
Abstract
DNA replication is a vital process in all living organisms. At each cell division, > 30,000 replication origins are activated in a coordinated manner to ensure the duplication of > 6 billion base pairs of the human genome. During differentiation and development, this program must adapt to changes in chromatin organization and gene transcription: its deregulation can challenge genome stability, which is a leading cause of many diseases including cancers and neurological disorders. Over the past decade, great progress has been made to better understand the mechanisms of DNA replication regulation and how its deregulation challenges genome integrity and leads to human disease. Growing evidence shows that gene transcription has an essential role in shaping the landscape of genome replication, while it is also a major source of endogenous replication stress inducing genome instability. In this review, we discuss the current knowledge on the various mechanisms by which gene transcription can impact on DNA replication, leading to genome instability and human disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Humans start their life as a single cell that has to repeatedly divide to create the ~ 40 trillion cells that comprise the human body (Bianconi et al. 2013). It is essential that all the genetic information contained in the zygote is reliably transmitted to all daughter cells to guarantee proper development. At each cell division, DNA replication involves the activation of tens of thousands of replication origins to ensure complete genome duplication. This program must be very robust and be able to adjust to the gene transcription programs and the chromatin organization of the different cell types that they are becoming. Furthermore, a huge level of DNA replication and cell division is necessary during the entire human life span to replace old, dead or damaged cells. If DNA replication fails, genome integrity is challenged and many diseases, such as cancers and neurological disorders, can arise (Ganier et al. 2019; Zeman and Cimprich 2014). It is therefore essential that this program is correctly accomplished. However, during the life span, large numbers of exogenous and endogenous replication stresses routinely challenge DNA integrity and lead to genome instability. In particular, growing evidence indicates that gene transcription itself is an important, yet unavoidable endogenous replication stress, which can either suppress replication initiation, or can generate conflicts with the DNA replication process. In this review, we focus on transcription-mediated replication stress and its impact on human diseases. First, we describe the mechanisms of DNA replication initiation and control, as well as its relation to gene transcription. We then discuss the different mechanisms by which transcription acts as a notable source of replication stress to induce genome instability. Finally, we explain how such transcription-mediated replication stresses are involved in various human diseases.
Origin licensing and firing: a two-step process
The nuclear genome must be correctly duplicated once and only once per mitotic cell division. To avoid genome re-replication, DNA replication is temporarily divided into two steps: (i) the origin licensing that takes place between mitosis and the beginning of the next interface, where all the possible replication origins are recognized and loaded with the pre-replication complex (pre-RC), and (ii) the origin firing that takes place during S phase (Fig. 1). Although most of the collected information comes from yeast, the major process seems to be highly conserved in other eukaryotes. In this section, we report mainly dynamics from budding yeast and integrate with information from other eukaryotes.

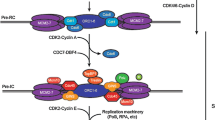
Origin licensing and firing occur over a two-step process. The first step of DNA replication, called origin licensing, consists of loading the pre-replication (pre-RC) complex onto chromatin on all the potential replication origins along the genome. This occurs between the end of mitosis and G1 phase. The second part of the process takes place in S phase, where replication origins are activated through the recruitment of limiting factors that lead to the conversion of pre-RC to pre-IC (pre-initiation complex). This transition is regulated by the replication timing program that marks the order of replication of the genome, with origins in the early-replicating regions fired before those in the late-replicating regions. To avoid re-replication, components of the pre-RC are segregated, exported or degraded, therefore impairing re-licensing
The loading of the pre-RCs onto chromatin starts with the recognition of origins by a hetero-hexamer called origin recognition complex (ORC, ORC1-6) (Bell and Stillman 1992), which further recruits other factors to form the pre-RCs. In yeast, replication origins are associated with specific sequences called autonomously replicating sequences (ARS) (Marahrens and Stillman 1992), while in higher eukaryotes the situation is less clear and replication origins are not defined by a specific sequence. Multiple techniques have been used to map replication origins and the results are not concordant, suggesting that we might be looking at different subsets of origins based on the limitations of the various approaches (see Prioleau and MacAlpine 2016; Ganier et al. 2019 for review). Origins identified with the small nascent strand (SNS) method seem to be enriched at transcriptional start sites (TSSs) (Sequeira-Mendes et al. 2009; Cadoret et al. 2008), origin G-rich repeated elements (OGRE) (Cayrou et al. 2012), G quadruplex (G4) (Besnard et al. 2012), high CpG and GC content regions (Cayrou et al. 2011; Delgado et al. 1998; Cadoret et al. 2008), while the OK-seq (Okazaki fragment sequencing) method has shown that origins preferentially position within the intergenic regions before and/or after gene bodies that are AT rich (Tubbs et al. 2018; Petryk et al. 2016) (Fig. 2).
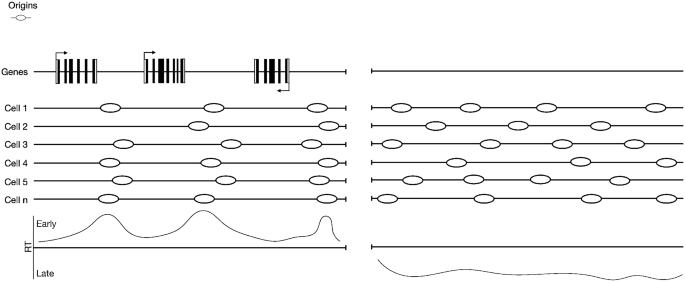
Origin distributions at early and late-replicating regions. Origin distribution differs between distinct regions of the genome. Within the early-replicating genome (Left panel), replication origins are enriched in intragenic regions between active genes. This effect might be because active transcription can cause disassembly of the pre-RC or sliding of the MCM complex away from the original loading position due to the passage of the RNA polymerase over transcribed genes. In the late-replicating regions (Right panel), which are frequently associated with regions that lack gene transcription, replication origins are almost randomly distributed
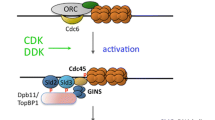
Replication origins are also marked by epigenetic signatures, such as H2A.Z and H4k20me2/3, the presence of which is needed for ORC1 recruitment (Beck et al. 2012; Long et al. 2020; Kuo et al. 2012). ORC binding is followed by the recruitment of cell division control protein 6 (CDC6), which stabilizes ORC binding (Speck and Stillman 2007), and CDC10-dependent transcript 1 (CDT1). CDT1 is loaded onto chromatin together with the mini-chromosome maintenance (MCM2-7) helicase complex through the interaction with ORC (Evrin et al. 2009; Maiorano et al. 2000; Nishitani et al. 2000). A second MCM complex is then loaded by ORC in an inverted orientation to form MCM double-hexamer formation (Miller et al. 2019). This process completes origin ‘’licensing” (Fig. 1). The pre-RC is not stable on chromatin, and recent studies in yeast and Drosophila have suggested that gene expression can alter origin licensing by disassembling the pre-RC or by sliding the MCM complex away from the original loading position due to the passage of the RNA polymerase over transcribed genes (Gros et al. 2015; Powell et al. 2015). This process might explain the preferential localization of replication initiation sites within intergenic regions between active genes.
To avoid re-replication when cells enter S phase, components of the pre-RC are made inaccessible through post-translational modifications that can cause their inactivation, export out of the nucleus and degradation, or as in the case of CDT1, segregation via an interaction with GEMININ (Ballabeni et al. 2013; Petersen et al. 2000, 1999; Nguyen et al. 2000; Li and DePamphilis 2002; Méndez et al. 2002). At the transition between G1 and S phase, fully formed pre-RCs are phosphorylated at specific sites by Dbf4-dependent kinase (DDK) and cyclin-dependent kinase (CDK). These phosphorylation events lead to CDC45, treslin and Mdm2-binding protein (MTBP) recruitment (Boos et al. 2013; Kumagai and Dunphy 2017; Heller et al. 2011; Ilves et al. 2010; Jares and Blow 2000). Treslin phosphorylation leads to the recruitment of topoisomerase 2-binding protein 1 (TOPBP1), RecQ-like helicase 4 (RECQL4), GINS complex and Pol ε and the subsequent conversion of pre-RCs into pre-initiation complexes (pre-ICs) (Tanaka et al. 2007; Kumagai et al. 2011; Boos et al. 2011; Muramatsu et al. 2010; Sangrithi et al. 2005). At this point, treslin, MTBP, RECQL4 and TOPBP1 are released and the active replisome is formed thanks to MCM10 loading (Kanke et al. 2012; Watase et al. 2012; Kanemaki and Labib 2006; Gambus et al. 2006). Finally, other proteins such as replication protein A (RPA), proliferating cell nuclear antigen (PCNA) and replication factor C (RFC) are loaded and DNA replication starts, called origin firing (MacNeill 2012) (Fig. 1).
Origins usage and replication timing
Of all the potential replication origins that are loaded with a pre-RC, only a subset will actually be fired. Most of these origins are licensed to work as a backup plan in case replicative stress stalls the replication forks (Ge et al. 2007; Woodward et al. 2006; Santocanale et al. 1999). Moreover, all replication origins do not fire at the same time, but instead they follow a cell-type specific spatio-temporal program, known as the replication timing (RT) program (Dimitrova and Gilbert 1999) (Fig. 2). In mammalian cells, this program is established during G1 phase, ~ 2 h after mitosis in a time window referred to as the time decision point (TDP) (Lu et al. 2010; Li et al. 2001, 2003; Wu et al. 2006; Dimitrova and Gilbert 1999). Interestingly, the establishment of the RT precedes the choice as to which origins are going to be used during S phase; that choice instead occurs later in G1 phase during the origin decision point (ODP) (Dimitrova and Gilbert 1999; Li et al. 2003). The relation between TDP and ODP, and the corresponding mechanism(s) need to be further investigated. RT establishment temporally corresponds to the re-establishment of an organized nuclear architecture after mitosis, with the anchoring of chromosomes to the nuclear periphery (Dimitrova and Gilbert 1999; Li et al. 2001) and the establishment of topologically associated domains (TADs) and the A/B compartments (Dileep et al. 2015). Likewise, RT domains have an extensive overlap with TADs and their being early or late-replicating corresponds to A or B compartments, respectively (Pope et al. 2014; Ryba et al. 2010).
Such a strong correlation between RT and the 3D genome structure led the field to hypothesize that these two processes might be coupled and that one might control the other. This hypothesis has been reinforced by the identification of Rif1 (Rap1-interacting factor 1) as a nuclear structural protein that has an important role in RT regulation (Foti et al. 2016). Conversely, knock-outs or knock-downs of several other nuclear structural proteins, such as cohesin or CTCF, alter chromatin structure but not RT (Oldach and Nieduszynski 2019; Rao et al. 2017; Sima et al. 2019; Nora et al. 2017). Moreover, it has been recently shown that Rif1 haploid cells show alterations in chromatin structure but normal RT, which indicates that although the two processes are coordinated, they can be uncoupled (Gnan et al. 2019).
For years, the field has investigated the possibility that RT could reflect gene transcription. In general, early-replicating regions are enriched with expressed genes, while late-replicating regions are not (Woodfine et al. 2004) (Fig. 2). In addition, during development, regions switching RT from early-to-late (or late-to-early) are associated with genes whose expression is switched off (or on) (Hiratani et al. 2008). However, there are numerous exceptions: for example, regions containing expressed genes can also be replicated late, which challenges a direct link between RT and gene transcription (Rivera-Mulia et al. 2015). In addition, switching off genes at the β-globin locus fails to alter RT when chromatin accessibility is not modified, which also seems to go against this model (Cimbora et al. 2000). Indeed, the change in RT at the β-globin locus is associated with changes in accessibility, which seems to support the idea that RT is associated with chromatin accessibility rather than gene expression (Cimbora et al. 2000). In fact, early-replicating regions are associated with open chromatin states (A compartment), while late-replicating regions are enriched in closed chromatin states (B compartment) (Pope et al. 2014). In a recent article, Dileep and colleagues showed that changes in RT can precede or follow changes in gene transcription or be totally independent from it (Dileep et al. 2019). It is therefore likely that both RT and gene transcription are regulated by some common factors shared between the two processes.
How RT and origin usage are regulated is not fully understood, but they can be explained through a model of differential affinity for limiting factors (Fig. 1). To date, limiting factors have been identified in some organisms and include proteins that are essential for the assembly of the pre-IC, such as CDC45, DBF4/CDC7 (regulatory/catalytic subunit of DDK), RecQL4, Treslin, TOPBP1 and MTBP orthologs (Mantiero et al. 2011; Wu and Nurse 2009; Collart et al. 2013; Wong et al. 2011; Tanaka et al. 2011). What regulates the affinity for these limiting factors to replication origins is still unclear, but probably multiple layers of regulation are in place. A first possibility lays on chromatin looping that clusters together origins being fired and leaving backup origins on the periphery of the loops (Courbet et al. 2008). Along the same line, the order of firing could be regulated through chromatin accessibility. As discussed previously, the early-replicating regions have a more open chromatin state than the late-replicating regions (Pope et al. 2014), which might make the late-replicating regions inaccessible at the beginning of the S phase. Moreover, some proteins globally regulate RT, in a way, controlling the accessibility of the limiting factors. One of these is RIF1, which is enriched at late-replicating regions: RIF1 counters DDK activity thanks to its interaction with PP1 (protein phosphatase 1), dephosphorylating components of the pre-RC and limiting origin firing until late S phase (Cornacchia et al. 2012; Mattarocci et al. 2014; Poh et al. 2014; Hiraga et al. 2014; Sukackaite et al. 2017). In fission yeast, the shelterin complex (also called telosome) is involved in RT regulation of a subgroup of late origins through Rif1. Shelterin can recruit Rif1 on telomeric DNA, as Taz1 does, and also brings late-replicating regions into the proximity of Rif1 (Tazumi et al. 2012; Ogawa et al. 2018; Kanoh and Ishikawa 2001). Moreover, Rap1 and Poz1 (two members of the shelterin complex) depletion can impact RT in an indirect manner. In fact, these mutants exhibit abnormal telomere elongation that delocalizes PP1 ortholog from the late Rif1-dependent and Taz1-independent regions to telomeres (Hasegawa et al. 2019). Fork head 1 and 2 (Fkh1/2) are two transcription factors that have also been reported to regulate RT in yeast. These factors group early origins into clusters to facilitate DDK activity (Knott et al. 2012; Fang et al. 2017) via a direct interaction between Fkh1/2 and Dbf4 (Fang et al. 2017). Similarly, Ctf19 and Swi6 recruit DDK to pericentromeric origins, allowing centromeres to replicate early in budding and fission yeasts (Hayashi et al. 2009; Natsume et al. 2013). In S. cerevisiae, two histone deacetylases, Sir2 and Rpd3, control the RT of origins located within the ribosomal DNA (rDNA) array by tuning their ability to compete with single-copy origins for limiting factors (Yoshida et al. 2014). Work is ongoing to identify additional factors and delineate the underlying mechanisms controlling the origin usage and RT. Such work will help us better understand the complex relationship between DNA replication, gene transcription and chromatin organization.
Transcription-mediated replication stresses and genome instability
As described earlier, replication initiation control is a multi-step process ensuring that the entire genome can be replicated once and only once for each cell division. Gene transcription can interplay with the DNA replication program at all stages, i.e., during the G1 phase for the origin setting (location, firing time etc.), or during the S phase for the origin activation, replication fork progress etc. Here, we describe in detail how gene transcription influences DNA replication that leads to genome instability in normal and pathological conditions, and in the contribution to human diseases.
Transcription–replication collision, R-loop formation and genome instability
Once replication forks have been deployed, their progression can be challenged by numerous factors. One such factor is the presence of active transcription along the genome. Collisions between the replication fork and the transcription machinery can either be co-directional (CD) or head-on (HO) (Fig. 3). The latter can be more dangerous for genome integrity (Hamperl et al. 2017). OK-Seq, which helps identify the direction of replication fork movement, has revealed that origin firing occurs more frequently upstream of the TSSs of active genes, ensuring co-directional replication of the most highly transcribed regions of the genome (Petryk et al. 2016). A wildly localized replication termination at the transcription termination sites (TTSs) of transcribed genes under unperturbed conditions was also revealed. Meanwhile, replication termination could redistribute to gene bodies under replication stress, causing increased gene 3′ end replication in an HO orientation (Chen et al. 2019), which strongly induces transcription–replication conflicts (TRCs).

Transcription–replication conflicts lead to fork stalling and genome instability. Replication and transcription machineries share the same DNA template, which causes replication–transcription conflicts (TRCs). These conflicts can occur in a head-on or co-directional manner. Head-On TRC is generally considered as more deleterious to genome stability, and preferentially occurs around gene transcription termination sites (TTS). The replication forks stall when they encounter RNA Pol II, which favors the transient formation of R-loops. Under normal conditions, harmful R-loop accumulation can be prevented by many factors, such as TOP1, SETX, BRCA1/2, and FANCM. Alternatively, this accumulation can be directly removed by RNase H, XRN2 and certain NER endonucleases like XPG/XPF. If the R-loops and stalled forks persist, the ATR-Chk1 pathway is activated and phosphorylates RPA at the stalled forks. Under topological stress, such as TOP1 depletion, DNA damage is induced, which leads to genome instability (Promonet et al. 2020). R-loops also frequently form at gene transcription start sites (TSS), while they do not seem to induce TRCs and are rather involved in other mechanisms, like transcription regulation
Recently, numerous studies have revealed that TRCs are frequently associated with a specific structure known as R-loops (Fig. 3). R-loops are formed when RNA polymerase progresses along the DNA double strands, with newly transcribed RNA re-annealed to the transiently accessible template strand: a DNA:RNA hybrid forms that displaces the non-template strand (Thomas et al. 1976) mainly in the presence of high GC content sequences (Sanz et al. 2016). Importantly, by analyzing the genome-wide distribution of R-loops by DNA:RNA hybrid immunoprecipitation and next-generation sequencing (DRIP-seq), Cimprich and colleagues revealed that R-loops form preferentially at regions with HO TRC (Hamperl et al. 2017). These data reinforce the idea that the CD bias of the human genome might help to minimize the accumulation of HO collisions and deleterious R-loops.
Cells can also regulate R-loops by opposing their formation. As a matter of fact, R-loops preferentially form in the presence of negative supercoils, such as those formed in concomitance with RNA transcription. To resolve these tensions, cells use topoisomerases that rescue normal DNA tension and reduce the accumulation of R-loops (El Hage et al. 2010; Yang et al. 2014). Recently, P. Pasero, C.L. Chen and colleagues discovered that R-loop formation is enriched at TTSs for a subset of highly expressed genes located at early-replicating regions. Here, a higher level of HO collision is frequently associated with the accumulation of phospho-RPA32 (S33), a hallmark of stalled forks. As a result, at these regions, an increase in DNA double-strand breaks (DSBs) and γ-H2AX, a histone mark around broken replication forks, have been observed in cells with topoisomerase 1 (Top1) depletion (Promonet et al. 2020).
It should be noted that although the presence of R-loops on HO TRC can be deleterious, R-loops can also have important physiological roles in many normal cellular processes, including the regulation of transcription termination, chromosome segregation and rearrangement events (Skourti-Stathaki and Proudfoot 2014; Kabeche et al. 2018; Skourti-Stathaki et al. 2011; Xu et al. 2017a). The R-loop balance in cells is therefore maintained via various strategies to protect genome stability. As mentioned, cells use topoisomerases to reduce topological stress and decrease harmful R-loop accumulation (Fig. 3). Cells also present RNase H 5′–3′ exonucleases that can digest RNA from DNA:RNA hybrids. R-loops can also be prevented or resolved through helicases, such as DHX9 and Aquarius (AQR) (Sollier et al. 2014; Chakraborty and Grosse 2011), senataxin (SETX) (Groh et al. 2017) and PIF1 (Zhou et al. 2014). R-loop formation is also tightly regulated via spliceosome binding to RNA (Li and Manley 2005; Gómez-González et al. 2011; Li et al. 2007; Pefanis et al. 2015), the presence of proteins coating RPA (Aguilera and García-Muse 2012; Nguyen et al. 2018) and the ATR-Chk1 pathway (Matos et al. 2020). Many studies have shown that mutations affecting these factors could induce R-loop-associated human diseases, which we discuss in more detail later.
Proteins of homologous recombination and non-homologous end joining on stalled forks
As obstacles to replication fork progression, R-loops can induce genome instability and thus inevitably activate the DNA damage repair pathway. In particular, stalled forks deriving from TRCs activate Fanconi anemia (FA) DSB pathway—a repair system involved in the resolution of R-loop-mediated replication fork collapse (Schwab et al. 2015; García-Rubio et al. 2015). The disruption of critical FA complex members FANCD2, FANCA and FANCM impairs the restarting of stalled forks, and leads to gene instability and DNA damage from R-loop-mediated replication fork collapse (Schwab et al. 2015; García-Rubio et al. 2015). These effects can be reverted by over-expressing RNase H1, a ribonuclease degrading DNA:RNA hybrid, reinforcing the idea that R-loops are responsible for fork stalling at the HO TRC sites (Schwab et al. 2015). Interestingly, a recent study revealed that SLX4, a tumor suppressor, drives (via its interaction with RTEL1) the recruitment of FANCD2 to RNA polymerase II to prevent endogenous transcription-induced replication stress (Takedachi et al. 2020).
Besides the core FA complex members, other factors involved in homologous recombination (HR) accumulate at DSBs, such as RAD52, RAD51, BRCA1 (also called FANCS), and BRCA2 (also called FANCD1), to regulate genome instability through R-loop resolution. Their recruitment can be reduced by RNase H overexpression at active transcription regions or through specific reporter systems (D’Alessandro et al. 2018; Yasuhara et al. 2018). For example, BRCA1 and BRCA2 prevent the potential harmful effects of R-loops by recruiting helicase SETX to R-loops (Hatchi et al. 2015; Zhang et al. 2017). In particular, BRCA1-dependent recruitment of SETX resolves R-loop structures preferentially at TTSs and suppresses DNA damage. Moreover, SETX depletion impairs RAD51 recruitment and favors 53BP1 accumulation, a key DNA damage response (DDR) factor in non-homologous end joining (NHEJ) (Cohen et al. 2018). These data suggest that DNA:RNA hybrids may favor HR factor accumulation to potentially facilitate the elimination of the hybrids so that HR could occur, likely counteracting NHEJ at DSBs within transcribed genes. Interestingly, a recent study revealed that 53BP1 and BRCA1 counteract each other to control the time-dependent switch of the fork restart pathways: here, 53BP1 promotes the fast and BRCA1 promotes the slow kinetics restart pathways, respectively (Xu et al. 2017b). On the other hand, BRCA2 depletion from cells also increases R-loop accumulation. BRCA2 might prevent R-loop formation by preventing replication fork collapse and recruiting the ssDNA binding protein, Rad51, to DSBs (Schlacher et al. 2011). Moreover, BRCA2 recruits RNA polymerase II-associated factor-1 (PAF1) to promoter-bound Pol II to enhance the pause and decrease of R-loop formation (Shivji et al. 2018).
G1 shortening induces abnormal initiation and genome instability within gene body
G1 phase is an important period for origin setting. Rapidly proliferating mammalian embryonic stem cells (ESCs) exhibit a short G1 phase that is < 2 h due to an unusual cell cycle structure (Savatier et al. 1994). Such a short G1 phase is considered a characteristic of ESCs that might help to inhibit differentiation and preserve their pluripotent state (Li et al. 2012). Several studies have reported that the short G1 phase in ESCs, before differentiating, is related to a unique mechanism of cell cycle regulation. In particular, ESCs express low cyclin D1 levels and no cyclin D2/D3, lack MAPK and pRB control (Jirmanova et al. 2002; Savatier et al. 1996; White et al. 2005), lack pathways of p53-p21 in response to DNA damage (Aladjem et al. 1998) and lack activity of cyclin E-Cdk2 and cyclin A-Cdk2 complexes throughout the cell (Stead et al. 2002; White et al. 2005). These findings highlight that cell proliferation control in ESCs is fundamentally different from that in differentiated somatic cell lineages (Coronado et al. 2013). Ample storage of the factors required for replication and relaxed chromatin structures in ESCs results in many more replication initiation sites in S phase. Despite their short G1 phase, ESCs can effectively tolerate an accumulation of replication stress by extensive fork reversal and replication-coupled repair. This feature allows these cells to preserve genome stability, demonstrating that fast proliferating ESCs do not exhibit mechanisms to delay G2/M and G1/S transitions on incomplete replication (Ahuja et al. 2016).
Conversely, somatic cells have a longer G1 phase, which might help to ensure proper origin licensing to guarantee complete genome duplication. Therefore, G1 shortening in somatic cells, e.g., by overexpressing cyclin E, associated with an altered G1-S transition, may lead to deregulation of replication fork progression and DNA damage (Jones et al. 2013). Cyclin E, a member of the cyclin family, has a critical role in controlling the G1-S transition. It binds CDK2 to form the cyclin E/CDK2 complex, which phosphorylates numerous downstream proteins (such as RB, p27, p21) to regulate multiple cellular processes, thus allowing replication initiation and S phase progression (Siu et al. 2012). Ekholm-Reed and colleagues demonstrated that overexpressing cyclin E can shorten the length of G1 phase from about 10–12 h to as little as 2–4 h (Ekholm-Reed et al. 2004). To deeply discern the detailed mechanisms related to the replication stress induced by cyclin E overexpression, Macheret and Halazonetis mapped DNA replication and transcription genome-wide in cells with abnormal cyclin E activation (Macheret and Halazonetis 2018). By investigating the DNA replication initiation profiles (HU-EdU-seq) from cells overexpressing cyclin E versus cells with normal cyclin E levels, they showed that cyclin E overexpression induces extra origins that are frequently located within intragenic regions (Fig. 4). In addition, analysis of newly synthesized transcript profiles through EU-seq has revealed that these novel origins induced by G1 shortening are often located at the 3′ ends of the gene body, showing lower levels of nascent transcripts in G1 cells due to G1 shortening. Importantly, a specific fork collapse has been observed around these origins that only appears under cyclin E overexpression, while fork collapse has not been observed for the constitutive origins (Fig. 4). Similar results have been obtained by overexpressing MYC. MYC-inducible activation leads to G1-phase shortening and to the firing of intragenic oncogene-induced (Oi) origins. Many of these Oi origins overlap with cyclin E-induced origins (Macheret and Halazonetis 2018). Moreover, overexpression of both genes can induce the firing of a novel set of replication origins within the 3′ gene body of highly transcribed genes that are usually suppressed by transcription during the G1 phase. The precocious entry into S phase, before all genic regions have been transcribed, allows the firing of origins within genes in cells with a short G1 phase (Macheret and Halazonetis 2018). Therefore, DNA replication stress resulted from extra intragenic origin firing caused by premature S phase entry is an important mechanism that leads to genomic instability in human cells.

G1 shortening induces abnormal origin firing within active genes leading to genome instability. In normal cell cycles, the length of G1 is sufficient for transcription to inactivate origins across the entire length of genes (Top panel). When the length of G1 is greatly reduced due to oncogene expression (Bottom panel), there is insufficient time for transcription to inactivate all intragenic origins. This effect allows for the activation of oncogene-induced extra-origins, located within intragenic regions, and leads to chromosome breakage. G1 shortening, e.g., induced by cyclin E or Myc, leads to abnormal replication and genome instability, which might contribute to early cancer development
Interestingly, under replication stress, i.e., under high dose HU treatment, cells can accumulate replication fork stalling and collapse within specific early-replicating regions known as early-replicating fragile sites (ERFSs) (Barlow et al. 2013). These sites are also enriched around replication origins containing long (> 20 bp) Poly(dA:dT) tracts (Tubbs et al. 2018). Whether similar or different mechanisms generate ERFSs is still unknown and thus warrants further investigation.
Transcription-mediated suppression of initiation within large genes lead to CFS instability
Transcription–replication collisions and R-loop formation are not the only ways in which transcription can interfere with DNA replication. Common fragile sites (CFSs) are an example of this. These sites are under-replicated during mild replication stress, for example, in response to aphidicolin, a DNA-polymerase inhibitor that slows the progression of replication forks (Glover et al. 1984). CFSs can be visualized on metaphase spreads as ultrafine bridges between chromatids, gaps or breaks (Chan et al. 2009; Glover et al. 1984) that are hotspots for chromatid exchange (Glover and Stein 1987), chromosome deletions (Bignell et al. 2010; Pichiorri et al. 2008) and amplifications (Hellman et al. 2002; Miller et al. 2006). These regions are preferential sites for chromosome lesions (such as deletion and/or rearrangement) involved in oncogenesis, neurological disorders and viral DNA integration (see Le Tallec et al. 2014; Ozeri-Galai et al. 2014; Sarni and Kerem 2016; Debatisse and Rosselli 2019 for review). The study of CFSs is challenging due to the lack of precise genomic mapping. Traditionally, they have been mapped by conventional cytogenetic screening at a megabase scale. In lymphocytes, the number of CFSs ranges from ~ 20 (with break frequency ≥ 1%) to 230 (including CFSs with lower frequency) (Mrasek et al. 2010). Only a few of them have been mapped on a fine scale (several hundred kb) by molecular cytogenetic analysis combined with fluorescence in situ hybridization (FISH) (Savelyeva and Brueckner 2014), which is very time-consuming. Therefore, most collected data derive from isolated CFSs, which has resulted in some controversial results. In a recent study, CFSs were mapped genome-wide at a high resolution by Repli-Seq technique. The authors compared the RT of cells exposed to a low dose of aphidicolin to the RT of control cells to define the significant delayed regions (SDRs), corresponding to CFSs (Brison et al. 2019). This first genome-wide analysis has shed light on the characteristics and mechanisms responsible for CFS instability, demonstrating that stress-induced delay/under-replication is a hallmark of CFSs (Brison et al. 2019).
CFSs were long believed to be associated with particular sequences, such as stretches of AT-rich sequences that can form a secondary structure that blocks replication fork progression, impedes replication completion and leads to DNA breaks. However, recent studies have shown that CFS instability is cell-type specific, which indicates that it is directed by epigenetic features rather than by specific sequence motifs (Le Tallec et al. 2011). It has indeed been shown that such sequences at FRA3B (a well-studied CFS on chr3) do not overlap with its break boundaries (Durkin et al. 2008). CFSs are mid-late and late-replicating regions, but this is not enough to mark them (Le Beau et al. 1998; Palakodeti et al. 2004; Pelliccia et al. 2008; Hellman et al. 2000; Brison et al. 2019) as there are many more late-replicating regions than CFSs. Interestingly, most fine-mapped CFS cores are replicated in mid-late S phase (instead of late) in non-treated cells, and they become the latest replicating regions only after aphidicolin treatment (Brison et al. 2019). This finding suggests that other mechanisms rather than late-replication per se are responsible for their instability. Remarkably, CFSs are frequently associated with very long expressed genes (> 300 kb) or large transcription domains (sometimes with two or three overlapping genes), although even this is not always the case (Mitsui et al. 2010; Ohta et al. 1996; Rozier et al. 2004; Zhu et al. 2006; Helmrich et al. 2007; Denison et al. 2003; Bednarek et al. 2000; Brison et al. 2019). It has been suggested that CFSs might be caused by R-loop formation resulting from TRC (Helmrich et al. 2011). However, TRC seems unlikely as the delay of replication decreases gradually, in most cases, around both sides of CFS cores in a symmetrical way that is independent of gene orientation but instead reflects the firing time of the flanking origins (Brison et al. 2019). In addition, R-loops and fork stalling positions seem to only accumulate within highly active genes located at early-replicating regions, but not at large late-replicating genes associated with CFSs showing a modest transcription level (Liu and Chen, unpublished results). More importantly, gene transcription–replication encounters are not necessary for CFS expression, as treatments with transcription inhibitors during S phase do not rescue CFS fragility (Brison et al. 2019). Taken together, these results indicate that mechanisms other than transcription–replication encounters are responsible for the strong correlation between large genes and CFSs.
Importantly, on FRA3B (Letessier et al. 2011) and FRA16C (a CFS on chr16) (Ozeri-Galai et al. 2011), there is no (or few) activation of dormant origins to rescue stalled or slowed replication forks. This lack of activation might actually be due to the removal of replication origins by transcription (Gros et al. 2015; Powell et al. 2015). Indeed, the occupancy of components of the pre-RC is low over large genes (> 300 kb) associated with CFSs (Miotto et al. 2016; Sugimoto et al. 2018). The genome-wide analyses of replication origin distribution obtained by OK-Seq or Bubble-Seq along fine-mapped CFSs also support a model by which transcription-dependent suppression of initiation across large genes generates ultra-long (several hundreds of kb) late-replicating origin-poor regions, which delays their replication upon stress (Brison et al. 2019) (Fig. 5a). Moreover, OK-Seq data have further revealed that, in most cases, two major initiation zones flank the large transcribed genes hosting CFSs, located immediately upstream or downstream of the gene, respectively. The unidirectional forks emanating from these initiation zones travel across several hundreds of kb to complete replication of the gene body (Brison et al. 2019) (Fig. 5b). Replication could not be completed when the fork speed was reduced by aphidicolin treatment. The distance separating the initiation zones flanking the genes is therefore a major parameter for CFS setting.

Transcription-dependent suppression of initiation across large genes lead to CFS instability. a Schematic showing how gene transcription shapes the replication landscape responsible for common fragile site (CFS) instability. CFSs are genomic regions that are replicated during mid-late S phase. They are nested within large genes (> 300 Kb) whose transcription leads to the removal of pre-RC complexes from the gene body, leaving it replicated by two long-travelling unidirectional replication forks arising from its flanking regions. Under replication stress, DNA replication might not be completed within these regions. This results in a cruciform structure that must be resolved, otherwise it will lead to the expression of CFSs and genome instability. b The replication fork directionality (RFD) profile detected by Okazaki fragment sequencing (OK-Seq) along FRA16D CFS containing the large gene, WWOX (1.1 Mb). Each point shows the RFD values computed in 1 kb windows. The red and blue points indicate the regions that are predominantly replicated by rightward and leftward replication forks, respectively. The RFD profile agrees with the model shown in (a), with two strong initiation zones (identified as upward transitions on the RFD profile, indicated by the blue box) located at both extremities of the WWOX gene, and the gene body is replicated by long-travelling unidirectional replication forks (red and blue arrows, respectively). The under-replicated CFS core overlaps with the termination zone (downward transition on the RFD profile, indicated by a red box) at the gene center. A similar RFD pattern is observed in most CFSs (Brison et al. 2019)
Independently from its molecular causes, at the end of S phase, cells containing under-replicated regions link together the two sister chromatids (Fig. 5a). At this point, the resolution of these structures could be due to a series of endonucleases (Guervilly et al. 2015; Naim et al. 2013; Ying et al. 2013) that could be recruited to disassemble the replication forks (Deng et al. 2019), and can create single and/or double-strand breaks that give the cells their last chance to repair the damage during the early stages of mitosis. Importantly, several recent studies have discovered that an E3 ubiquitin-protein ligase, TRAIP (TRAF interacting protein), makes an important contribution to driving replisome disassembly during mitosis and promoting fork breakage (Sonneville et al. 2019; Wu et al. 2019; Deng et al. 2019). This event might allow factors involved in mitotic DNA synthesis (MiDAS) (Minocherhomji et al. 2015), a form of break-induced replication (BIR), to have access to the under-replicated CFSs (see Ovejero et al. 2020 for a review). The CFS is expressed if the broken DNA is not properly repaired.
Transcription-mediated replication stresses and human diseases
Defects in DNA replication processes can lead to various diseases. In the following sections, we will focus on some of the most common diseases.
Neurological disorders
R-loops can occur from a variety of cellular stresses, and lead to deleterious complications such as transcriptional irregularities, replication defects and genomic instability, relating to numerous pathologic conditions (reviewed in Richard and Manley 2017). Among them, various neurological disorders, have been linked to R-loops and gene-specific repeat expansions (Table 1).
Trinucleotide repeat expansions within intergenic regions provide additional risk for harmful R-loop formation that disrupts proper transcription and normal gene expression. For example, diseases like Huntingtin (HTT; Huntington’s disease), ataxin 1/2 (ATXN1/ATXN2; spinocerebellar ataxias) and frataxin (FXN; Friedreich ataxia), all contain GC-rich or GAA trinucleotide expansions that form R-loops in vitro and associate with disease (Reddy et al. 2011; Loomis et al. 2014). The mechanism of fragile X syndrome (FXS) is also related to the trinucleotide expansion in the 5′ UTR (Untranslated Transcribed Region) of the FMR1 gene, which leads to DNA methylation-mediated silencing of this locus (Groh et al. 2014; Colak et al. 2014). It favors the transcription-dependent R-loops, which are resistant to degradation and co-localize with repressive H3K9me2 chromatin mark. By performing a nascent nuclear run-on analysis, Groh and colleagues showed that in FXS patient cells, R-loop over-expanded repeats can block RNA polymerase II transcription of the FXN gene. In affected patients, the FMR1 allele with a (CGG)n>200 expansion in the 5′ UTR is completely methylated and transcriptionally silenced (Santoro et al. 2012; Groh et al. 2014). To test the role of such R-loop formation in trinucleotide expansion diseases, FMR1 transcription has been reactivated by using the DNA methylation inhibitor 5-aza-29-deoxycytidine (5-azadC) (Groh et al. 2014). A fourfold increase in R-loops has been observed over the exon 1 region upstream of the expansion in FXS cells, while in control cells, changes are not significant. This specificity of R-loop formation has been confirmed by RNase H treatment. These findings suggest that transcription-dependent R-loops are localized to the expanded (CGG) repeat region to regulate the expression of the FMR1 gene. Meanwhile, increasing R-loop formation leads to transcriptional repression of the FXN gene, suggesting a direct molecular association between R-loop formation and the pathology of Friedreich ataxia (FRDA) (Groh et al. 2014). The formation of R-loops over expanded repeats might, therefore, favor FXN and FMR1 silencing, and might represent a common feature of nucleotide expansion-associated diseases, contributing to the corresponding pathology in vivo (Groh et al. 2014). Interestingly, FXS cells exhibit high levels of chromosome breaks, in particular, under replication stress (Chakraborty et al. 2019). More importantly, the FMRP, the protein product of FMR1, is required for abating R-loop accumulation, thereby preventing chromosome breakage (Chakraborty et al. 2019). These data provide a detailed mechanism on the direct link between R-loop formation, replication stress and genome instability in FXS.
Active pathways that have a role in avoiding transcription–replication collisions and R-loop accumulation could be altered, leading to DNA damage and human diseases including neurological disorders (reviewed in Zeman and Cimprich 2014). For example, dysfunctional TREX1 or RNase H is responsible for Aicardi–Goutières syndrome that is characterized by severe neurological dysfunction and a congenital infection-like phenotype (Lim et al. 2015). Mutations in aprataxin (APTX), a protein present in the same pathway as RNase H, induce the neurological disorder apraxia oculomotor ataxia 1 (AOA1), characterized by cerebellar degeneration (Tumbale et al. 2014). Neurodegenerative disorders have also been associated with the loss of DNA helicase that has a clear role in the replication stress response. Of note, loss of SMARCAL1, which functions at the interface of replication and transcription (Baradaran-Heravi et al. 2012), leads to Schimke immuno-osseous dysplasia (SIOD), a multisystem disorder characterized by notable neurologic manifestations. Another example is the loss of the helicase SETX, which is involved in avoiding the formation of aberrant DNA:RNA hybrids. SETX has been associated with juvenile amyotrophic lateral sclerosis (ALS4) and ataxia–ocular apraxia (Moreira et al. 2004; Lavin et al. 2013). It should be noted that mature neurons are non-cycling cells; therefore, R-loops would either act on neurons in a replication-independent manner, or on neuron precursors link to DNA replication process. The extent by which R-loops contribute to these diseases via a replication-dependent and/or independent mechanism needs to be further investigated.
DSB repair through canonical NHEJ is important for the development of primary neural stem/progenitor cells (NSPCs) (Gao et al. 1998). Previous studies have demonstrated the presence of recurrent endogenous DSBs using genome-wide translocation sequencing (HTGTS) (Chiarle et al. 2011; Frock et al. 2015), which is a sensitive DNA break joining assay using “bait” DNA breaks introduced on different chromosomes to reveal endogenous “prey” DNA breaks. Recurrent DSB clusters (RDCs) have been mapped in NSPCs in response to replication stress induction (Wei et al. 2016, 2018). The NSPC-RDCs are enriched in the gene bodies of large (> 100 kb), late-replicating genes. Considering that these characteristics (i.e., large active genes at late-replicating regions) are often associated with CFSs, and most RDCs only present after aphidicolin treatment to induce a mild replication stress (Wei et al. 2016), a common mechanism (i.e., transcription-dependent suppression of initiation across large genes) might underlie these events.
Other studies have suggested that TRC might also function in RDC formation (reviewed in Bouwman and Crosetto 2018). Importantly, several neurodevelopmental and neuropsychiatric disorders have been linked to NSPC RDC-containing genes and the activity of neural cell adhesion and/or regulation of synapse formation. For example, molecules involved in cell–cell adhesion and neural development and growth—including the cadherin-associated proteins Ctnna2 and Ctnnd2, Cdh13 Cadherin, Cadm2, the membrane proteins Csmd1 and Csmd3, the glycoprotein Lsamp, cell adhesion molecules Mdga2, Ntm, Sdk1, Npas3, members of the neurexin family Nrxn 1/3, and the excitatory neurotransmitter receptor Grik2—are associated with numerous diseases, including attention deficit hyperactivity disorder (ADHD) (Lesch et al. 2008), intellectual disabilities (Belcaro et al. 2015; Motazacker et al. 2007), schizophrenia (Børglum et al. 2014; Donohoe et al. 2013), bipolar disorder (Ferreira et al. 2008; Nurnberger et al. 2014; Noor et al. 2014) and autism spectrum disorder (ASD) (Turner et al. 2015; Hu-Lince et al. 2005; Vaags et al. 2012; Casey et al. 2012). Interestingly, mutations linked to cerebellar ataxia and microcephaly syndrome have been found in the WW domain-containing oxidoreductase (WWOX) gene, within FRAD16, a well-studied CFS (Abdel-Salam et al. 2014; Mallaret et al. 2014). Likewise, the PARKIN (PARK2) gene, located within another CFS locus, FRA6E, is involved (via germline mutation) in Parkinson’s disease pathogenesis (Denison et al. 2003). Thus, the formation of RDCs and the CFS loci are highly associated with the gene fragility that underlies the most frequent neuronal disorders.
Cancer
The conflicts between replication and transcription are related to oncogene-induced replication stress and consequently to genomic instability, which is a hallmark of cancer (Gaillard et al. 2015; Kotsantis et al. 2016; Jones et al. 2013) (Table 2). For example, increased transcriptional activity induced by H-RAS overexpression causes replication stress, which depends on R-loop accumulation (Kotsantis et al. 2016). Using estrogen receptor-positive (ER +) breast cancer cells, Stork and colleagues showed that treating human breast cancer cells with estrogen (E2) promotes E2-activated transcription and an increase in DSBs together with R-loop formation, which colocalize particularly in regions of the genome containing estrogen-activated genes (Stork et al. 2016). In addition, replication stress induced by oncogene activation during tumorigenesis is associated with increased replication initiation within intragenic regions, leading to conflicts between replication, transcription and genomic instability (Jones et al. 2013). As described earlier, cyclin E and its subunit CDK2 form the cyclin E/CDK2 complex, the activity of which can be regulated at multiple levels and seems to be involved in triggering DNA replication initiation and in regulating genes important for proliferation and progression through the S phase (Ekholm-Reed et al. 2004). When deregulated, cyclin E is involved in tumorigenesis, and is overexpressed in many cancer types (Cooley et al. 2010; Fukuse et al. 2000; Niu et al. 2015). Importantly, somatic cells can tolerate the replication stress induced by oncogenes such as cyclin E, for several cell cycles before going through chromosomal breakage (Neelsen et al. 2013) that could constitute an initiating event in cancer. Together with cyclin E, cyclin A2 (encoded by CCNE1 and CCNA2 genes respectively) shows alterations that have been identified in a subgroup of hepatocellular carcinoma (HCC), named CCN-HCC: here, rearrangements of CCNE1 promoter regions and recurrent fusions involving CCNA2 have been identified. CCN-HCC is characterized by the accumulation of hundreds of tandem duplications and templated insertion cycles (Bayard et al. 2018). Under cyclin E overexpression, BIR, which is involved in DSB and damaged replication fork repair, is required for cell cycle progression (Costantino et al. 2014). Because chromosome rearrangements often occur during BIR upon oncogene activation (Smith et al. 2007), the rearrangements found in CCN-HCC together with the enrichment of breakpoints in early-replicated and actively transcribed regions might be associated with BIR mechanisms caused by replication stress.
The contribution of loss of BRCA1 and BRCA2 function on cancer development has been well established, particularly in breast and ovarian cancers. Tandem duplications (~ 10 kilobase length) frequently observed in BRCA1 mutant breast and ovarian cancers generated by a replication restart-bypass mechanism, which is completed by end joining or by microhomology-mediated template switching (Willis et al. 2017). This finding supports that BRCA1 and BRCA2 have an important role in protecting the replication forks (Xu et al. 2017b; Schlacher et al. 2011). When lacking the protective effects that these genes confer against replication fork collapse, cells show an increase in DSBs. These cancer cells lacking BRCA1/2 are therefore more sensitive to PARP (poly ADP ribose polymerase) inhibitors such as olaparib, rucaparib, niraparib or talazoparib, which can block another alternative repair pathway used by cells (reviewed in Ubhi and Brown 2019). PARP inhibitors are now used frequently as a targeted therapy for cancers with defective BRACA1/2 or other critical HR components, such as Rad51. Interestingly, the cancer-associated genotoxic stress that arises from mutations in BRCA1/2 can be partially rescued by overexpressing RNase H1 in cancer cell lines, suggesting that aberrant R-loop formation also contributes to malignancy (Hill et al. 2014; Hatchi et al. 2015; Zhang et al. 2017).
In addition, Ewing’s sarcoma has been linked to damage-induced transcription, an accumulation of R-loops related to transcriptional stress, and subsequent depletion of functional BRCA1, all of which ultimately results in DNA damage (Gorthi et al. 2018). Moreover, R-loops might have a role in the oncogenic c-MYC-Igh translocation commonly seen in Burkitt's lymphoma and multiple myeloma. Here, the Tudor domain-containing protein 3 (TDRD3) forms a complex with TOP3B, is recruited to the c-MYC CpG island promoter to avoid R-loop accumulation and suppresses chromosomal translocations (Küppers and Dalla-Favera 2001; Shou et al. 2000; Yang et al. 2014). Finally, cancer-derived somatic SLX4 mutations and HHS-associated germline RTEL1 mutations, abrogating the SLX4–RTEL1 interaction, affect the recruitment of FANCD2 at RNA Pol II to resolve R-loops from transcription-induced replication stress and contribute to cancer development (Takedachi et al. 2020).
As described previously, large genes expressed in NSPCs are prone to DSBs and translocations. Genes identified within RDCs are also frequently altered in different tumors (Wei et al. 2016). For example, LSAMP is contained in a small region that is frequently deleted and it has been assigned a tumor suppressor role (Kresse et al. 2009). CDH13 cadherin is involved in cell–cell adhesion activity and neural growth, and is deleted in different tumor types (Kawakami et al. 1999; Kadota et al. 2010; Sato et al. 1998). The NRXN3 synaptic cell surface protein is altered in the medulloblastoma. In prostate cancer, CADM2 and CSMD3 are rearranged and DGKB is involved in inter-chromosomal gene fusions (Berger et al. 2011; Maher et al. 2009). Moreover, a recent report found that CSMD3 and CSMD1 are included in a group of genes identified as the most frequently mutated in stomach adenocarcinoma (Wang et al. 2020). NPAS3, which helps to regulate genes that are involved in neurogenesis, is deleted in high-grade astrocytoma and glioblastoma (Moreira et al. 2011). Finally, the cell adhesion molecule BAI3 has been implicated in glioma progression (Kee et al. 2004).
Deletions in CFSs are considered as one of the major common genetic variations observed during tumor development. The first large gene discovered to be spanned by a highly unstable CFS region was fragile histidine triad (FHIT) that is located within FRA3B. FHIT alterations, such as deletions or loss of expression, have been observed in various tumors, including breast and B-cell lymphoma (Pandis et al. 1997; Kameoka et al. 2004). Another example gene spanned by the CFS region is WWOX, which is located within the second most active common fragile site FRA16D (Bednarek et al. 2000; Ludes-Meyers et al. 2003) and is frequently deleted in several tumors (Krummel et al. 2000; Paige et al. 2000). The third most frequent CFS locus is FRA6E, which contains the E3 ubiquitin gene PARK2: here, its inactivation can accelerate cell-cycle progression and induce cyclin D1 accumulation (reviewed in Glover et al. 2017). Like FHIT and WWOX, PARK2 is a tumor suppressor. Deletion of PARK2 has been described in various cancers and causes a loss of its activity (Letessier et al. 2007; Iwakawa et al. 2012; Denison et al. 2003). Loss of PARK2 activity can induce chromosome instability related to tumor formation. This effect might be due to an alteration of several mitosis regulators, such as Plk1, Aurora A/B, Cyclin B1, Cdc20, and UbcH10, which are normally controlled by PARK2. These alterations can lead to mitotic defects, such as prometaphase-like arrest, anaphase and cytokinesis failure. Given that loss of PARK2 induces multiple chromosomal defects, it seems that PARK2 has an important role in maintaining genomic stability (Lee et al. 2015).
Several CFS-associated genes have protective roles by promoting the DDR, which is a critical mechanism to maintain genome stability. Indeed, the inactivation of several tumor suppressors located within CFSs induces DDR de-regulation. In particular, the tumor suppressor FHIT as well as WWOX has a role in the DDR in regulating apoptosis, which is achieved through interactions with the pro-apoptotic p53 family of transcription factors. Thus, loss of function of these tumor suppressors, together with other gene mutations, such as in p53, have an important role in enhancing the uncontrolled proliferation that promotes genome instability (reviewed in Hazan et al. 2016). Many other genes, such as CTNNA1/3, DLG2, DMD, GRID2, IL1RAPL1, LRP1B, NBEA and RORA, which span CFS regions, are well described and linked to different tumor types (reviewed in Gao and Smith 2015). The high number of large genes contained in the CFS regions can be explained by the transcription-mediated suppression of replication initiation within these large genes (Brison et al. 2019) (see previous section for detail), creating large regions without replication initiations, and leading to genome instability under replication stress, as frequently observed in cancer.
Other pathological conditions
Transcription-mediated replication stress is also involved in a number of other pathological conditions, such as immunodeficiencies, infertility, Prader–Willi and facial anomalies syndromes (Table 3). In particular, genome instability induced by the co-transcriptional R-loop formation has been linked to FA, a genetic disease characterized by bone marrow failure and a strong predisposition to cancer. FA occurs following germline mutations that can occur in up to 22 FA genes, including BRCA1/2 (Yamamoto et al. 2005; van Twest et al. 2017; Nepal et al. 2017). Of note, FANCD2, a core FA gene, accumulates at transcribed genes and has a role in resolving R-loop and transcription–replication conflicts by recruiting RNA processing factors (Schwab et al. 2015; García-Rubio et al. 2015). Particularly, mono-ubiquitination of the FANCI–FANCD2 (ID2) heterodimer complex is due to FANCL ubiquitin E3 ligase activity occurring during S phase and under conditions of replication stress (van Twest et al. 2017; Rajendra et al. 2014). Several reports have shown the presence of increased R-loops in FA mutant cells (Schwab et al. 2015; García-Rubio et al. 2015; Liang et al. 2019), demonstrating that FANCD2 mono-ubiquitination is required to prevent their accumulation and colocalization with R-loops in an actively transcribed genomic region. Although BRCA1 and BRCA2 also belong to the FA gene family, surprisingly, breast or ovarian cancer rarely, if ever, develop in FA patients. It should be noted that FA is primarily an autosomal recessive genetic disorder, in which two mutated alleles are required to cause the disease, while BRCA1/2 defects linked to breast or ovarian cancer are mostly found in heterozygote carriers. Patients with homozygous BRCA2 depletion (BRCA2−/−) generally die from complications of aplastic anemia well before the age of developing breast or ovarian cancer. In addition, FA patients carrying BRCA1 biallelic mutations have not been identified, suggesting biallelic loss of BRCA1 might be lethal to the embryo (reviewed in D’Andrea 2010). It is not completely clear how the loss of a single DNA-repair pathway can induce bone marrow failure, developmental abnormalities and a predisposition to cancer in FA patients; we anticipate that this point will continue to be a hot topic in the field.
Prader–Willi syndrome (PWS) is a genetic disorder that is caused by the loss of paternal gene expression in the 15q11-q13 chromosomal region, due to small deletions of the SNORD116 locus. Interestingly, R-loops form within the G-rich repeats of the SNORD116 locus, inducing nucleosome displacement in a transcription-dependent manner and chromatin decondensation of the paternal allele (Powell et al. 2013). The SNORD116 locus mediates the effects of topotecan, which induces an increase in R-loops and stalling of transcriptional progression. Among the genetic syndromes characterized by immunodeficiency related to R-loop formation, centromeric region instability and facial anomalies syndrome (ICF) have been described. This syndrome is caused by mutations in the DNA methyltransferase 3B (DNMT3B) and sub-telomeric hypomethylation associated with atypically short telomere length. Transcription of telomeric repeat-containing RNA (TERRA) has an important role in regulating telomere length and its replication. Mature TERRA RNA forms DNA:RNA hybrids with the C-rich DNA template: these telomeric hybrids are present in telomerase-positive cancers (Arora et al. 2014). Moreover, in ICF cells, telomere shortening or loss, increases TERRA transcription levels, indicating that telomere hybrids are involved in promoting instability at the telomeric ICF regions. Indeed, Sagie and colleagues demonstrated that telomere hybrids enhance telomere shortening together with other unknown factors that regulate the length of telomeres, suggesting the contribution of epigenetic modifications (e.g., compromised methylation by DNMT3B) in telomere-specific length regulation (Sagie et al. 2017). Understanding the relationship between DNA:RNA hybrids, replication stress and genome instability in these disorders, and how to use such relationships to find additional targeted therapies, need to be further investigated in future studies.
Conclusion and perspectives
In conclusion, studies over the past few years have provided new and important insights into replication stress and genome instability. Increasing evidence supports that gene transcription has an essential role in shaping the landscape of human genome replication, while it is also a major source of endogenous replication stress inducing genome instability and leading to human diseases. Transcription-mediated replication stresses present at both early and late-replicating regions via two major mechanisms: head-on transcription replication conflicts frequently occur at the transcription termination sites of highly expressed genes in the early-replicating regions, while transcription-dependent suppression of initiation across large genes creating large origin-poor regions is responsible for CFS instability in the late-replicating regions. Due to technical limitations, most studies have only used cell lines as their model system. Ongoing development on high-throughput single-molecule (Müller et al. 2019; Klein et al. 2017) and single-cell (Dileep and Gilbert 2018; Takahashi et al. 2019) approaches to study the DNA replication program will provide novel tools to directly address these questions using patient samples. We expect that this advancement will bring new insights into the detailed mechanisms by which transcription-mediated replication stress impacts on genome instability and human diseases. These will help to better select the patients who will likely respond to a given targeted therapy (such as PARP, ATR or TOP1 inhibitors) targeting factors involving in the corresponding processes, and further develop new targeted therapies to better fight against cancers and other human diseases.
Abbreviations
- ARS:
-
Autonomously replicating sequence
- BIR:
-
Break-induced replication
- BRCA1/2:
-
Breast-related cancer antigen 1/2
- CD:
-
Co‑directional
- CDC45/6:
-
Cell division control protein 45/6
- CDK:
-
Cyclin-dependent kinase
- CDT1:
-
CDC10-dependent transcript 1
- CFS:
-
Common fragile site
- DDK:
-
Dbf4-Dependent Kinase;
- DDR:
-
DNA damage response
- DSB:
-
DNA double-strand break
- ER:
-
Estrogen receptor
- ERFS:
-
Early-replicating fragile site
- ESC:
-
Embryonic stem cell
- FA:
-
Fanconi anemia
- FISH:
-
Fluorescence in situ hybridization
- FXS:
-
Fragile X syndrome
- G4:
-
G quadruplex
- HO:
-
Head-on
- HR:
-
Homologous recombination
- MCM:
-
Mini-chromosome maintenance
- MiDAS:
-
Mitotic DNA synthesis
- NHEJ:
-
Non-homologous end joining
- NSPC:
-
Neural stem/progenitor cell
- ODP:
-
Origin decision point
- OGRE:
-
Origin G-rich repeated elements
- OK-seq:
-
Okazaki fragment sequencing
- ORC:
-
Origin recognition complex
- PCNA:
-
Proliferating cell nuclear antigen
- pre-IC:
-
Pre-initiation complex
- pre-RC:
-
Pre‑replication complex
- RDC:
-
Recurrent DSB cluster
- RFC:
-
Replication factor C
- Rif1:
-
Rap1-interacting factor 1
- RPA:
-
Replication protein A
- RT:
-
Replication timing
- SDR:
-
Significant delayed region
- SNS:
-
Small nascent strand
- TAD:
-
Topologically associated domain
- TDP:
-
Time decision point
- TRC:
-
Transcription–replication conflict
- TSS:
-
Transcriptional start site
- TTS:
-
Transcription termination site
- UTR:
-
Untranslated transcribed region
References
Abdel-Salam, G., Thoenes, M., Afifi, H. H., Körber, F., Swan, D., & Bolz, H. J. (2014). The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet Journal of Rare Diseases, 9, 1–7.
Aguilera, A., & García-Muse, T. (2012). R Loops: From transcription byproducts to threats to genome stability. Molecular Cell, 46, 115–124.
Ahuja, A. K., Jodkowska, K., Teloni, F., Bizard, A. H., Zellweger, R., Herrador, R., et al. (2016). A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nature Communications, 7, 1–11.
Aladjem, M. I., Spike, B. T., Rodewald, L. W., Hope, T. J., Klemm, M., Jaenisch, R., et al. (1998). ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Current Biology, 8, 145–155.
Arora, R., Lee, Y., Wischnewski, H., Brun, C. M., Schwarz, T., & Azzalin, C. M. (2014). RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nature Communications, 5, 1–11.
Ballabeni, A., Zamponi, R., Moore, J. K., Helin, K., & Kirschner, M. W. (2013). Geminin deploys multiple mechanisms to regulate Cdt1 before cell division thus ensuring the proper execution of DNA replication. Proceedings of the National Academy of Sciences U S A, 110, E2848–E2853.
Baradaran-Heravi, A., Cho, K. S., Tolhuis, B., Sanyal, M., Morozova, O., Morimoto, M., et al. (2012). Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expression. Human Molecular Genetics, 21, 2572–2587.
Barlow, J. H., Faryabi, R. B., Callén, E., Wong, N., Malhowski, A., Chen, H. T., et al. (2013). Identification of early replicating fragile sites that contribute to genome instability. Cell, 152, 620–632.
Bayard, Q., Meunier, L., Peneau, C., Renault, V., Shinde, J., Nault, J. C., et al. (2018). Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nature Communications, 9, 5235.
Beck, D. B., Burton, A., Oda, H., Ziegler-Birling, C., Torres-Padilla, M. E., & Reinberg, D. (2012). The role of PR-Set7 in replication licensing depends on Suv4-20h. Genes & Development, 26, 2580–2589.
Bednarek, A. K., Laflin, K. J., Daniel, R. L., Liao, Q., Hawkins, K. A., & Aldaz, C. M. (2000). WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Research, 60, 2140–2145.
Belcaro, C., Dipresa, S., Morini, G., Pecile, V., Skabar, A., & Fabretto, A. (2015). CTNND2 deletion and intellectual disability. Gene, 565, 146–149.
Bell, S. P., & Stillman, B. (1992). ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature, 357, 128–134.
Berger, M. F., Lawrence, M. S., Demichelis, F., Drier, Y., Cibulskis, K., Sivachenko, A. Y., et al. (2011). The genomic complexity of primary human prostate cancer. Nature, 470, 214–220.
Besnard, E., Babled, A., Lapasset, L., Milhavet, O., Parrinello, H., Dantec, C., et al. (2012). Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nature Structural & Molecular Biology, 19, 837–844.
Bianconi, E., Piovesan, A., Facchin, F., Beraudi, A., Casadei, R., Frabetti, F., et al. (2013). An estimation of the number of cells in the human body. Annals of Human Biology, 40, 463–471.
Bignell, G. R., Greenman, C. D., Davies, H., Butler, A. P., Edkins, S., Andrews, J. M., et al. (2010). Signatures of mutation and selection in the cancer genome. Nature, 463, 893–898.
Boos, D., Sanchez-Pulido, L., Rappas, M., Pearl, L. H., Oliver, A. W., Ponting, C. P., et al. (2011). Regulation of DNA replication through Sld3-Dpb11 interaction is conserved from yeast to humans. Current Biology, 21, 1152–1157.
Boos, D., Yekezare, M., & Diffley, J. F. X. (2013). Identification of a heteromeric complex that promotes DNA replication origin firing in human cells. Science, 340, 981–984.
Børglum, A. D., Demontis, D., Grove, J., Pallesen, J., Hollegaard, M. V., Pedersen, C. B., et al. (2014). Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Molecular Psychiatry, 19, 325–333.
Bouwman, B. A. M., & Crosetto, N. (2018). Endogenous DNA double-strand breaks during DNA transactions: Emerging insights and methods for genome-wide profiling. Genes, 9, 632.
Brison, O., El-Hilali, S., Azar, D., Koundrioukoff, S., Schmidt, M., Nähse, V., et al. (2019). Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nature Communications, 10, 5693.
Cadoret, J.-C., Meisch, F., Hassan-Zadeh, V., Luyten, I., Guillet, C., Duret, L., et al. (2008). Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proceedings of the National Academy of Sciences U S A, 105, 15837–15842.
Casey, J. P., Magalhaes, T., Conroy, J. M., Regan, R., Shah, N., Anney, R., et al. (2012). A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Human Genetics, 131, 565–579.
Cayrou, C., Coulombe, P., Vigneron, A., Stanojcic, S., Ganier, O., Peiffer, I., et al. (2011). Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Research, 21, 1438–1449.
Cayrou, C., Coulombe, P., Puy, A., Rialle, S., Kaplan, N., Segal, E., et al. (2012). New insights into replication origin characteristics in metazoans. Cell Cycle, 11, 658–667.
Chakraborty, P., & Grosse, F. (2011). Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair (Amst), 10, 654–665.
Chakraborty, A., Jenjaroenpun, P., McCulley, A., Li, J., El, H. S., Haarer, B., et al. (2019). Fragile X Mental Retardation Protein regulates R-loop formation and prevents global chromosome fragility. BioRxiv. https://doi.org/10.1101/601906.
Chan, K. L., Palmai-Pallag, T., Ying, S., & Hickson, I. D. (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nature Cell Biology, 11, 753–760.
Chen, Y.-H., Keegan, S., Kahli, M., Tonzi, P., Fenyö, D., Huang, T. T., et al. (2019). Transcription shapes DNA replication initiation and termination in human cells. Nature Structural & Molecular Biology, 26, 67–77.
Chiarle, R., Zhang, Y., Frock, R. L., Lewis, S. M., Molinie, B., Ho, Y. J., et al. (2011). Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell, 147, 107–119.
Cimbora, D. M., Schübeler, D., Reik, A., Hamilton, J., Francastel, C., Epner, E. M., et al. (2000). Long-distance control of origin choice and replication timing in the human beta-globin locus are independent of the locus control region. Molecular and Cellular Biology, 20, 5581–5591.
Cohen, S., Puget, N., Lin, Y.-L., Clouaire, T., Aguirrebengoa, M., Rocher, V., et al. (2018). Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nature Communications, 9, 533.
Colak, D., Zaninovic, N., Cohen, M. S., Rosenwaks, Z., Yang, W. Y., Gerhardt, J., et al. (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science, 343, 1002–1005.
Collart, C., Allen, G. E., Bradshaw, C. R., Smith, J. C., & Zegerman, P. (2013). Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science, 341, 893–896.
Cooley, A., Zelivianski, S., & Jeruss, J. S. (2010). Impact of cyclin E overexpression on Smad3 activity in breast cancer cell lines. Cell Cycle, 9, 4900–4907.
Cornacchia, D., Dileep, V., Quivy, J.-P., Foti, R., Tili, F., Santarella-Mellwig, R., et al. (2012). Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO Journal, 31, 3678–3690.
Coronado, D., Godet, M., Bourillot, P. Y., Tapponnier, Y., Bernat, A., Petit, M., et al. (2013). A short G1 phase is an intrinsic determinant of naïve embryonic stem cell pluripotency. Stem Cell Research, 10, 118–131.
Costantino, L., Sotiriou, S. K., Rantala, J. K., Magin, S., Mladenov, E., Helleday, T., et al. (2014). Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science, 343, 88–91.
Courbet, S., Gay, S., Arnoult, N., Wronka, G., Anglana, M., Brison, O., et al. (2008). Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature, 455, 557–560.
D’Alessandro, G., Whelan, D. R., Howard, S. M., Vitelli, V., Renaudin, X., Adamowicz, M., et al. (2018). BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nature Communications, 9, 1–17.
D’Andrea, A. D. (2010). Susceptibility pathways in Fanconi’s anemia and breast cancer ed. R.S. Schwartz. The New England Journal of Medicine, 362, 1909.
Debatisse, M., & Rosselli, F. (2019). A journey with common fragile sites: From S phase to telophase. Genes, Chromosomes and Cancer, 58, 305–316.
Delgado, S., Gomez, M., Bird A., & Antequera F. (1998). Initiation of DNA replication at CpG islands in mammalian chromsomes. EMBO Journal, 17, 2426–2435.
Deng, L., Wu, R. A., Sonneville, R., Kochenova, O. V., Labib, K., Pellman, D., et al. (2019). Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Molecular Cell, 73, 915–929.e6.
Denison, S. R., Callahan, G., Becker, N. A., Phillips, L. A., & Smith, D. I. (2003). Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Genes, Chromosomes and Cancer, 38, 40–52.
Dileep, V., & Gilbert, D. M. (2018). Single-cell replication profiling to measure stochastic variation in mammalian replication timing. Nature Communications, 9, 427.
Dileep, V., Ay, F., Sima, J., Vera, D. L., Noble, W. S., & Gilbert, D. M. (2015). Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Research, 25, 1104–1113.
Dileep, V., Wilson, K. A., Marchal, C., Lyu, X., Zhao, P. A., Li, B., et al. (2019). Rapid Irreversible Transcriptional Reprogramming in Human Stem Cells Accompanied by Discordance between Replication Timing and Chromatin Compartment. Stem Cell Reports, 13, 193–206.
Dimitrova, D. S., & Gilbert, D. M. (1999). The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Molecular Cell, 4, 983–993.
Donohoe, G., Walters, J., Hargreaves, A., Rose, E. J., Morris, D. W., Fahey, C., et al. (2013). Neuropsychological effects of the CSMD1 genome-wide associated schizophrenia risk variant rs10503253. Genes, Brain and Behaviour, 12, 203–209.
Durkin, S. G., Ragland, R. L., Arlt, M. F., Mulle, J. G., Warren, S. T., & Glover, T. W. (2008). Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proceedings of the National Academy of Sciences, 105, 246–251.
Ekholm-Reed, S., Méndez, J., Tedesco, D., Zetterberg, A., Stillman, B., & Reed, S. I. (2004). Deregulation of cyclin E in human cells interferes with prereplication complex assembly. Journal of Cell Biology, 165, 789–800.
El Hage, A., French, S. L., Beyer, A. L., & Tollervey, D. (2010). Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes & Development, 24, 1546–1558.
Evrin, C., Clarke, P., Zech, J., Lurz, R., Sun, J., Uhle, S., et al. (2009). A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proceedings of the National Academy of Sciences, 106, 20240–20245.
Fang, D., Lengronne, A., Shi, D., Forey, R., Skrzypczak, M., Ginalski, K., et al. (2017). Dbf4 recruitment by forkhead transcription factors defines an upstream rate-limiting step in determining origin firing timing. Genes & Development, 31, 2405–2415.
Ferreira, M. A. R., Donovan, M. C. O., Meng, Y. A., Jones, I. R., Ruderfer, D. M., Jones, L., et al. (2008). UKPMC Funders Group for ANK3 and CACNA1C in bipolar disorder. Nature Genetics, 40, 1056–1058.
Foti, R., Gnan, S., Cornacchia, D., Dileep, V., Bulut-Karslioglu, A., Diehl, S., et al. (2016). Nuclear architecture organized by Rif1 underpins the replication-timing program. Molecular Cell, 61, 260–273.
Frock, R. L., Hu, J., Meyers, R. M., Ho, Y.-J., Kii, E., & Alt, F. W. (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology, 33, 179–186.
Fukuse, T., Hirata, T., Naiki, H., Hitomi, S., & Wada, H. (2000). Prognostic significance of cyclin E overexpression in resected non-small cell lung cancer. Cancer Research, 60, 242–244.
Gaillard, H., Garcia-Muse, T., & Aguilera, A. (2015). Replication stress and cancer. Nature Reviews Cancer, 15, 276–289.
Gambus, A., Jones, R. C., Sanchez-Diaz, A., Kanemaki, M., van Deursen, F., Edmondson, R. D., et al. (2006). GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nature Cell Biology, 8, 358–366.
Ganier, O., Prorok, P., Akerman, I., & Méchali, M. (2019). Metazoan DNA replication origins. Current Opinion in Cell Biology, 58, 134–141.
Gao, G., & Smith, D. I. (2015). WWOX, large common fragile site genes, and cancer. Experimental Biology and Medicine, 240, 285–295.
Gao, Y., Sun, Y., Frank, K. M., Dikkes, P., Fujiwara, Y., Seidl, K. J., et al. (1998). A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell, 95, 891–902.
García-Rubio, M. L., Pérez-Calero, C., Barroso, S. I., Tumini, E., Herrera-Moyano, E., Rosado, I. V., et al. (2015). The Fanconi anemia pathway protects genome integrity from R-loops ed. J. Sekelsky. PLoS Genetics, 11, e1005674.
Ge, X. Q., Jackson, D. A., & Blow, J. J. (2007). Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes & Development, 21, 3331–3341.
Glover, T. W., & Stein, C. K. (1987). Induction of sister chromatid exchanges at common fragile sites. American Journal of Human Genetics, 41, 882–890.
Glover, T. W., Berger, C., Coyle, J., & Echo, B. (1984). DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Human Genetics, 67, 136–142.
Glover, T. W., Wilson, T. E., & Arlt, M. F. (2017). Fragile sites in cancer: More than meets the eye. Nature Reviews Cancer, 17, 489–501.
Gnan, S., Flyamer, I. M., Klein, K. N., Castelli, E., Turner, J. L., Weber, P., et al. (2019). Nuclear organisation and replication timing are coupled through RIF1-PP1 interaction. bioRxiv. https://doi.org/10.1101/812156.
Gómez-González, B., García-Rubio, M., Bermejo, R., Gaillard, H., Shirahige, K., Marín, A., et al. (2011). Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO Journal, 30, 3106–3119.
Gorthi, A., Romero, J. C., Loranc, E., Cao, L., Lawrence, L. A., Goodale, E., et al. (2018). EWS-FLI1 increases transcription to cause R-Loops and block BRCA1 repair in Ewing sarcoma. Nature, 555, 387–391.
Groh, M., Lufino, M. M. P., Wade-Martins, R., & Gromak, N. (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich Ataxia and Fragile X Syndrome. PLoS Genetics, 10, e1004318.
Groh, M., Albulescu, L. O., Cristini, A., & Gromak, N. (2017). Senataxin: genome guardian at the interface of transcription and neurodegeneration. Journal of Molecular Biology, 429, 3181–3195.
Gros, J., Kumar, C., Lynch, G., Yadav, T., Whitehouse, I., & Remus, D. (2015). Post-licensing specification of eukaryotic replication origins by facilitated Mcm2-7 sliding along DNA. Molecular Cell, 60, 797–807.
Guervilly, J. H., Takedachi, A., Naim, V., Scaglione, S., Chawhan, C., Lovera, Y., et al. (2015). The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Molecular Cell, 57, 123–137.
Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., & Cimprich, K. A. (2017). Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell, 170, 774–786.e19.
Hasegawa, Y., Yamamoto, M., Miyamori, J., & Kanoh, J. (2019). Telomere DNA length-dependent regulation of DNA replication timing at internal late replication origins. Scientific Report, 9, 1–9.
Hatchi, E., Skourti-Stathaki, K., Ventz, S., Pinello, L., Yen, A., Kamieniarz-Gdula, K., et al. (2015). BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Molecular Cell, 57, 636–647.
Hayashi, M. T., Takahashi, T. S., Nakagawa, T., Nakayama, J. I., & Masukata, H. (2009). The heterochromatin protein Swi6/HP1 activates replication origins at the pericentromeric region and silent mating-type locus. Nature Cell Biology, 11, 357–362.
Hazan, I., Hofmann, T. G., & Aqeilan, R. I. (2016). Tumor suppressor genes within common fragile sites are active players in the DNA damage response. PLoS Genetics, 12, 1–19.
Heller, R. C., Kang, S., Lam, W. M., Chen, S., Chan, C. S., & Bell, S. P. (2011). Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell, 146, 80–91.
Hellman, A., Rahat, A., Scherer, S. W., Darvasi, A., Tsui, L.-C., & Kerem, B. (2000). Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Molecular and Cellular Biology, 20, 4420–4427.
Hellman, A., Zlotorynski, E., Scherer, S. W., Cheung, J., Vincent, J. B., Smith, D. I., et al. (2002). A role for common fragile site induction in amplification of human oncogenes. Cancer Cell, 1, 89–97.
Helmrich, A., Stout-Weider, K., Matthaei, A., Hermann, K., Heiden, T., & Schrock, E. (2007). Identification of the human/mouse syntenic common fragile site FRA7K/Fra12C1—Relation of FRA7K and other human common fragile sites on chromosome 7 to evolutionary breakpoints. International Journal of Cancer, 120, 48–54.
Helmrich, A., Ballarino, M., & Tora, L. (2011). Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Molecular Cell, 44, 966–977.
Hill, S. J., Rolland, T., Adelmant, G., Xia, X., Owen, M. S., Dricot, A., et al. (2014). Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes & Development, 28, 1957–1975.
Hiraga, S. I., Alvino, G. M., Chang, F., Lian, H. Y., Sridhar, A., Kubota, T., et al. (2014). Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7- mediated phosphorylation of the MCM complex. Genes & Development, 28, 372–383.
Hiratani, I., Ryba, T., Itoh, M., Yokochi, T., Schwaiger, M., Chang, C.-W., et al. (2008). Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biology, 6, e245.
Hu-Lince, D., Craig, D. W., Huentelman, M. J., & Stephan, D. A. (2005). The Autism Genome Project: Goals and strategies. American Journal of PharmacoGenomics, 5, 233–246.
Ilves, I., Petojevic, T., Pesavento, J. J., & Botchan, M. R. (2010). Activation of the MCM2-7 Helicase by Association with Cdc45 and GINS Proteins. Molecular Cell, 37, 247–258.
Iwakawa, R., Okayama, H., Kohno, T., Sato-Otsubo, A., Ogawa, S., & Yokota, J. (2012). Contribution of germline mutations to PARK2 gene inactivation in lung adenocarcinoma. Genes, Chromosomes and Cancer, 51, 462–472.
Jares, P., & Blow, J. J. (2000). Xenopus Cdc7 function is dependent on licensing but not on XORC, XCdc6, or CDK activity and is required for XCdc45 loading. Genes & Development, 14, 1528–1540.
Jirmanova, L., Afanassieff, M., Gobert-Gosse, S., Markossian, S., & Savatier, P. (2002). Differential contributions of ERK and PI3-kinase to the regulation of cyclin D1 expression and to the control of the G1/S transition in mouse embryonic stem cells. Oncogene, 21, 5515–5528.
Jones, R. M., Mortusewicz, O., Afzal, I., Lorvellec, M., García, P., Helleday, T., et al. (2013). Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene, 32, 3744–3753.
Kabeche, L., Nguyen, H. D., Buisson, R., & Zou, L. (2018). A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science, 359, 108–114.
Kadota, M., Yang, H. H., Gomez, B., Sato, M., Clifford, R. J., Meerzaman, D., et al. (2010). Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS ONE, 5, 1–10.
Kameoka, Y., Tagawa, H., Tsuzuki, S., Karnan, S., Ota, A., Suguro, M., et al. (2004). Contig array CGH at 3p14.2 points to the FRA3B/FHIT common fragile region as the target gene in diffuse large B-cell lymphoma. Oncogene, 23, 9148–9154.
Kanemaki, M., & Labib, K. (2006). Distinct roles for Sld3 and GINS during establishment and progression of eukaryotic DNA replication forks. EMBO Journal, 25, 1753–1763.
Kanke, M., Kodama, Y., Takahashi, T. S., Nakagawa, T., & Masukata, H. (2012). Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO Journal, 31, 2182–2194.
Kanoh, J., & Ishikawa, F. (2001). spRap1 and spRif1, recruited to telomeres by Taz1, are essential for telomere function in fission yeast. Current Biology, 11, 1624–1630.
Kawakami, M., Staub, J., Cliby, W., Hartmann, L., Smith, D. I., & Shridhar, V. (1999). Involvement of H-cadherin (CDH13) on 16q in the region of frequent deletion in ovarian cancer. International Journal of Oncology, 15, 715–720.
Kee, H. J., Ahn, K. Y., Choi, K. C., Won Song, J., Heo, T., Jung, S., et al. (2004). Expression of brain-specific angiogenesis inhibitor 3 (BAI3) in normal brain and implications for BAI3 in ischemia-induced brain angiogenesis and malignant glioma. FEBS Letters, 569, 307–316.
Klein, K., Wang, W., Borrman, T., Chan, S., Zhang, D., Weng, Z., et al. (2017). Genome-wide identification of early-firing human replication origins by optical replication mapping. bioRxiv. https://doi.org/10.1101/214841.
Knott, S. R. V., Peace, J. M., Ostrow, A. Z., Gan, Y., Rex, A. E., Viggiani, C. J., et al. (2012). Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell, 148, 99–111.
Kotsantis, P., Silva, L. M., Irmscher, S., Jones, R. M., Folkes, L., Gromak, N., et al. (2016). Increased global transcription activity as a mechanism of replication stress in cancer. Nature Communications, 7, 13087.
Kresse, S. H., Ohnstad, H. O., Paulsen, E. B., Bjerkehagen, B., Szuhai, K., Serra, M., et al. (2009). LSAMP, a novel candidate tumor suppressor gene in human osteosarcomas, identified by array comparative genomic hybridization. Genes, Chromosomes and Cancer, 48, 679–693.
Krummel, K. A., Roberts, L. R., Kawakami, M., Glover, T. W., & Smith, D. I. (2000). The characterization of the common fragile site FRA16D and its involvement in multiple myeloma translocations. Genomics, 69, 37–46.
Kumagai, A., & Dunphy, W. G. (2017). MTBP, the partner of Treslin, contains a novel DNA-binding domain that is essential for proper initiation of DNA replication. Molecular Biology of the Cell, 28, 2998–3012.
Kumagai, A., Shevchenko, A., Shevchenko, A., & Dunphy, W. G. (2011). Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. Journal of Cell Biology, 193, 995–1007.
Kuo, A. J., Song, J., Cheung, P., Ishibe-Murakami, S., Yamazoe, S., Chen, J. K., et al. (2012). The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature, 484, 115–119.
Küppers, R., & Dalla-Favera, R. (2001). Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene, 20, 5580–5594.
Lavin, M., Yeo, A., & Becherel, O. (2013). Senataxin protects the genome: Implications for neurodegeneration and other abnormalities. Rare Diseases, 1, e25230.
Le Beau, M. M., Rassool, F. V., Neilly, M. E., Espinosa, R., Glover, T. W., Smith, D. I., et al. (1998). Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Human Molecular Genetics, 7, 755–761.
Le Tallec, B., Dutrillaux, B., Lachages, A.-M., Millot, G. A., Brison, O., & Debatisse, M. (2011). Molecular profiling of common fragile sites in human fibroblasts. Nature Structural & Molecular Biology, 18, 1421–1423.
Le Tallec, B., Koundrioukoff, S., Wilhelm, T., Letessier, A., Brison, O., & Debatisse, M. (2014). Updating the mechanisms of common fragile site instability: how to reconcile the different views? Cellular and Molecular Life Sciences, 71, 4489–4494.
Lee, S. B., Kim, J. J., Nam, H. J., Gao, B., Yin, P., Qin, B., et al. (2015). Parkin Regulates Mitosis and Genomic Stability through Cdc20/Cdh1. Molecular Cell, 60, 21–34.
Lesch, K. P., Timmesfeld, N., Renner, T. J., Halperin, R., Röser, C., Nguyen, T. T., et al. (2008). Molecular genetics of adult ADHD: Converging evidence from genome-wide association and extended pedigree linkage studies. Journal of Neural Transmission, 115, 1573–1585.
Letessier, A., Garrido-Urbani, S., Ginestier, C., Fournier, G., Esterni, B., Monville, F., et al. (2007). Correlated break at PARK2/FRA6E and loss of AF-6/Afadin protein expression are associated with poor outcome in breast cancer. Oncogene, 26, 298–307.
Letessier, A., Millot, G. A., Koundrioukoff, S., Lachagès, A.-M., Vogt, N., Hansen, R. S., et al. (2011). Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature, 470, 120–123.
Li, C.-J., & DePamphilis, M. L. (2002). Mammalian Orc1 Protein Is Selectively Released from Chromatin and Ubiquitinated during the S-to-M Transition in the Cell Division Cycle. Molecular and Cellular Biology, 22, 105–116.
Li, X., & Manley, J. L. (2005). Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell, 122, 365–378.
Li, F., Chen, J., Izumi, M., Butler, M. C., Keezer, S. M., & Gilbert, D. M. (2001). The replication timing program of the Chinese hamster β-globin locus is established coincident with its repositioning near peripheral heterochromatin in early G1 phase. Journal of Cell Biology, 154, 283–292.
Li, F., Chen, J., Solessio, E., & Gilbert, D. M. (2003). Spatial distribution and specification of mammalian replication origins during G1 phase. Journal of Cell Biology, 161, 257–266.
Li, X., Niu, T., & Manley, J. L. (2007). The RNA binding protein RNPS1 alleviates ASF/SF2 depletion-induced genomic instability. RNA, 13, 2108–2115.
Li, V. C., Ballabeni, A., & Kirschner, M. W. (2012). Gap 1 phase length and mouse embryonic stem cell self-renewal. Proceedings of National Academy of Sciences U S A, 109, 12550–12555.
Liang, Z., Liang, F., Teng, Y., Chen, X., Liu, J., Longerich, S., et al. (2019). Binding of FANCI-FANCD2 Complex to RNA and R-Loops Stimulates Robust FANCD2 Monoubiquitination. Cell Reports, 26, 564–572.e5.
Lim, Y. W., Sanz, L. A., Xu, X., Hartono, S. R., & Chédin, F. (2015). Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutières syndrome. Elife, 4, e08007.
Long, H., Zhang, L., Lv, M., Wen, Z., Zhang, W., Chen, X., et al. (2020). H2A.Z facilitates licensing and activation of early replication origins. Nature, 577, 576–581.
Loomis, E. W., Sanz, L. A., Chédin, F., & Hagerman, P. J. (2014). Transcription-associated R-Loop formation across the human FMR1 CGG-repeat region. PLoS Genetics, 10, e1004294.
Lu, J., Li, F., Murphy, C. S., Davidson, M. W., & Gilbert, D. M. (2010). G2 phase chromatin lacks determinants of replication timing. Journal of Cell Biology, 189, 967–980.
Ludes-Meyers, J. H., Bednarek, A. K., Popescu, N. C., Bedford, M., & Aldaz, C. M. (2003). WWOX, the common chromosomal fragile site, FRA16D, cancer gene. Cytogenetic and Genome Research, 100, 101–110.
Macheret, M., & Halazonetis, T. D. (2018). Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature, 555, 112–116.
MacNeill, S. (2012). Composition and dynamics of the eukaryotic replisome: A brief overview. SubCellular Biochemistry, 62, 1–17.
Maher, C. A., Kumar-Sinha, C., Cao, X., Kalyana-Sundaram, S., Han, B., Jing, X., et al. (2009). Transcriptome sequencing to detect gene fusions in cancer. Nature, 458, 97–101.
Maiorano, D., Moreau, J., & Méchali, M. (2000). XCDT1 is required for the assembly of pre-replicative complexes in Xenopus laevis. Nature, 404, 622–625.
Mallaret, M., Synofzik, M., Lee, J., Sagum, C. A., Mahajnah, M., Sharkia, R., et al. (2014). The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain, 137, 411–419.
Mantiero, D., MacKenzie, A., Donaldson, A., & Zegerman, P. (2011). Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO Journal, 30, 4805–4814.
Marahrens, Y., & Stillman, B. (1992). A yeast chromosomal origin of DNA replication defined by multiple functional elements. Science, 255, 817–823.
Matos, D. A., Zhang, J. M., Ouyang, J., Nguyen, H. D., Genois, M. M., & Zou, L. (2020). ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Molecular Cell, 77, 514–527.e4.
Mattarocci, S., Shyian, M., Lemmens, L., Damay, P., Altintas, D. M., Shi, T., et al. (2014). Rif1 Controls DNA replication timing in yeast through the PP1 Phosphatase Glc7. Cell Reports, 7, 62–69.
Méndez, J., Zou-Yang, X. H., Kim, S. Y., Hidaka, M., Tansey, W. P., & Stillman, B. (2002). Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Molecular Cell, 9, 481–491.
Miller, C. T., Lin, L., Casper, A. M., Lim, J., Thomas, D. G., Orringer, M. B., et al. (2006). Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene, 25, 409–418.
Miller, T. C. R., Locke, J., Greiwe, J. F., Diffley, J. F. X., & Costa, A. (2019). Mechanism of head-to-head MCM double-hexamer formation revealed by cryo-EM. Nature, 575, 704–710.
Minocherhomji, S., Ying, S., Bjerregaard, V. A., Bursomanno, S., Aleliunaite, A., Wu, W., et al. (2015). Replication stress activates DNA repair synthesis in mitosis. Nature, 528, 286–290.
Miotto, B., Ji, Z., & Struhl, K. (2016). Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proceedings of the National Academy of Sciences U S A, 113, E4810–E4819.
Mitsui, J., Takahashi, Y., Goto, J., Tomiyama, H., Ishikawa, S., Yoshino, H., et al. (2010). Mechanisms of Genomic Instabilities Underlying Two Common Fragile-Site-Associated Loci, PARK2 and DMD, in Germ Cell and Cancer Cell Lines. American Journal of Human Genetics, 87, 75–89.
Moreira, M. C., Klur, S., Watanabe, M., Németh, A. H., Le Ber, I., Moniz, J. C., et al. (2004). Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nature Genetics, 36, 225–227.
Moreira, F., Kiehl, T. R., So, K., Ajeawung, N. F., Honculada, C., Gould, P., et al. (2011). NPAS3 demonstrates features of a tumor suppressive role in driving the progression of astrocytomas. American Journal of Pathology, 179, 462–476.
Motazacker, M. M., Rost, B. R., Hucho, T., Garshasbi, M., Kahrizi, K., Ullmann, R., et al. (2007). A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. American Journal of Human Genetics, 81, 792–798.
Mrasek, K., Schoder, C., Teichmann, A.-C., Behr, K., Franze, B., Wilhelm, K., et al. (2010). Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. International Journal of Oncology, 36, 929–940.
Müller, C. A., Boemo, M. A., Spingardi, P., Kessler, B. M., Kriaucionis, S., Simpson, J. T., et al. (2019). Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads. Nature Methods, 16, 429–436.
Muramatsu, S., Hirai, K., Tak, Y. S., Kamimura, Y., & Araki, H. (2010). CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ε, and GINS in budding yeast. Genes & Development, 24, 602–612.
Naim, V., Wilhelm, T., Debatisse, M., & Rosselli, F. (2013). ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nature Cell Biology, 15, 1008–1015.
Natsume, T., Müller, C. A., Katou, Y., Retkute, R., Gierliński, M., Araki, H., et al. (2013). Kinetochores coordinate pericentromeric cohesion and early DNA replication by Cdc7-Dbf4 kinase recruitment. Molecular Cell, 50, 661–674.
Neelsen, K. J., Zanini, I. M. Y., Herrador, R., & Lopes, M. (2013). Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. Journal of Cell Biology, 200, 699–708.
Nepal, M., Che, R., Ma, C., Zhang, J., & Fei, P. (2017). FANCD2 and DNA damage. International Journal of Molecular Sciences, 18, 1–9.
Nguyen, H. D., Leong, W. Y., Li, W., Reddy, P. N. G., Sullivan, J. D., Walter, M. J., et al. (2018). Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Research, 78, 5363–5374.
Nguyen, V. Q., Co, C., Irie, K., & Li, J. J. (2000). Clb/Cdc28 kinases promote nuclear export of the replication initiator proteins Mcm2-7. Current Biology, 10, 195–205.
Nishitani, H., Lygerou, Z., Nishimoto, T., & Nurse, P. (2000). The Cdt1 protein is required to license DNA for replication in fission yeast. Nature, 404, 625–628.
Niu, D., Wang, G., & Wang, X. (2015). Up-regulation of cyclin E in breast cancer via estrogen receptor pathway. International Journal of Clinical and Experimental Medicine, 8, 910–915.
Noor, A., Lionel, A. C., Cohen-Woods, S., Moghimi, N., Rucker, J., Fennell, A., et al. (2014). Copy number variant study of bipolar disorder in Canadian and UK populations implicates synaptic genes. American Journal of Medical Genetics Part B: Neuropsychiatrics Genetics, 165, 303–313.
Nora, E. P., Goloborodko, A., Valton, A. L., Gibcus, J. H., Uebersohn, A., Abdennur, N., et al. (2017). Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell, 169, 930–944.e22.
Nurnberger, J. I., Koller, D. L., Jung, J., Edenberg, H. J., Foroud, T., Guella, I., et al. (2014). Identification of pathways for bipolar disorder. JAMA Psychiatry, 71, 657.
Ogawa, S., Kido, S., Handa, T., Ogawa, H., Asakawa, H., Takahashi, T. S., et al. (2018). Shelterin promotes tethering of late replication origins to telomeres for replication-timing control. EMBO Journal, 37, e98997.
Ohta, M., Inoue, H., Cotticelli, M. G., Kastury, K., Baffa, R., Palazzo, J., et al. (1996). The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell, 84, 587–597.
Oldach, P., & Nieduszynski, C. A. (2019). Cohesin-mediated genome architecture does not define DNA replication timing domains. Genes, 10, 196.
Ovejero, S., Bueno, A., & Sacristán, M. P. (2020). Working on genomic stability: From the S-phase to mitosis. Genes, 11, 225.
Ozeri-Galai, E., Lebofsky, R., Rahat, A., Bester, A. C., Bensimon, A., & Kerem, B. (2011). Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Molecular Cell, 43, 122–131.
Ozeri-Galai, E., Tur-Sinai, M., Bester, A. C., & Kerem, B. (2014). Interplay between genetic and epigenetic factors governs common fragile site instability in cancer. Cellular and Molecular Life Sciences, 71, 4495–4506.
Paige, A. J. W., Taylor, K. J., Stewart, A., Sgouros, J. G., Gabra, H., Sellar, G. C., et al. (2000). A 700-kb physical map of a region of 16q23.2 homozygously deleted in multiple cancers and spanning the common fragile site FRA16D. Cancer Research, 60, 1690–1697.
Palakodeti, A., Han, Y., Jiang, Y., & Le Beau, M. M. (2004). The role of late/slow replication of the FRA16D in common fragile site induction. Genes, Chromosomes and Cancer, 39, 71–76.
Pandis, N., Bardi, G., Mitelman, F., & Heim, S. (1997). Deletion of the short arm of chromosome 3 in breast tumors. Genes, Chromosomes and Cancer, 18, 241–245.
Pefanis, E., Wang, J., Rothschild, G., Lim, J., Kazadi, D., Sun, J., et al. (2015). RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell, 161, 774–789.
Pelliccia, F., Bosco, N., Curatolo, A., & Rocchi, A. (2008). Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenetic and Genome Research, 121, 196–200.
Petersen, B. O., Lukas, J., Sørensen, C. S., Bartek, J., & Helin, K. (1999). Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO Journal, 18, 396–410.
Petersen, B. O., Wagener, C., Marinoni, F., Kramer, E. R., Melixetian, M., Denchi, E. L., et al. (2000). Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes & Development, 14, 2330–2343.
Petryk, N., Kahli, M., D’Aubenton-Carafa, Y., Jaszczyszyn, Y., Shen, Y., Silvain, M., et al. (2016). Replication landscape of the human genome. Nature Communications, 7, 10208.
Pichiorri, F., Ishii, H., Okumura, H., Trapasso, F., Wang, Y., & Huebner, K. (2008). Molecular parameters of genome instability: Roles of fragile genes at common fragile sites. Journal of Cellular Biochemistry, 104, 1525–1533.
Poh, W. T., Chadha, G. S., Gillespie, P. J., Kaldis, P., & Blow, J. J. (2014). Xenopus Cdc7 executes its essential function early in S phase and is counteracted by checkpoint-regulated protein phosphatase 1. Open Biology, 4, 130138–130138.
Pope, B. D., Ryba, T., Dileep, V., Yue, F., Wu, W., Denas, O., et al. (2014). Topologically associating domains are stable units of replication-timing regulation. Nature, 515, 402–405.
Powell, W. T., Coulson, R. L., Gonzales, M. L., Crary, F. K., Wong, S. S., Adams, S., et al. (2013). R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proceedings of the National Academy of Sciences U S A, 110, 13938–13943.
Powell, S. K., MacAlpine, H. K., Prinz, J. A., Li, Y., Belsky, J. A., & MacAlpine, D. M. (2015). Dynamic loading and redistribution of the Mcm2-7 helicase complex through the cell cycle. EMBO Journal, 34, 531–543.
Prioleau, M.-N., & MacAlpine, D. M. (2016). DNA replication origins? Where do we begin? Genes & Development, 30, 1683–1697.
Promonet, A., Padioleau, I., Liu, Y., Sanz, L., Biernacka, A., Schmitz, A., et al. (2020). Topoisomerase 1 prevents R-loop mediated replication stress at transcription termination sites. Nature Communications. https://doi.org/10.1038/s41467-020-17858-2.
Rajendra, E., Oestergaard, V. H., Langevin, F., Wang, M., Dornan, G. L., Patel, K. J., et al. (2014). The genetic and biochemical basis of FANCD2 monoubiquitination. Molecular Cell, 54, 858–869.
Rao, S. S. P., Huang, S. C., Glenn St Hilaire, B., Engreitz, J. M., Perez, E. M., Kieffer-Kwon, K. R., et al. (2017). Cohesin loss eliminates all loop domains. Cell, 171, 305–320.e24.
Reddy, K., Tam, M., Bowater, R. P., Barber, M., Tomlinson, M., Nichol Edamura, K., et al. (2011). Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Research, 39, 1749–1762.
Richard, P., & Manley, J. L. (2017). R loops and links to human disease. Journal of Molecular Biology, 429, 3168–3180.
Rivera-Mulia, J. C., Buckley, Q., Sasaki, T., Zimmerman, J., Didier, R. A., Nazor, K., et al. (2015). Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Research, 25, 1091–1103.
Rozier, L., El-Achkar, E., Apiou, F., & Debatisse, M. (2004). Characterization of a conserved aphidicolin-sensitive common fragile site at human 4q22 and mouse 6C1: Possible association with an inherited disease and cancer. Oncogene, 23, 6872–6880.
Ryba, T., Hiratani, I., Lu, J., Itoh, M., Kulik, M., Zhang, J., et al. (2010). Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Research, 20, 761–770.
Sagie, S., Edni, O., Weinberg, J., Toubiana, S., Kozlovski, T., Frostig, T., et al. (2017). Non-random length distribution of individual telomeres in immunodeficiency, centromeric instability and facial anomalies syndrome, type I. Human Molecular Genetics, 26, 4244–4256.
Sangrithi, M. N., Bernal, J. A., Madine, M., Philpott, A., Lee, J., Dunphy, W. G., et al. (2005). Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell, 121, 887–898.
Santocanale, C., Sharma, K., & Diffley, J. F. X. (1999). Activation of dormant origins of DNA replication in budding yeast. Genes & Development, 13, 2360–2364.
Santoro, M. R., Bray, S. M., & Warren, S. T. (2012). Molecular mechanisms of fragile X syndrome: A twenty-year perspective. Annual Review of Pathology Mechanisms of Disease, 7, 219–245.
Sanz, L. A., Hartono, S. R., Lim, Y. W., Steyaert, S., Rajpurkar, A., Ginno, P. A., et al. (2016). Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Molecular Cell, 63, 167–178.
Sarni, D., & Kerem, B. (2016). The complex nature of fragile site plasticity and its importance in cancer. Current Opinion in Cell Biology, 40, 131–136.
Sato, M., Mori, Y., Sakurada, A., Fujimura, S., & Horii, A. (1998). The H-cadherin (CDH13) gene is inactivated in human lung cancer. Human Genetics, 103, 96–101.
Savatier, P., Huang, S., Szekely, L., Wiman, K. G., & Samarut, J. (1994). Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene, 9, 809–818.
Savatier, P., Lapillonne, H., Van Grunsven, L. A., Rudkin, B. B., & Samarut, J. (1996). Withdrawal of differentiation inhibitory activity/leukemia inhibitory factor up-regulates D-type cyclins and cyclin-dependent kinase inhibitors in mouse embryonic stem cells. Oncogene, 12, 309–322.
Savelyeva, L., & Brueckner, L. M. (2014). Molecular characterization of common fragile sites as a strategy to discover cancer susceptibility genes. Cellular and Molecular Life Sciences, 71, 4561–4575.
Schlacher, K., Christ, N., Siaud, N., Egashira, A., Wu, H., & Jasin, M. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell, 145, 529–542.
Schwab, R. A., Nieminuszczy, J., Shah, F., Langton, J., Lopez Martinez, D., Liang, C.-C., et al. (2015). The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Molecular Cell, 60, 351–361.
Sequeira-Mendes, J., Díaz-Uriarte, R., Apedaile, A., Huntley, D., Brockdorff, N., & Gómez, M. (2009). Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genetics, 5, e1000446.
Shivji, M. K. K., Renaudin, X., Williams, Ç. H., & Venkitaraman, A. R. (2018). BRCA2 regulates transcription elongation by RNA polymerase II to prevent R-loop accumulation. Cell Reports, 22, 1031–1039.
Shou, Y., Martelli, M. L., Gabrea, A., Qi, Y., Brents, L. A., Roschke, A., et al. (2000). Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proceedings of the National Academy of Sciences U S A, 97, 228–233.
Sima, J., Chakraborty, A., Dileep, V., Michalski, M., Klein, K. N., Holcomb, N. P., et al. (2019). Identifying cis elements for spatiotemporal control of mammalian DNA replication. Cell, 176, 816–830.e18.
Siu, K. T., Rosner, M. R., & Minella, A. C. (2012). An integrated view of cyclin E function and regulation. Cell Cycle, 11, 57–64.
Skourti-Stathaki, K., & Proudfoot, N. J. (2014). A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes & Development, 28, 1384–1396.
Skourti-Stathaki, K., Proudfoot, N. J., & Gromak, N. (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Molecular Cell, 42, 794–805.
Smith, C. E., Llorente, B., & Symington, L. S. (2007). Template switching during break-induced replication. Nature, 447, 102–105.
Sollier, J., Stork, C. T., García-Rubio, M. L., Paulsen, R. D., Aguilera, A., & Cimprich, K. A. (2014). Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Molecular Cell, 56, 777–785.
Sonneville, R., Bhowmick, R., Hoffmann, S., Mailand, N., Hickson, I. D., & Labib, K. (2019). TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. Elife, 8, e48686.
Speck, C., & Stillman, B. (2007). Cdc6 ATPase activity regulates ORC·Cdc6 stability and the selection of specific DNA sequences as origins of DNA replication. Journal of Biological Chemistry, 282, 11705–11714.
Stead, E., White, J., Faast, R., Conn, S., Goldstone, S., Rathjen, J., et al. (2002). Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene, 21, 8320–8333.
Stork, C. T., Bocek, M., Crossley, M. P., Sollier, J., Sanz, L. A., Chédin, F., et al. (2016). Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife, 5, e17548.
Sugimoto, N., Maehara, K., Yoshida, K., Ohkawa, Y., & Fujita, M. (2018). Genome-wide analysis of the spatiotemporal regulation of firing and dormant replication origins in human cells. Nucleic Acids Research, 46, 6683–6696.
Sukackaite, R., Cornacchia, D., Jensen, M. R., Mas, P. J., Blackledge, M., Enervald, E., et al. (2017). Mouse Rif1 is a regulatory subunit of protein phosphatase 1 (PP1). Scientific Reports, 7, 2119.
Takahashi, S., Miura, H., Shibata, T., Nagao, K., Okumura, K., Ogata, M., et al. (2019). Genome-wide stability of the DNA replication program in single mammalian cells. Nature Genetics, 51, 529–540.
Takedachi, A., Despras, E., Scaglione, S., Guérois, R., Guervilly, J. H., Blin, M., et al. (2020). SLX4 interacts with RTEL1 to prevent transcription-mediated DNA replication perturbations. Nature Structural & Molecular Biology, 27, 438–449.
Tanaka, S., Umemori, T., Hirai, K., Muramatsu, S., Kamimura, Y., & Araki, H. (2007). CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature, 445, 328–332.
Tanaka, S., Nakato, R., Katou, Y., Shirahige, K., & Araki, H. (2011). Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Current Biology, 21, 2055–2063.
Tazumi, A., Fukuura, M., Nakato, R., Kishimoto, A., Takenaka, T., Ogawa, S., et al. (2012). Telomere-binding protein Taz1 controls global replication timing through its localization near late replication origins in fission yeast. Genes & Development, 26, 2050–2062.
Thomas, M., White, R. L., & Davis, R. W. (1976). Hybridization of RNA to double stranded DNA: Formation of R loops. Proceedings of the National Academy of Sciences U S A, 73, 2294–2298.
Tubbs, A., Sridharan, S., van Wietmarschen, N., Maman, Y., Callen, E., Stanlie, A., et al. (2018). Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell, 174, 1127–1142.e19.
Tumbale, P., Williams, J. S., Schellenberg, M. J., Kunkel, T. A., & Williams, R. S. (2014). Aprataxin resolves adenylated RNA–DNA junctions to maintain genome integrity. Nature, 506, 111–115.
Turner, T. N., Sharma, K., Oh, E. C., Liu, Y. P., Collins, R. L., Sosa, M. X., et al. (2015). Loss of δ-catenin function in severe autism. Nature, 520, 51–56.
Ubhi, T., & Brown, G. W. (2019). Exploiting DNA replication stress for cancer treatment. Cancer Research, 79, 1730–1739.
Vaags, A. K., Lionel, A. C., Sato, D., Goodenberger, M., Stein, Q. P., Curran, S., et al. (2012). Rare deletions at the neurexin 3 locus in autism spectrum disorder. American Journal of Human Genetics, 90, 133–141.
van Twest, S., Murphy, V. J., Hodson, C., Tan, W., Swuec, P., O’Rourke, J. J., et al. (2017). Mechanism of ubiquitination and deubiquitination in the Fanconi anemia pathway. Molecular Cell, 65, 247–259.
Wang, H., Shen, L., Li, Y., & Lv, J. (2020). Integrated characterisation of cancer genes identifies key molecular biomarkers in stomach adenocarcinoma. Journal of Clinical Pathology. https://doi.org/10.1136/jclinpath-2019-206400.
Watase, G., Takisawa, H., & Kanemaki, M. T. (2012). Mcm10 plays a role in functioning of the eukaryotic replicative DNA helicase, Cdc45-Mcm-GINS. Current Biology, 22, 343–349.
Wei, P. C., Chang, A. N., Kao, J., Du, Z., Meyers, R. M., Alt, F. W., et al. (2016). Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell, 164, 644–655.
Wei, P. C., Lee, C. S., Du, Z., Schwer, B., Zhang, Y., Kao, J., et al. (2018). Three classes of recurrent DNA break clusters in brain progenitors identified by 3D proximity-based break joining assay. Proceedings of the National Academy of Sciences U S A, 115, 1919–1924.
White, J., Stead, E., Faast, R., Conn, S., Cartwright, P., & Dalton, S. (2005). Developmental activation of the Rb–E2F pathway and establishment of cell cycle-regulated cyclin-dependent kinase activity during embryonic stem cell differentiation. Molecular Biology of the Cell, 16, 2018–2027.
Willis, N. A., Frock, R. L., Menghi, F., Duffey, E. E., Panday, A., Camacho, V., et al. (2017). Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature, 551, 590–595.
Wong, P. G., Winter, S. L., Zaika, E., Cao, T. V., Oguz, U., Koomen, J. M., et al. (2011). Cdc45 limits replicon usage from a low density of prercs in mammalian cells. PLoS ONE, 6, e17533.
Woodfine, K., Fiegler, H., Beare, D. M., Collins, J. E., McCann, O. T., Young, B. D., et al. (2004). Replication timing of the human genome. Human Molecular Genetics, 13, 191–202.
Woodward, A. M., Göhler, T., Luciani, M. G., Oehlmann, M., Ge, X., Gartner, A., et al. (2006). Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. Journal of Cell Biology, 173, 673–683.
Wu, P. Y. J., & Nurse, P. (2009). Establishing the program of origin firing during S phase in fission yeast. Cell, 136, 852–864.
Wu, R., Singh, P. B., & Gilbert, D. M. (2006). Uncoupling global and fine-tuning replication timing determinants for mouse pericentric heterochromatin. Journal of Cell Biology, 174, 185–194.
Wu, R. A., Semlow, D. R., Kamimae-Lanning, A. N., Kochenova, O. V., Chistol, G., Hodskinson, M. R., et al. (2019). TRAIP is a master regulator of DNA interstrand crosslink repair. Nature, 567, 267–272.
Xu, W., Xu, H., Li, K., Fan, Y., Liu, Y., Yang, X., et al. (2017a). The R-loop is a common chromatin feature of the Arabidopsis genome. Nature Plants, 3, 704–714.
Xu, Y., Ning, S., Wei, Z., Xu, R., Xu, X., Xing, M., et al. (2017b). 53BP1 and BRCA1 control pathway choice for stalled replication restart. Elife, 6, e30523.
Yamamoto, K., Hirano, S., Ishiai, M., Morishima, K., Kitao, H., Namikoshi, K., et al. (2005). Fanconi anemia protein FANCD2 promotes immunoglobulin gene conversion and DNA repair through a mechanism related to homologous recombination. Molecular and Cellular Biology, 25, 34–43.
Yang, Y., McBride, K. M., Hensley, S., Lu, Y., Chedin, F., & Bedford, M. T. (2014). Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Molecular Cell, 53, 484–497.
Yasuhara, T., Kato, R., Hagiwara, Y., Shiotani, B., Yamauchi, M., Nakada, S., et al. (2018). Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell, 175, 558–570.e11.
Ying, S., Minocherhomji, S., Chan, K. L., Palmai-Pallag, T., Chu, W. K., Wass, T., et al. (2013). MUS81 promotes common fragile site expression. Nature Cell Biology, 15, 1001–1007.
Yoshida, K., Bacal, J., Desmarais, D., Padioleau, I., Tsaponina, O., Chabes, A., et al. (2014). The histone deacetylases sir2 and rpd3 act on ribosomal DNA to control the replication program in budding yeast. Molecular Cell, 54, 691–697.
Zeman, M. K., & Cimprich, K. A. (2014). Causes and consequences of replication stress. Nature Cell Biology, 16, 2–9.
Zhang, X., Chiang, H. C., Wang, Y., Zhang, C., Smith, S., Zhao, X., et al. (2017). Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat Commun, 8, 15908.
Zhou, R., Zhang, J., Bochman, M. L., Zakian, V. A., & Ha, T. (2014). Periodic DNA patrolling underlies diverse functions of Pif1 on R-loops and G-rich DNA. Elife, 3, e02190.
Zhu, Y., McAvoy, S., Kuhn, R., & Smith, D. I. (2006). RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene, 25, 2901–2908.
Acknowledgements
The authors would like to thank Dr. Philippe Pasero, Pr. David M. Gilbert and Dr. Jessica Tamanini for their critical comments on the manuscript. Work of the C.L.C lab was supported by grants from the I. Curie YPI program, the ATIP-Avenir program from CNRS and Plan Cancer from INSERM, CNRS 80|Prime inter-disciplinary program, the Agence Nationale pour la Recherche (ANR) “ReDeFINe” and “TELOCHROM”, the Institut National du Cancer (INCa) PLBIO19-076, and the I. Curie PIC3i and ICGex Equipex program. Y.L. would like to thank the ANR for the PhD fellowship; M.S. would like to thank the I. Curie for the international post-doc fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gnan, S., Liu, Y., Spagnuolo, M. et al. The impact of transcription-mediated replication stress on genome instability and human disease. GENOME INSTAB. DIS. 1, 207–234 (2020). https://doi.org/10.1007/s42764-020-00021-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42764-020-00021-y