next step, the CMV-EGFP-Neo gene was cut from the pEGFP-C1 vector (Clontech, CA, USA) using VspI and ApaLI (Fastdigest enzymes, Thermo Fisher Scientific, Waltham, MA, USA). It was then blunted using T4 DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA). In parallel, the pBeloBAC11 vector (BAC; NEB, MA, USA) was digested using BamHI enzyme (Thermo Fisher Scientific, Waltham, MA, USA) and blunted. The blunt pBeloBAC11 and CMV-EGFP-Neo genes were ligated to create the pBAC-G vector. This plasmid was then verified by restriction enzyme digestion and sequencing. Two loxp sequences were added to pBAC-G by PCR amplification of the plasmid using the primer set listed in Table 1. Finally, the pSP-TM-BG plasmid was created by inserting the loxp-pBAC-CMV-EGFP-Neo-loxp vector between ORF2 and ORF3 of the pSP-TM23 vector using blunt-end ligation. Enhanced green fluorescence protein (EGFP) expression of the final construct was verified in HEK293FT cells after transfection of the cells using lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions in a 24-well plate 72 h post-transfection.
To create recombinant BoHV-4 viruses containing the BAC vector (BoHV-4-BAC), we used homologous recombination (HR) in mammalian cells [30].
Briefly, 24 h after virus (Movar 33/63 strain) inoculation at 1 moi in MDBK cells, the infected cells were collected. The circular pSP-TM-BG vector and salmon sperm DNA at 10:1 ratio were transfected into the cells using electroporation in a 2-mm cuvette by CelljecT Duo (Thermo Fisher Scientific, Waltham, MA, USA) under the established conditions of 270 V and 1500µF capacitor of the infinite shunt resistor. The electroporated cells were then returned to the flask and fed for about 12 days to develop complete cytopathic effects (CPEs). The recombinant viruses were separated from the pool of non-recombinant and recombinant viruses by three rounds of virus passages in the presence of geneticin antibiotic (G418; Thermo Fisher Scientific, Waltham, MA, USA) at a final concentration of 700 µg/ml in the BEK cells, followed by 8 rounds of plaque purification in MDBK cells. The collected recombinant viruses were titrated by plaque assay and stored at − 80 °C. Circular intermediate recombinant herpesviral DNAs were extracted from the MDBK-infected cells using the classic Hirt extraction method [33] and transformed into electro-competent SW102 bacteria using a MicroPulser electroporator (BioRad, Hercules, CA, USA) in a 1-mm cuvette, according to the manufacturer’s protocol. The constructs were verified by sequencing. Finally, the MDBK cells were transfected with the BoHV-4-BAC DNAs, which were extracted from SW-102 bacteria to reconstruct the infectious recombinant viruses (infectious BAC; iBAC).
pC-tgD construction
Truncated glycoprotein D (tgD) of BoHV-1 [34] was amplified from the DNAs of BoHV-1 Cooper strain using the primer set listed in Table 1.
The PCR reaction steps were as follows: initial denaturation at 98 °C for 30 s, 30 cycles of denaturation at 98 °C for 10 s, annealing at 52 °C for 15 s, extension at 72 °C for 45 s, final extension at 72 °C for 10 min. The pC-tgD construct was created by insertion of this PCR product into the pCDNA3.1 myc/His A vector (Invitrogen, Carlsbad, CA, USA) using EcoRI and XhoI enzymes (Thermo Fisher Scientific, Waltham, MA, USA). The final construct was verified by sequencing.
Recombineering in SW102 bacteria to create BoHV-4-tgD∆TK
To perform recombineering in SW102 bacteria [30], truncated glycoprotein D of BoHV-1 flanked by thymidine kinase of BoHV-4 (TK1-CMV-tgD-Amp-TK2) was amplified from the pC-tgD construct using the TM1 and TM2 primer sets listed in Table 1. These primers contained 50 bp homologous arms to the thymidine kinase gene (access number: AF318573.1) of the BoHV-4 Movar 33/63 strain. The recombineering protocol is freely available elsewhere (https://redrecombineering.ncifcrf.gov/protocols/) [35]. Briefly, the gel-purified TK1-CMV-tgD-Amp-TK2 was electroporated into heat-induced SW102 bacteria (42 °C for 15 min) containing BoHV-4-BAC followed by overnight incubation at 32 °C for colony growth on the LB agar containing the ampicillin (50 µg/µL) and chloramphenicol (12.5 µg/µL) antibiotics. The constructs were verified by partial sequencing. Finally, the infectious BoHV-4-tgD∆TK viruses were constructed by transfection of extracted BoHV-4-tgD∆TK DNAs into the MDBK cells. The loxp-BAC-CMV-EGFP-loxp cassette was eliminated from the final constructs by virus propagation in MDBK-cre cells [30].
Indirect immunofluorescence assay
Indirect immunofluorescence assay (IIFA) was performed in BHK21-C13 cells to confirm in vitro expression of the tgD protein from the pC-tgD construct, as previously described [36, 37].
Briefly, the cells were cultivated in 24-well plates before the plasmid was transfected into the cells using Lipofectamine3000 (Thermo Fisher Scientific, Waltham, MA, USA). The transfected cells were then fixed using 3.7% formaldehyde 48 h post-DNA delivery and blocked with 5% skimmed milk (Cell Signaling, Leiden, The Netherlands) in 1 × tris buffered saline (TBS) buffer containing 0.2% Tween-20 (1xTBST) for 1 h at room temperature. Next, anti-BoHV-1-gD antibody (clone 1B8-F11; VRMD, Inc., Pullman, WA) at 1:250 dilution was added to each well, which was then incubated for 90 min at room temperature (RT). Finally, the assay was evaluated by examining the cells in an Axio Vert A1 Microscope (Ziess, Oberkochen, Germany) after incubation with 1:750 dilution of FITC-labelled anti-mouse IgG secondary antibody (Sigma, St. Louis, MO, USA).
Western blot assay
Expression of the tgD protein from the BoHV-4-tgD∆TK and pC-tgD constructs was evaluated, respectively, 72 h after inoculation of MDBK cells or transfection of BHK21-C13 cells by Western blot assay in 6-well plates [38]. The total protein of the infected/transfected cells was collected using PRO-PREP™ Protein Extraction Solution (iNtRON Biotechnology, Burlington, MA, USA), according to the manufacturer’s instructions, separated in a 4–12% gradient SDS page, and transferred to polyvinylidene difluoride (PVDF) membrane using the Trans-Blot Turbo Transfer System (BioRad, Hercules, CA, USA, USA). After blocking in the presence of 5% skimmed milk solution, the membrane was subjected to 2 h of incubation with the primary antibody of anti-BoHV-1-gD (clone 1B8-F11; VRMD, Inc., Pullman, WA) at a dilution of 1:500 for 90 min at RT. After incubation in the presence of the secondary antibody of HRP-Anti-Mouse IgG (whole molecule) at a dilution of 1:750 for 1 h at RT, the bands were visualized by adding Clarity Western ECL substrate solution (BioRad, Hercules, CA, USA, USA) using ChemiDoc MP System (BioRad, Hercules, CA, USA, USA). For controls, we used Cooper-infected cells and beta-actin.
Immunization schedule
We used 5 groups of 8–12-week-old female BALB/c mice with 4 healthy animals in each group. For immunization, 50 µg of the DNA constructs (pC-tgD and pCDNA3.1 myc/His A) and 100TCID50 of BoHV-4-tgD∆TK and Movar 33/63 were injected via intramuscular (thigh muscle) and intraperitoneal routes, respectively. The booster dose was administered on day 14 and the experiment was ended on day 28 by euthanasia of the immunized mice (Table 2). The immunized mice were observed daily for the onset of any clinical symptoms, including allergic reactions. Blood samples were collected through the tail vein on days 0, 14, and 28. The collected serum samples were stored at − 80 °C.
Lymphocyte proliferation responses
To determine the total splenocyte cell proliferation effect of the vaccine constructs, the spleens were aseptically collected from the immunized mice on day 28 post-immunization and plated in the 96-well plates, as described previously [39]. BoHV-1 Cooper strain (10 moi) was added to all wells in triplicate, including the splenocyte cells from vaccinated and normal saline immunized mice. After stimulation for 48 h, the total splenocyte cell proliferation level was measured with MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) kit (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. Briefly, 10 µl of 12 mM MTT stock solution was added to each well and incubated for 4 h at 37 °C / 5% CO2. Then, after 100 µl of SDS-HCl solution was added to each well, and after 16 h of incubation at 37 °C, the plate was read at 570 nm with an ELISA reader (Titertek Multiskan PLUS MK II Microplate Reader, Midland, ON, Canada).
Cytotoxicity assay
This assay was performed using the lactate dehydrogenase enzyme release assay (LDH) kit (Thermo Fisher Scientific, Waltham, MA, USA) [40].
Briefly, the total splenocyte cells of the immunized mice, collected on day 28 post-immunization, were used as the effector cells along with MDBK cells infected with BoHV-1 Cooper strain, which acted as the target cells. Four hours after the inoculation of the MDBK cells by BoHV-1 at moi of 1, the splenocyte cells from vaccinated and control mice were added at effector-to-target cell ratios of 1:10, 1:25, and 1:50, followed by 4 h of incubation at 37 °C. Then, 10X lysis buffer was added to Maximum LDH Activity Control triple wells before the cells were reincubated at 37° C for 45 min. Next, 50 µl of the medium from each well was transferred to a new 96-well plate and mixed with an equal amount of reaction buffer before incubation at RT for 30 min protected from light. Finally, the reaction was terminated by adding 50 µl of stop solution and read at 490 nm.
Cytokine assay
The cytokine responses in the supernatant of the virus-stimulated splenocyte cells were measured by stimulating the cells with 1 moi of Cooper strain on days 2 and 3 post-inoculation and in the serum samples (collected on days 0 and 28) of the immunized mice by LEGENDplex™ bead Mouse Th Cytokine Panel (8-plex) kit (BioLegend, San Diego, CA, USA), using BD Bioscience FacsCanto II Flow Cytometer (BD Bioscience, Franklin Lakes, NJ, USA), as previously described [36]. The results were analyzed by the LEGENDplexTM Data Analysis Software. As a control, we added the naïve and virus-stimulated splenocyte cells of the negative (normal saline) group.
Total antibody isotyping
To differentiate between the total antibody isotypes (IgG1, IgG2a, and iGg2b) produced as a consequence of the vaccine construct injection, we used the Mouse ELISA Total Antibody Isotyping kit (Thermo Fisher Scientific, Waltham, MA, USA) to analyze the serum samples collected from each group on days 0 (before immunization) and 28 (2 weeks after the booster dose), as previously described [36].
Briefly, 50 μl of diluted serum (1:500 in DMEM) was mixed with an equal volume of goat anti-mouse IgG + IgA + IgM HRP conjugate and kept at room temperature for 1 h. Finally, 75 µl of the 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate was added, followed by 15 min incubation. The reaction was then stopped with TMB stop solution and read at 450 nm.
Virus neutralization assay
Heat-inactivated serum samples were 1/2 diluted (1/50, 1/100, and 1/200), an equal volume of Cooper strain (10TCID50) was added, and the mixture was incubated at 37ºC for 1 h before plating onto MDBK cells in a 96-well plate. The infected cells were further incubated at 37 °C for 2 days to complete the CPEs in the virus control wells. The results were analyzed by the Reed-Muench method [41].
Statistical analysis
Antibody isotype data and cytokine levels in the different groups were evaluated using a two-way (Sidak’s post hoc correction) ANOVA by SPSS (IBM SPSS Statistics for Windows, Version 22.0, Armonk, NY, USA). One-way (Tukey’s post hoc correction) ANOVA was also used for analyzing the neutralizing antibody data. All data are presented as mean ± SD. Graphs were produced using GraphPad Prism version 7.0 (GraphPad Software, San Diego, CA, USA; www.graphpad.com). Differences were considered statistically significant when p < 0.05. All molecular biology procedures were simulated using the SnapGene Viewer software (www.snapgene.com).
Results
Generation of BoHV-4-BAC
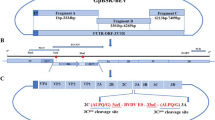
The pSP-TM-BG vector (Fig. 1A) was verified by sequencing and restriction enzyme analysis. The correct pattern was observed (data not shown). Green fluorescence protein (GFP) expression from this construct was verified in HEK293FT cells on day 3 post-DNA delivery (Fig. 1B). The pSP-TM-BG vector was then transfected into MDBK cells infected with BoHV-4 (Movar 33/63 strain) to create the crude stock of BoHV-4-BAC by homologous recombination (Fig. 1D). To guarantee purification of the recombinant viruses, we used two different strategies based on our designed vector, namely GFP expression and geneticin (G418) resistance in the infected MDBK and BEK cells, respectively. After the last purification step, the crude virus stock contained a large proportion of the recombinant BoHV-4-BAC infected BEK cells, as confirmed by GFP expression (Fig. 1B-iv). The stability of the viral DNAs of the BoHV-4-BAC extracted from SW-102 bacteria after seven serial passages was confirmed using EcoRI enzyme. The results were compared to the digested DNAs of the Movar 33/63 strain. As shown in Fig. 1D, DNAs of the recombinant viruses possessed an extra band of 7400 bp in comparison to the Movar 33/63 strain.

Generation and verification of BoHV-4-BAC. A) pSP-TM-BG vector. This plasmid was created by ligation of ORF2-loxp-BAC-CMV-EGFP-Neo-loxp-ORF3 and pSP72 vector. B) i: phase contrast, ii: fluorescence: detection of green fluorescence protein (EGFP) expression in the pSP-TM-BG transfected HEK293FT cells (× 40). iii: phase contrast, iv: fluorescence: pure BoHV-4-BAC virus expressing GFP, obtained after 3 rounds of G418 (700 µg/µl) selection followed by 8 rounds of plaque purification in the infected BEK cells (× 40). C) Overall strategy of recombinant BoHV-4-BAC generation. D) Stability analysis of BoHV-4-BAC extracted from SW102 bacteria using EcoRI enzyme digestion. C1: passage 1 in the bacteria, C2: passage 2, C3: passage 3, C6: passage 6, and C7: passage 7. The different passages were compared to Movar33/63
BoHV-4-tgD∆TK construct
We exploited recombineering in the SW-102 bacteria to create the BoHV-4-tgD∆TK virus as the vaccine construct. This process occurred among TK1-CMV-tgD-Amp-TK2 and BoHV-4-BAC to create BoHV-4-tgD∆TK (Fig. 2A). In this experiment, we obtained 100% positive colonies. All contained the truncated glycoprotein D of BoHV-1 (tgD), as assessed by colony PCR, restriction enzyme analysis, and partial sequencing (data not shown). The DNAs of BoHV-4-tgD∆TK were then extracted from the bacteria and transfected into MDBK cells to reconstitute the infectious recombinant viruses carrying tgD. The obtained recombinant viruses were inoculated onto the MDBK cells to analyze GFP expression and cytopathic effect (CPE) in the infected cells (Fig. 2B-i and ii). Finally, the BAC cassette (loxp-BAC-CMV-EGFP-loxp) was eliminated from the recombinant virus by passaging in MDBK-Cre cells, generating BoHV-4-tgD∆TK as the final vaccine construct (data not shown).

BoHV-4-tgD∆TK Generation. A) Recombineering between TK1-CMV-tgD-Amp-TK2 and BoHV-4-BAC in SW102 bacteria to generate the BoHV-4-tgD∆TK construct. B) i: fluorescence, ii:: phase-contrast images of BoHV-4-tgD∆TK infected MDBK cells on day 5 post-inoculation. Magnification × 100. iii: fluorescence, iv: phase-contrast images of Indirect Immunofluorescence Assay (IIFA) for the tgD expression detection in the pC-tgD-transfected BHK21-C13 cells on the second day post-transfection in IIFA. As a control, we included pCDNA3.1 myc/His A plasmid transfected BHK21-C13 cells in the IIFA experiment (v: fluorescence, vi: phase contrast). Magnification × 100. C) Western blot assay. i) Lane 1: BoHV-4-tgD∆TK-infected MDBK on day 3 post-infection. Lane 2: pC-tgD-transfected BKH-21-C13 on day 3 post-transfection. Lane 3: uninfected MDBK cells on day 3 post-transfection. Lane 4: untransfected BHK-13-C13 on day 3 post-transfection. Truncated gD detected as a 64 kDa protein. ii) Beta-actin protein (43 kDa) as the control, detected by Anti-beta Actin antibody (St John’s Laboratory, USA) at a dilution of 1/10000
Generation of pC-tgD
The DNA vaccine construct was created by classic ligation between the PCR product (tgD) and the vector and verified by sequencing (data not shown).
In vitro expression of tgD from both constructs
Expression of tgD from pC-tgD was verified by indirect immunofluorescence assay (IIFA) in transfected BHK21-C13 cells on day 2 post-DNA delivery (Fig. 2B-iii and iv). As a control, we included pCDNA3.1 myc/HisA transfected cells in the experiment (Fig. 2B (v) and (vi)). We conducted Western blot analyses from BoHV-4-tgD∆TK-infected MDBK cells and pC-tgD-transfected BHK21-C13 cells, respectively. tgD expression was confirmed in both transfected and infected cells by detection of the 64 kDa protein on the membrane (Fig. 2C-i). As a control, we included beta-actin detection in the Western blot experiment (Fig. 2C-ii).
Proliferation responses
Splenocytes from all the mice groups were tested for proliferation responses. The stimulation index increased significantly in both the BoHV-4-tgD∆TK (p < 0.001) and pC-tgD (p < 0.001) groups (Fig. 3A). Although the virus moi increased during the experiment, this index remained unchanged within each group. However, the value for 100 moi of the pC-tgD group was slightly higher than for the other moi levels within this group (p < 0.01) and also in comparison to the BoHV-4-tgD∆TK group.

Proliferation and Cytotoxicity Assays in the Splenocytes of Immunized Mice. A) MTT assay. The stimulation index of BoHV-4-tgD∆TK and pC-tgD was high at different moi. There were no significant differences between the different moi within each group. The data are shown as mean ± SD; p < 0.05 was considered significant (ns: non-significant); *p < 0.05, **p < 0.01, and ***p < 0.001. B) LDH assay. The cytotoxicity percentage in BoHV-4-tgD∆TK is slightly higher than pC-tgD for different effector-to-target-cell ratios. However, the data shows that both constructs can stimulate significant cytotoxicity. The data are shown as mean ± SD; p < 0.05 was considered significant (ns: non-significant); *p < 0.05, **p < 0.01, and ***p < 0.001
Cytotoxicity assay
The cytotoxicity response in immunized mice was examined using the LDH release assay. As shown in Fig. 3B, the cytotoxicity response in the BoHV-4-tgD∆TK group (p < 0.001) was higher than in the pC-tgD group (p < 0.001) for all effector-to-target cell ratios. In each group, this response was elevated by increasing the ratio. The highest response was in the BoHV-4-tgD∆TK group at a ratio of 1:50 (p < 0.05).
Total antibody response
As indicated in Figs. 4A and B, IgG1 (BoHV-4-tgD∆TK; p < 0.05 and pC-tgD; p < 0.01) and IgG2a (BoHV-4-tgD∆TK; p < 0.05) responses were significantly higher than their respective controls in the BoHV-4-tgD∆TK and pC-tgD groups. The IgG2b response in the pC-tgD group (p < 0.05) was slightly higher than in the BoHV-4-tgD∆TK group (p < 0.01) (Fig. 4C). On the other hand, BoHV-4-tgD∆TK was more capable of eliciting IgG3 than pC-tgD (Fig. 4D).

Total and neutralization antibody responses in the immunized mice. A) IgG1. Both vaccine constructs elicited significant IgG1 antibody responses in immunized BALB/c mice. B) IgG2a. Although the pC-tgD and BoHV-4-tgD∆TK IgG2a responses were both high, the BoHV-4-tgD∆TK response was slightly higher than pC-tgD. C) IgG2b. Both the pC-tgD and BoHV-4-tgD∆TK constructs stimulated significant IgG2b responses. However, the pC-tgD group’s response was slightly stronger. D) IgG3. Both constructs induced significant responses. E) Neutralization Antibody response on day 14. To perform the neutralization assay, 1/2 serially diluted serum samples (1/50, 1/100, and 1/200) were mixed with an equal volume of BoHV-1 Cooper strain virus with a known titer, followed by incubation at 37 °C for 1 h and inoculation onto MDBK cells in a 96-well plate. The results were determined after 72 h by comparing them to the negative and positive controls. While both constructs elicited a significant response after both the first and second injection, the neutralization antibody level was slightly higher in BoHV-4-tgD∆TK than pC-tgD. F) Neutralization Antibody response on day 28. At this point also, the level of the neutralization antibodies in both immunized groups is significant but the BoHV-4-tgD∆TK group is slightly higher than pC-tgD. The data are shown as mean ± SD; p < 0.05 was considered significant (ns: non-significant); *p < 0.05, **p < 0.01, and ***p < 0.001
Neutralization assay
In the virus neutralization assay, using the collected serum samples from immunized mice, we showed that BoHV-4-tgD∆TK and pC-tgD triggered significant production of neutralization antibodies at 1/50, 1/100, and 1/200 dilutions, and at two different time points: 14 (Fig. 4E) and 28-day post-immunization (Fig. 4F). However, the titer in BoHV-4-tgD∆TK was slightly higher than for pC-tgD at all dilutions and both days. The neutralization antibody titer 3 weeks after the second immunization increased slightly in comparison to the titer obtained after the first injection on day 14 (Fig. 4E).
Cytokine assay
In comparison to pC-tgD, BoHV-4-tgD∆TK elicited higher cytokine responses in the supernatant of the virus-stimulated splenocyte cells on days 2 and 3 post-inoculation for all tests, namely IFN-gamma, IL-2, IL-4, IL-5, IL-6, IL-10, IL-13, and TNF-alpha. Cytokine release increased in a time-dependent manner, being higher at 72 h post-stimulation than at 48 h. pC-tgD did not stimulate the production of IL-4 and IL-10. As a control, we included virus-stimulated splenocyte cells from normal saline-injected mice to assess the cytokine profile baseline of the virus in vitro (Fig. 5).

Cytokine responses from virus-stimulated splenocyte cells of immunized BALB/c mice on days 2 and 3 post stimulation. A) IFN-gamma. B) IL-2. C) IL-4 response. D) IL-5. E) IL-13. F) IL-6. G) IL-10. H) TNF-alpha. BoHV-4-tgD∆TK responses in all the cytokines were higher than pC-tgD at 48 and 72 h post-stimulation. The data are shown as mean ± SD; p < 0.05 was considered significant (ns: non-significant); *p < 0.05, **p < 0.01, and ***p < 0.001
In the serum samples collected on day 28 (2 weeks after the booster dose), the pC-tgD construct stimulated IFN-gamma (Fig. 6A) and IL-13 (Fig. 6E) responses slightly more than the other vaccine construct whereas BoHV-4-tgD∆TK was dominant in IL-6 (Fig. 6F) and IL-10 (Fig. 6G) cytokine responses. Both vaccine constructs induced significant levels of IL-5 (Fig. 6D), IL-2 (Fig. 6B), IL-4 (Fig. 6C), and TNF-alpha (Fig. 6H) cytokines in the serum samples, with no significant differences between them. BoHV-4-tgD∆TK did not induce an IL-13 response.

Cytokine responses in the serum samples of immunized BALB/c mice were collected on day 0 (before immunization) and 28 (2 weeks after the booster dose). A) IFN-gamma. pC-tgD response was slightly higher than BoHV-4-tgD∆TK. B) IL-2. Both constructs elicited significant cytokine levels. C) IL-4. While both levels are high, the level for BoHV-4-tgD∆TK was slightly higher than pC-tgD. D) IL-5. The result is the same as for IL-4. E) IL-13. pC-tgD elicited a significant response. BoHV-4-tgD∆TK elicited no response. F) IL-6. BoHV-4-tgD∆TK stimulated the highest response. G) IL-10. The result is the same as for IL-4 and IL-5. H) TNF-alpha. BoHV-4-tgD∆TK response was higher than pC-tgD. However, pC-tgD induced a slightly greater response than BoHV-4-tgD∆TK. All data are shown as mean ± SD; p < 0.05 was considered significant (ns: non-significant); *p < 0.05, **p < 0.01, and ***p < 0.001
Discussion
Several studies have demonstrated that Bovine herpesvirus 4 (BoHV-4) is an appropriate gene delivery vector to combat various diseases, such as BoHV-1, Bovine Viral Diarrhea Virus (BVDV), Bluetongue virus (BTV), Nipah, Ebola virus, and Peste des Petits Ruminants Virus (PPRV). This viral vector can also elicit adequate immune responses against the gene of interest in diverse animal models, including mouse, rat, rabbit, sheep, swine, and goat [13, 21,22,23, 30, 41, 42]
In the present study, we created a new viral vector based on the Movar 33/63 (European) strain of BoHV-4 using bacterial artificial chromosome plasmid (BAC) to facilitate recombineering in the bacteria as an efficient recombination method. BAC vectors can carry more than 300 kb with low toxicity because the plasmid is only copied once. The area between ORF2 and ORF3 of this virus was selected for BAC insertion to create the recombinant BoHV-4 viral vector, as previously described [21] The BAC cassette was inserted into the BoHV-4 genome by exploiting homologous recombination (HR) in the MDBK cells [22] However, due to low HR rate in mammalian cells, we used two different selection genes (green fluorescence protein and G418 antibiotic resistance) to guarantee propagation of the recombinant viruses in the pool of non-recombinant and recombinant viruses. This approach facilitated the selection process and can be considered as an efficient way for future studies to purify the recombinant viruses. We used the created viral vector to develop a new vaccine against Bovine herpesvirus 1 (BoHV-4-tgD∆TK) by selecting its truncated glycoprotein D (tgD) as an immunodominant gene. To create this construct, we chose the thymidine kinase (TK) area of BoHV-4 as a non-essential gene to insert the gene of interest (tgD) during recombineering in the SW-102 bacteria. Although previous studies have demonstrated that it is essential to use homologous arms of 150–200 bp to ensure the recombineering of a large insert (above 2000 bp) [43], we used 50 bp homologous arms targeting TK. We demonstrated that these short arms are sufficient for recombineering in the bacterial system with 100% efficiency. After the creation of BoHV-4-tgD∆TK, we compared the immunogenicity potential of this platform with a DNA vector expressing the same antigen (pC-tgD). Based on LDH assay, we demonstrated that the BoHV-4-tgD∆TK construct is more capable of eliciting cytotoxicity response than pC-tgD. In addition, analysis of the supernatant of virus-stimulated splenocyte cells and serum samples of immunized mice showed that cytokine responses were significantly higher in both the BoHV-4-tgD∆TK and pC-tgD groups than the control group.
In the present study, IgG1 (Th2), IgG2a (Th1), IgG2b, and IgG3 subclasses were sufficiently produced in both groups (pC-tgD and BoHV-4-tgD∆TK), indicating the stimulation of B cell response in the vaccinated animals. This finding was verified by the presence of high levels of neutralizing antibodies (NABs) in both immunized mice groups. Van Drunen et al. [44] reported a direct correlation between the titer of the neutralizing antibodies against the gD of BoHV-1 and virus shedding after challenge assay while Ioannou et al. [45] demonstrated that the most important part of the immune response in protecting against BoHV-1 challenge is the induction of neutralizing antibodies. Finally, Donofrio et al. [21] created a BoHV-4 based vaccine against BoHV-1 using non-truncated glycoprotein D as the immunodominant antigen and evaluated immune responses in a rabbit model. They reported that one injection of the vaccine was enough to elicit the binding antibody in the ELISA assay while serum-neutralizing antibodies appeared with kinetics like antibodies detectable by ELISA. Our data demonstrated that both vaccine constructs elicited neutralizing antibodies 2 weeks after the first and second injection.
One of the major concerns in the development of new expression platforms is pre-existing anti-vector immunity in the host organism. However, in the case of BoHV-4, Gillet et al. [20] showed that this immunity should not be a problem because this virus does not elicit serum neutralizing antibody production in infected animals. Even after introducing a foreign gene (gD of BoHV-1) into the BoHV-4 viral vector, they detected no serum neutralizing antibodies against BoHV-4.
In conclusion, the BoHV-4 viral vector generated in this study is almost as immunogenic as the DNA vaccine platform (pCDNA3.1 myc/His A) when both express tgD of BoHV-1. The DNA- and BoHV-4-based vaccine candidates in this experiment both showed promising results; hence, both can be considered in future experiments. This viral vector has great potential to stimulate both total and specific humoral and cellular responses with none of the drawbacks of the routinely used expression platforms in vaccination, such as pre-existing immunity and integration into the host genome. Although the efficacy of a BoHV-4-based vaccine for BoHV-1 needs to be corroborated by an in-vivo challenge study with a lethal BoHV-1 strain in cattle, our results with the mouse model provide a beneficial demonstration of the efficacy of this platform. Thus, it is worth considering this virus as an interesting viral vector due to its advantages as a rival to currently well-known viral and DNA vectors, as discussed above.
Data availability
The data that supports the findings of this study and the materials developed in this experiment are available on request from the corresponding author.
References
Kramberger P, Urbas L, Štrancar A (2015) Downstream processing and chromatography based analytical methods for production of vaccines, gene therapy vectors, and bacteriophages. Hum Vaccin Immunother 11(4):1010–1021
Dorange F, Le Bec C (2018) Latest developments in vral & non-viral vector manufacturing. Cell GeneTherapy Insights 4(2):119–129
Artusi S, Miyagawa Y, Goins WF, Cohen JB, Glorioso JC (2018) Herpes simplex virus vectors for gene transfer to the central nervous system. Diseases (Basel, Switzerland) 6(3):74. https://doi.org/10.3390/diseases6030074
Ehlers B, Buhk HJ, Ludwig H (1985) Analysis of bovine cytomegalovirus genome structure: cloning and mapping of the monomeric polyrepetitive DNA unit, and comparison of European and American strains. J Gen Virol 66(1):55–68
Bublot M, Van Bressem MF, Thiry E, Dubuisson J, Pastoret PP (1990) Bovine herpesvirus 4 genome: cloning, mapping and strain variation analysis. J Gen Virol 71(1):133–142. https://doi.org/10.1099/0022-1317-71-1-133
Dagalp SB, Demir AB, Gungor E, Alkan F (2007) The seroprevalence of Bovine Herpes Virus Type 4 (BHV4) infection in dairy herds in Turkey and possible interaction with reproductive disorders. Rev Méd Vét 158(4):201
Bilge-Dagalp S, Güngör E, Demir A, Pinar-Muz D, Vilmaz V, Oguzoglu TC, Ataseven VS, Alkan F (2010) The investigation of the presence of bovine herpesvirus type 4 (BoHV-4) in cows with metritis in a dairy herd. Ankara Üniv Vet Fak Derg 57:87–91
Bilge Dagalp SB, Babaoglu AR, Doğan F, Farzani TA, Alkan F (2020) An assessment of bovine herpes virus 4 as a causative agent in abortions and neonatal death. Onderstepoort J Vet Res 87(1):e1–e5. https://doi.org/10.4102/ojvr.v87i1.1761
Wellenberg GJ, Bruschke CJM, Wisselink HJ, Barkema HW, Van Oirschot JT (2002) Simultaneous intramammary and intranasal inoculation of lactating cows with bovine herpesvirus 4 induce subclinical mastitis. Vet Microbiol 86(1–2):115–129. https://doi.org/10.1016/S0378-1135(01)00496-5
Zimmermann W, Broll H, Ehlers B, Buhk HJ, Rosenthal A, Goltz M (2001) Genome sequence of bovine herpesvirus 4, a bovine Rhadinovirus, and identification of an origin of DNA replication. J Virol 75(3):1186–1194. https://doi.org/10.1128/JVI.75.3.1186-1194.2001
Thiry E, Bublot M, Dubuisson J, Van Bressem MF, Lequarre AS, Lomonte P, ... Pastoret PP (1992) Molecular biology of bovine herpesvirus type 4. Vet Microbiol 33(1-4): 79-92. https://doi.org/10.1016/0378-1135(92)90037-T
Redaelli M, Mucignat-Caretta C, Cavaggioni A, Caretta A, D’Avella D, Denaro L, Donofrio G (2010) Bovine herpesvirus 4 based vector as a potential oncolytic-virus for treatment of glioma. Virol J 7(1):298. https://doi.org/10.1186/1743-422X-7-298
Pedrera M, Macchi F, McLean RK, Franceschi V, Thakur N, Russo L, Medfai L, Todd S, Tchilian E, Audonnet JC, Chappell K, Isaacs A, Watterson D, Young PR, Marsh G, Bailey D, Graham S, Donofrio G (2020) Bovine herpesvirus-4-vectored delivery of Nipah virus glycoproteins enhances T cell immunogenicity in pigs. Vaccines 8(1):115
Aligholipour Farzani T, Bilge Dagalp S, Ozkul A et al (2021) Assessment of replication of bovine herpesvirus type 4 in human glioblastoma and breast cancer cells as a potential oncolytic virus. Virus Genes 57:31–39. https://doi.org/10.1007/s11262-020-01802-z
Wagner M, Ruzsics Z, Koszinowski UH (2002) Herpesvirus genetics has come of age. Trends Microbiol 10(7):318–324. https://doi.org/10.1016/S0966-842X(02)02394-6
Suter M, Lew AM, Grob P, Adema GJ, Ackermann M, Shortman K, Fraefel C (1999) BAC-VAC, a novel generation of (DNA) vaccines: a bacterial artificial chromosome (BAC) containing a replication-competent, packaging-defective virus genome induces protective immunity against herpes simplex virus 1. Proc Natl Acad Sci 96(22):12697–12702. https://doi.org/10.1073/pnas.96.22.12697
Meseda CA, Schmeisser F, Pedersen R, Woerner A, Weir JP (2004) DNA immunization with a herpes simplex virus 2 bacterial artificial chromosome. Virology 318(1):420–428. https://doi.org/10.1016/j.virol.2003.09.033
Donofrio G, Cavirani S, Simone T, van Santen VL (2002) Potential of bovine herpesvirus 4 as a gene delivery vector. J Virol Methods 101(1–2):49–61. https://doi.org/10.1016/S0166-0934(01)00419-0
Donofrio G, Galli C, Lazzari G, Van Santen VL, Cavirani S, Flammini CF (2003) Interaction of a green recombinant bovine herpesvirus 4 with in vitro-produced bovine embryos. Vet Res Commun 27(5):415–424. https://doi.org/10.1023/A:1024889606158
Gillet L, Dewals B, Farnir F, de Leval L, Vanderplasschen A (2005) Bovine herpesvirus 4 induces apoptosis of human carcinoma cell lines in vitro and in vivo. Can Res 65(20):9463–9472. https://doi.org/10.1158/0008-5472.CAN-05-1076
Donofrio G, Cavirani S, Vanderplasschen A, Gillet L, Flammini CF (2006) Recombinant bovine herpesvirus 4 (BoHV-4) expressing glycoprotein D of BoHV-1 is immunogenic and elicits serum-neutralizing antibodies against BoHV-1 in a rabbit model. Clin Vaccine Immunol 13(11):1246–1254. https://doi.org/10.1128/CVI.00200-06
Donofrio G, Sartori C, Ravanetti L, Cavirani S, Gillet L, Vanderplasschen A, ... Flammini CF (2007) Establishment of a bovine herpesvirus 4 based vector expressing a secreted form of the bovine viral diarrhoea virus structural glycoprotein E2 for immunization purposes. BMC Biotechnol 7(1): 68. https://doi.org/10.1186/1472-6750-7-68
Donofrio G, Franceschi V, Lovero A, Capocefalo A, Camero M, Losurdo M, Tempesta M (2013) Clinical protection of goats against CpHV-1 induced genital disease with a BoHV-4-based vector expressing CpHV-1 gD. PLoS One 8(1):e52758. https://doi.org/10.1371/journal.pone.0052758
Graham DA (2013) Bovine herpes virus-1 (BoHV-1) in cattle–a review with emphasis on reproductive impacts and the emergence of infection in Ireland and the United Kingdom. Ir Vet J 66(1):15. https://doi.org/10.1186/2046-0481-66-15
Alkan F, Burgu I, Bilge-Dagalp S, Yildirim Y, Gencay A, Gungor B, Akça Y (2005) The seroprevalence of BHV-1 infection on selected dairy cattle herds in Turkey. Rev Med Vet 156(3):166–169
Weiss M, Brum MCS, Anziliero D, Weiblen R, Flores EF (2015) A glycoprotein E gene-deleted bovine herpesvirus 1 as a candidate vaccine strain. Braz J Med Biol Res 48(9):843–851. https://doi.org/10.1590/1414-431X20154243
Perrin B, Perrin M, Moussa A, Coudert M (1996) Evaluation of a commercial gE blocking ELISA test for detection of antibodies to infectious bovine rhinotracheitis virus. Vet Rec. https://doi.org/10.1136/vr.138.21.520
Van Drunen Littel-Van Den Hurk S, Braun RP, Lewis PJ, Karvonen BC, Baca-Estrada ME, Snider M et al Intradermal immunization with a bovine herpesvirus-1 DNA vaccine induces protective immunity in cattle. J Gen Virol 79: 831–839. https://doi.org/10.1099/0022-1317-79-4-831
Reddy PS, Idamakanti N, Pyne C, Zakhartchouk AN, Godson DL, Papp Z, Tikoo SK (2000) The immunogenicity and efficacy of replication-defective and replication-competent bovine adenovirus-3 expressing bovine herpesvirus-1 glycoprotein gD in cattle. Vet Immunol Immunopathol 76(3–4):257–268. https://doi.org/10.1016/S0165-2427(00)00217-8
Rosamilia A, Jacca S, Tebaldi G, Tiberti S, Franceschi V, Macchi F, Donofrio G (2016) BoHV-4-based vector delivering Ebola virus surface glycoprotein. J Transl Med 14(1):1–9. https://doi.org/10.1186/s12967-016-1084-5
Aligholipour Farzani T, Földes K, Hanifehnezhad A, Yener Ilce B, Bilge Dagalp S, Amirzadeh Khiabani N, Ergünay K, Alkan F, Karaoglu T, Bodur H, Ozkul A (2019a) Bovine Herpesvirus Type 4 (BoHV-4) Vector Delivering nucleocapsid protein of Crimean-Congo hemorrhagic fever virus induces comparable protective immunity against lethal challenge in IFNα/β/γR-/- mice models. Viruses 11(3):237. https://doi.org/10.3390/v11030237
Sambrook HC (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor, 2001
Hirt B (1967) Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365
Toussaint JF, Coen L, Letellier C, Dispas M, Gillet L, Vanderplasschen A, Kerkhofs P (2005) Genetic immunisation of cattle against bovine herpesvirus 1: glycoprotein gD confers higher protection than glycoprotein gC or tegument protein VP8. Vet Res 36(4):529–544. https://doi.org/10.1051/vetres:2005015
Thomason LC, Sawitzke JA, Li X, Costantino N, Court DL (2014) Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol 106(1):1–16. https://doi.org/10.1002/0471142727.mb0116s106
Aligholipour Farzani T, Hanifehnezhad A, Földes K, Ergünay K, Yilmaz E, Hashim Mohamed Ali H, Ozkul A (2019c) Co-delivery effect of CD24 on the immunogenicity and lethal challenge protection of a DNA vector expressing nucleocapsid protein of Crimean Congo hemorrhagic fever virus. Viruses 11(1):75. https://doi.org/10.3390/v11010075
Pasandideh R, Seyfi Abad Shapouri MR, Beigi Nassiri MT (2018) Immunogenicity of a plasmid DNA vaccine encoding G1 epitope of bovine ephemeral fever virus G glycoprotein in mice. Onderstepoort J Vet Res 85(1):e1–e6. https://doi.org/10.4102/ojvr.v85i1.1617
Muthumani K, Griffin BD, Agarwal S, Kudchodkar SB, Reuschel EL, Choi H, ... Kim YK (2016) In vivo protection against ZIKV infection and pathogenesis through passive antibody transfer and active immunisation with a prMEnv DNA vaccine.Npj Vaccines 1(1): 1–11. https://doi.org/10.1038/npjvaccines.2016.21
Aligholipour Farzani T, Földes K, Ergünay K, Gurdal H, Bastug A, Ozkul A (2019b) Immunological analysis of a CCHFV mRNA vaccine candidate in mouse models. Vaccines (Basel) 7(3):115. Published 2019 Sep 16. https://doi.org/10.3390/vaccines7030115
Liu C, Xie Y, Sun B, Geng F, Zhang F, Guo Q, Wu H, Yu B, Wu J, Yu X, Kong W, Zhang H (2018) MUC1- and survivin-based DNA vaccine combining immunoadjuvants CpG and interleukin-2 in a bicistronic expression plasmid generates specific immune responses and antitumour effects in a murine colorectal carcinoma model. Scand J Immunol 87:63–72. https://doi.org/10.1111/sji.12633
Reed LJ, Muench H (1938) A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27(3):493–497. https://doi.org/10.1093/oxfordjournals.aje.a118408
Franceschi V, Capocefalo A, Calvo-Pinilla E, Redaelli M, Mucignat-Caretta C, Mertens P, ... Donofrio G (2011) Immunization of knock-out α/β interferon receptor mice against lethal bluetongue infection with a BoHV-4-based vector expressing BTV-8 VP2 antigen. Vaccine 29(16): 3074-3082.https://doi.org/10.1016/j.vaccine.2011.01.075
Liu P, Jenkins NA, Copeland NG (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13(3):476–484. https://doi.org/10.1101/gr.749203
van Drunen Littel-van den Hurk S, Van Donkersgoed J, Kowalski J, van den Hurk JV, Harland R, Babiuk LA (1998) A subunit gIV vaccine, produced by transfected mammalian cells in culture, induces mucosal immunity against bovine herpesvirus-1 in cattle. Vaccine 1994. https://doi.org/10.1016/S0264-410X(94)80055-5
Ioannou XP, Griebel P, Hecker R, Babiuk LA (2002) The immunogenicity and protective efficacy of bovine herpesvirus 1 glycoprotein D plus Emulsigen are increased by formulation with CpG oligodeoxynucleotides. J Virol 76(18):9002–9010. https://doi.org/10.1128/JVI.76.18.9002-9010.2002
Acknowledgements
The findings partly constitute the second author’s dissertation for her Doctorate of Science degree in Virology at the graduate school of health sciences, Ankara University of Turkey. The authors would like to thank Prof. Dr. Donald L. Court, Chief, Molecular Control Genetics Section, RNA Biology Lab, Frederick, MD, USA, for his kind support in providing SW102 cells, and Prof. Dr. L. Egyed, Budapest University, Hungary for providing the BoHV-4 Movar 33/63 strain.
Funding
This study was supported by a grant from Scientific Research Projects, Ankara University (BAP; Project number: 17B0239004).
Author information
Authors and Affiliations
Contributions
S.B.D. and T.A.F. designed the study; T.A.F. conducted the experiments and analyzed the data and prepared the figures; T.A.F., F.D., and S.B.D. wrote the draft manuscript; and T.A.F., S.B.D., F.D., A.O., F.A, Z.A-Y. edited it and G.D. intellectually contributed. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All animal experiments were performed with the official permission of the Ankara University Ethical Committee for Animal Experiments (17/12/2014; 2014–23-155). All animal sampling was conducted according to national regulations on the operation and procedure of animal experiments ethics committees (Regulation Nr.26220, Date: 09.7.2006). During these studies, humane endpoint scores were considered, and the mice were humanely euthanized by CO2 exposure and cervical dislocation. Multiple observations per day were conducted to confirm the animals’ welfare. Constant access to sterilized water and food was provided. The BALB/c mice were purchased from Kobay DHL A.Ş (Ankara, Turkey).
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Flavio Guimaraes Fonseca
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bilge-Dagalp, S., Farzani, T.A., Dogan, F. et al. Development of a BoHV-4 viral vector expressing tgD of BoHV-1 and evaluation of its immunogenicity in mouse model. Braz J Microbiol 52, 1119–1133 (2021). https://doi.org/10.1007/s42770-021-00525-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-021-00525-z