
Abstract
The rapid advancements in the computational abilities of numerical simulations have attracted researchers to work on the area of reactive transport in porous media to improve the hydrocarbon production processes from mature reservoirs. In the hydrology community, reactive transport is well developed where the main research focuses on studying the movement of groundwater and contaminants in aquifers, and quantifying the effect of chemical reactions between the rocks and water. Recently, great efforts have been made to adapt similar models for petroleum applications where multiphase flow is experienced in the subsurface reservoirs. In such systems, thermodynamic and chemical equilibrium is key in establishing an accurate description of the states of the fluids existing in the reservoir. This paper presents a detailed and comprehensive review on the concepts of geochemical modeling, and how it can be mathematically adapted to petroleum multiphase flow problems in porous media. We introduce key physical concepts outlining the treatment of chemical reactions in experimental trials and then explain in detail how a network of chemical reactions can be modeled mathematically for numerical simulation applications. The steps of characterizing the physical behavior of the fluid flow—through a set of governing equations by either natural or molar variables formulations, and the methodology to simplify and incorporate the numerical algorithms into an existing reservoir simulation scheme are shown as well. We finally present two numerical cases from the literature to highlight the key variations between the different variable formulations and comment on the advantages and disadvantages of each approach.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The topic of reactive transport has attracted the efforts of researchers from different disciplines for the past four decades [17, 21, 26, 27, 73, 98, 99] due to its role in understanding the composition of natural water aquifers and the formation and dissolution of rocks and minerals in geologic formations such as deep hydrocarbon reservoirs. The general framework of a reactive transport model was mainly developed to understand the rock–fluid interactions in groundwater movement. At early stages, the modeling process was restricted to single components in the hydrology community, but was later extended to include multicomponents after Litchner established the theoretical basis of the continuum model [73]. This introduced new capabilities to treat the coupling of various transport and biochemical processes. Such an advancement paved the path for many alterations of the original framework to be adapted for reactive contaminant transport, non-isothermal water–rock interactions with kinetic reactions and many more [14, 25, 77, 100, 120].
Traditionally, the solution schemes in numerical codes for reactive transport problems can be divided into two families [90]. The first consists of codes that utilize the sequential approach and can be broken down into two main subcategories: sequential iterative (SIA) and sequential non-iterative (SNIA). In the SNIA approach, the mass transport and chemical reactions are decoupled and solved separately where constant parameters are used when computing the pressure gradient. This is referred to as operator splitting, where the flow equations are solved at each time step without the reaction terms and then followed by the reaction step. This approach introduces splitting errors that is usually reduced by utilizing the SIA. The latter approach is quite similar in the fact that the mass and chemistry equations are decoupled, but the mole fractions and phase velocities from the transport step are used in a chemical submodel to update the concentrations of the components while iterating between solutions of transport and chemistry until an error tolerance is met. The operator splitting techniques have been utilized in many simulators such as HYDROGEOCHEM [122], THOUGHREACT [121], PARSIM [15] and PHREEQC [28], as these approaches are characterized by their easy implementation in a working simulator and fewer demands of computational resources in terms of memory and CPU time. Moreover, legacy codes can be coupled for each phenomenon individually since both chemistry and transport are treated as operators that are modeled independently of one another. However, the SNIA and SIA require small time steps to be used, which could lead to slow convergence rates. This issue can be overcome by introducing the global implicit approach (GIA), which we classify as the second family of solution techniques and is mainly utilized by CrunchFlow [19], MIN3P [101] and PFLOTRAN [56] simulators. For simple models with a limited number of chemical components, the fully implicit/coupled integration method allows solving the system directly. The scheme is often accurate as both the transport and chemical steps are solved simultaneously using a Newton–Raphson numerical approach with less restrictions on time step size, providing stability and robustness [42]. However, if the number of components in the system is large, this method suffers from an excessive computation time and memory requirements, especially in two- and three-dimensional simulations. The difference in the time scales of the transport and the chemistry steps and degree of coupling might impose additional hurdles to the efficiency of the approach. A summary of the properties of the GIA and SIA approaches is presented in the flowchart in Fig. 1, and a detailed comparison of key numerical capabilities of reactive transport modeling codes can be found in the work of [100].
In groundwater simulations, many codes pioneer in modeling reactive transport on the basis of the continuum model, such as HYDROGEOCHEM [122], TOUGHREACT [121], MIN3P[101], PFLOTRAN [56] and CrunchFlow[19]. A famous package is the one developed by Lawrence Berkeley National Laboratory, TOUGHREACT [121], and has been tested extensively for problems with multiple phases and various rock–fluid interactions. It mainly employs the sequential iteration scheme discussed earlier and can handle thermal conditions in the reservoir. Another package is CrunchFlow [19] that utilizes fully implicit schemes for 1-D and 2-D problems but lacks the treatment of multiphase flow. However, the PFLOTRAN package developed by Litchner [56] can deal with multicomponent and multiphase systems but is limited to a defined set of problems. The phase behavior is fully characterized using Henry’s law approach, and it supports parallel computations. Although these sets of codes are relatively mature, they cannot be applied directly to oil production scenarios because of the absence of the hydrocarbon phase treatment in the system.
On the other hand, and in the context of petroleum reservoir simulation, very limited models have been developed to couple transport and chemistry for hydrocarbon systems. Amongst these packages are STARS [53], ECLIPSE 300 [76] and UTCHEM [83], where the first two are commercialized, while the third serves as an open-source research simulator. STARS [53] is a module incorporated in the Computer Modelling Group simulator, GEM [52], and it is a generalized thermal-compositional simulator with a full equation of state characterization for phase behavior coupled with mineral dissolution and precipitation kinetics. Similar features are observed in Schlumberger’s simulator ECLIPSE 300 [76] with its robust performance in modeling the compositional behavior of multiphase systems. Both codes employ a fully implicit scheme in their numerical algorithms but lack comprehensive chemical reactions implementations. On the contrary, UTCHEM [83], a simulator developed at The University of Texas at Austin, offers full reactions characterization with abilities to model chemical flooding scenarios in hydrocarbon reservoirs with full thermodynamic capabilities. Despite these features, the simulator lacks in the area that only SIA is employed, which might cause accuracy problems to arise, especially for multicomponent and multiphase systems in petroleum reservoirs. A fully general GIA implementation of coupled transport and chemistry for petroleum reservoir simulation applications does not yet exist.

Characteristics of SA and GIA numerical procedures for solving chemical reactions systems
It is important to distinguish between reactive transport modeling and geochemical modeling as most people refer to both terms interchangeably. In geochemical modeling, the interactions between water and earth material are analyzed and interpreted in the light of the chemical reactions in the system to provide useful insights on the composition and the thermodynamic behavior of such studied systems. The analysis is usually carried out in a 0-D environment to focus on the effects of temperature, ion speciation, sorption and solubility on the concentration of the various minerals in the system. On the other hand, reactive transport modeling focuses on more complex physical processes involving fluid flow and mass transfer, which require establishing geochemical equilibrium and understanding the thermodynamical state of the system [57, 58]. This major distinction challenged the abilities of traditional reactive transport models to deal with multiphase flow of fluids in heterogeneous porous media, especially in hydrocarbon reservoirs. When multiple fluids are present in the same space, determining the geochemical equilibrium of a multicomponent system and the phase split ratios can be gruesome. Thus, incorporating geochemical reactions with the transport of fluids in a hydrocarbon reservoir is not a straightforward process but rather an extensive procedure at the physical and numerical level.
Recent advancements were able to modernize the approach of reactive transport modeling, thus coupling the flow with a comprehensive thermodynamic description of the system and the kinetic framework [43,44,45, 62, 75, 95, 108]. However, these models cannot be treated as a traditional black-box simulator. In fact, special treatments should always be applied to the mathematical formulation of the problem based on the physical application at hand. The skill of selectively filtering the processes that affect your given system and neglecting the impact of minor processes requires a deep understanding of the geological and chemical settings of the system. This essential understanding highlights the complexity of reactive transport modeling as the input of the system is highly dependent on sources and information from various fields including but not limited to chemistry, thermodynamics, fluid dynamics and rock properties. This in turn requires researchers and scientists to have high expertise in multiple fields to be able to fully characterize and describe the problem at hand.
The objective of this work is to provide a central source describing previous key breakthroughs in the area of reactive transport modeling with a special focus on its applications in multiphase flow in porous media. We will give great attention to the different mathematical formulations used to couple flow and reactions in a reservoir simulation framework and critically analyze the performance of each approach. We will also present the current challenges facing the development of comprehensive geochemical reservoir simulations for different practical applications and finish our review with concluding remarks on the potential of the implementation of reactive transport simulators in the field.
2 Experimental reactive transport
Typically, a reactive transport system is studied quantitatively through modeling tools that rely on mathematical formulations of the problem. However, it is often challenging to account for complex reaction systems in a simple yet physically comprehensive mathematical formulation. This complexity in the modeling highlights the need to establish an experimental framework for controlled laboratory experiments to better identify the processes and reaction paths in a reactive transport system. The experimental results contribute massively in improving existing models through providing high-resolution data sets.
The main key in designing laboratory experiments for reactive transport is to ensure a deep knowledge and understanding of the conceptual model of the physical process. This involves studying the degree of heterogeneity in the porous media and how would state variables in the governing equations vary with the progression of the reactions [24, 88]. A prime focus in this case would be to understand the physics of the flow at both the pore scale and the continuum (Darcy) scale.
Flow tests at laboratory scales with varying configurations are usually carried out to examine the interactions between rock–fluid systems. Core flooding tests are performed to acquire petrophysical data on the properties of the porous media through specific flow cells, pumps and sensors, see Fig. 2 for a core flooding setup by [60]. The fluids in this setup are injected into the sample core, while confining stress is applied. The difference in the pressure is recorded in the chamber, and back pressure is applied to regulate the pressure at the core outlet. In some cases, using actual rock samples in reactive transport experiments might yield inconsistent results due to the extreme heterogeneity present in the rock. Hence, analog materials with consistent properties are used to facilitate the fundamental understating of the physical processes under study as the fabricated gypsum specimens by [34] and the textured glass (non-reactive) with a polished, flat, transparent rock crystal by [37].

A schematic illustration of a core flooding setup by [60]
In some cases of core flooding when a back pressure regulator is used, it is possible to carry out a chemical analysis on the effluent stream. The experiments in [94] focused on the dissolution of Marly Dolostone in \(\hbox {CO}_{2}\) rich brine where the concentration of the calcium ions, magnesium ions and inorganic carbon was measured through the effluent sampling from a back pressure regulator. It is also possible to monitor the effluent chemistry directly through sampling probes across the flow cells, and such experiments are generally performed in 1-D and 2-D domains. Although 3-D experiments represent the boundaries and the conditions of the reservoir to an accurate degree, they are rarely performed due to the associated operational complexity. The experimental design for continuous sampling of the fluid is composed of a flow chamber that is filled with a material to resemble the porous media under investigation. The chamber has a typical maximum length or width of 10 cm and is connected with a feed line that matches the inlet components of your system. The final mixture after the reaction process ceases is collected at the other side of the chamber. The sampling ports are needed to take samples to measure the distribution of the solute concentration spatially and temporally. The measurements are further used to cross-check and validate the accuracy of the numerical models. It is important to note that only a limited number of samples are viable to avoid disturbing the dynamics of the fluid flow in the cell, see Fig. 3.

A schematic illustration of an experimental setup used to characterize the reactive transport phenomena in flow cells (Adapted from [64])
The work of [69] presented a comprehensive investigation for diffusive and advective processes to observe the effect of porosity changes on the transport parameters. This was accomplished through injecting a reactive solution (acidic zinc sulfate) into the pore space causing gypsum and smithsonite formation. The experiments were later used to benchmark the reactive transport code Hytec [105]. Later came the work of [103] where he combined laboratory experiments with a deep investigation on the changes of rock properties at the pore level. The information obtained allowed for a better representation of the continuum-scale model and introduced specific mixing parameters to the advection–dispersion reaction equations to account for highly non-uniform pore-scale concentration gradients. Similarly, [64] performed a laboratory experiment in a 2-D flow cell with multiple sampling ports and demonstrated the shortcomings of the reactive continuum model to reproduce the concentration of the solute measured at the sampling ports.
However, selective sampling does not efficiently contribute to the understanding of how the reaction rate progresses with space and time [29, 35, 39, 41, 51, 54, 93]. Only specific systems where the reaction might cause the products to change colors might be measured, but this only provides a local understating of the rate and does not correlate with the change in properties of the porous medium. To counter such a shortcoming, a computer-controlled triaxial system (CCTS) was designed and built in MIT Geotechnical Engineering Laboratory which provides a continuous effluent chemistry monitoring system. The initial CCTS development of the system was inspired by the setup of [68] in which he designed a triaxial pedestal with integrated sensors for effluent chemistry analysis. The CCTS setup was progressed and updated over the years by [10, 72, 102], and it is extremely efficient in providing long-term continuous measurements of temperature and concentration and on the back-pressurized effluent stream, see Fig. 4.

Diagram of the triaxial setup adapted for coreflood tests from [72])
In brief, laboratory experiments are essential to gain a deep understating of the conceptual and numerical model of the reactive transport process. However, the experimental setup can be challenging in many cases and upscaling problems might arise when mapping the data from flow cells to field-scale systems. Moreover, geochemical reactions are mainly controlled by processes occurring at the pore scale, which might alter the connectivity of the porous media and substantially affect the flow of fluids. The evolution of the porous media especially in heterogeneous rocks poses a challenge in representing reactive transport in a continuum approach. Traditionally, reactions and transport have been described on the basis of empirical relationships [104] and thus severely limiting the simulation results due to the loss of information in the upscaled parameters. Recent efforts have been utilizing the technological advancement in the area of CT-scanning and X-ray micro-tomography to provide a greater focus on the fluctuations of such parameters at the pore level of the rock.
The work of [18, 38, 82, 107] has succeeded in improving pore-scale modeling formulation thankfully due to the increasing computational power of the computer machines, allowing for processing of very accurate and detailed images of the pore network that are obtained experimentally [7, 8, 89]. Although multiple efforts have been made in the recent years to advance pore-scale experiments, the process of upscaling the findings from a pore-scale level to a continuum level that extends in order of km’s is still not completely feasible [59, 84, 87, 115]. This is mainly attributed to the complicated experimental procedure and low reproducibility.
3 Assessing the role of \(CO_2\) in geochemical reactions
Typically, crude oil production goes through three stages. The first stage is the primary recovery where the oil is ousted into the wellbore from the reservoir using its existing energy, such as compaction, gravity drainage, gas drive and water drive mechanisms. The next recovery stage is called the secondary oil recovery in which an external fluid is injected into the reservoir to support the depleted reservoir pressure from the primary recovery. The injected fluid can be either gas or water, and the widely used secondary oil recovery technique is waterflooding. After the secondary oil recovery process, a substantial volume of oil is still trapped in the pores of the reservoir rock and is economical to produce, hence leading to the tertiary oil recovery stage which is also known as enhanced oil recovery (EOR). A major focus in experimental efforts was directed toward understanding the role of geochemical reactions in \(\hbox {CO}_{2}\) injection scenarios for such EOR schemes.
In EOR applications, when \(\hbox {CO}_{2}\) is injected, the geochemical equilibrium is disturbed and the \(\hbox {CO}_{2}\) can then flow in two phases: liquid and vapor. As the \(\hbox {CO}_{2}\) contacts the formation water, it gets dissolved in the aqueous phase, while the rest of the \(\hbox {CO}_{2}\) can either be dissolved in the hydrocarbon phase or roam freely as a vapor, Eq. 1. From a geochemical perspective, when \(\hbox {CO}_{2}\) is dissolved in the aqueous phase, a dissolution process might occur, while ionic species such as \(\hbox {HCO}_{3}^{-}\) and \(\hbox {CO}_{3}^{2-}\) are formed, which subsequently impacts the porosity evolution in the reservoir. In other words, \(\hbox {CO}_{2}\) reacts with water to produce carbonic acid (\(\hbox {H}_{2}\hbox {CO}_{3}\)), which then dissociates into bicarbonate (\(\hbox {HCO}_{3}^{-}\)) and hydrogen ions (\(\hbox {H}^{+}\)), Eq. 2. Generally, mineral interactions are dictated by the formation of the reservoir, where calcite is the main mineral in case of carbonate rocks. However, as the reservoir starts producing and the pressure decreases, the \(\hbox {CO}_{2}\) in the gas phase will expand causing the \(\hbox {CO}_{2}\) in the aqueous phase to exsolve. This causes the minerals to re-precipitate on the rock surface and might lead to significant changes in porosity and wettability of the porous media, and hence affecting hydrocarbon recovery. The set of equations below describe how gaseous \(\hbox {CO}_{2}\) gas in the aqueous phase interacts with water to produce a number of different compounds:
The schematic in Fig. 5 illustrates how pressurization affects the behavior of \(\hbox {CO}_{2}\) in high pressure and low pressure environments. With pressure being low, the \(\hbox {CO}_{2}\) might escape the gaseous phase and interact with the minerals in the aqueous phase causing their dissolution. As pressure is increased, \(\hbox {CO}_{2}\) exsolves from the aqueous phase and the dissociated minerals begin to re-precipitate governed by the reversible reaction in Eq. 2. Other reactions might arise as well depending on the mineralogy of the reservoir rock and how it reacts with the present \(\hbox {CO}_{2}\).

A schematic illustration of the pore and the reservoir-scale levels of \(\hbox {CO}_{2}\) mineral dissolution and precipitation with (a) and without pressure (b) [119]
The injection of \(\hbox {CO}_{2}\) in carbonate reservoirs causes the dissolution of the formation which greatly affects the capillary pressure and the relative permeability of the system. However, such changes have not been experimentally quantified and documented in well-established studies. The mechanism at which the intrinsic rock properties change is attributed to the enlargement of the pore throats. This creates new flow channels that enhances the connectivity of the system, as shown in Fig. 6.

The mineral dissolution changes the pore structure by enlarging the throat diameters which is shown in the system to the right. As the pressure decreases, minerals start to deposit again on the rock surface [119]
The methods used to quantify and assess the interaction between the rock and carbon dioxide can be categorized into four groups:
3.1 Experimental studies
The experiments to investigate the rock–\(\hbox {CO}_{2}\) interactions are performed on large and small scales and named total impact and specific experiments, respectively. In total impact experiments, the aim is to assess the immediate effects of \(\hbox {CO}_{2}\) in the short term. For this purpose, \(\hbox {CO}_{2}\) is typically injected to a host rock under controlled temperature and pressure, and the evolution of new species due the reactions in the system is observed. The work of [12] focused on evaluating the sealing efficiency of a caprock upon \(\hbox {CO}_{2}\) injections to handle the natural migration of \(\hbox {CO}_{2}\) rich pore fluid through the caprock and in fault zones, while [22] studied a specific shale sample (Muderong Shale, Australia) with different clay minerals to obtain information on its sealing integrity. Many other similar experiments were also carried out and modeled in the laboratory through batch reactors [32, 63, 116, 117] to understand the capture and geological CO2 storage (CCGS) in deep aquifers and depleted oil and gas fields. On the other hand, specific experiments target selective minerals to be examined as opposed to the whole heterogeneous rock in the total impact studies. This approach is popular as it is scientifically more accurate when quantifying the effect of \(\hbox {CO}_{2}\) on certain minerals. The results are highly advantageous in determining the reaction kinetics in the system and measuring the thermodynamic equilibrium constants associated with a specific type of mineral [33, 86]. The experimental work of many scientists [9, 70, 91, 113, 114] provided great insight on how different minerals interact with the rock, but the developed results and models suffer from scalability issues to the field conditions. The measured rates of the kinetics of the reactions fail to represent field cases to date, as the research on this topic is still ongoing.
3.2 Natural analogues
The information obtained from natural \(\hbox {CO}_{2}\) analogues could result in an better understating of \(\hbox {CO}_{2}\)–water–rock interactions at the geological time scale [13, 92]. Although such comparisons can provide important information about the interaction process, it is rather generic and qualitative. The studies can be broadly split into two types: studying the effect of \(\hbox {CO}_{2}\) on the reservoir rock formation [16, 50, 118] or tracking the \(\hbox {CO}_{2}\) source as it migrates throughout the subsurface [46, 50, 111]. However, it is almost impossible to replicate a geologic setting in different fields, as each location has its own set of unique properties and boundary conditions. This deems the analogue studies insufficient in providing critical analysis of the \(\hbox {CO}_{2}\) role in rock–fluid interactions.
3.3 Field observations
The observation of already existing producing fields could provide insights on how \(\hbox {CO}_{2}\) would react with the formation water and the reservoir rock. The data gathered about the rock properties, and the produced fluid is deeply investigated and analyzed to provide information on the potential behavior of \(\hbox {CO}_{2}\) upon injection. Moreover, dedicated field-scale tests can be performed to track mineralogy changes and \(\hbox {CO}_{2}\) migration in real time at different time scales. Pilot \(\hbox {CO}_{2}\) injections have been carried out in many locations in the North Sea, Algeria, Canada [11, 48, 55, 67, 79], and the results were used to further support numerical modeling of reactive transport processes.
3.4 Numerical modeling
Since other methods come short when quantifying rock–fluid interactions at a large scale, numerical simulation is deemed essential. The models can be used to track the flow of \(\hbox {CO}_{2}\), investigate porosity evolution and quantify dissolution/precipitation of minerals in the porous media. The main attraction in using numerical techniques for fluid–rock interaction scenarios is the capacity of the simulators to model sophisticated physical phenomena temporally and spatially [36], a feature that laboratory experiments lack in. Many studies have been performed to benchmark mathematical models to physical processes in the subsurface, specifically in the work of [30, 61]. Multiple parameters were fine tuned in these studies such as the vertical permeability and the system boundary conditions, and the effects of \(\hbox {CO}_{2}\) solubility in water were also considered [40, 96].
The path to develop an efficient and robust geochemical model is not well paved. It requires a deep understating of the integrated processes and factors involved in the system under focus. Each scientist will look at the same system from his own background and point of view to determine the important features to be preserved by the conceptual model. But in order to understand how the coupling of reactions and transport is important to model a physical system, we first have to touch on some important concepts in reaction modeling.
4 Geochemical modeling: basics and concepts
4.1 What is chemical equilibrium?
When dealing with chemical reactions, we have to introduce three important concepts: chemical kinetics, equilibrium and chemical equilibrium. The chemical kinetics can be referred to as the science of studying and determining (both experimentally and theoretically) the rates of a reaction network. This reaction network can be formed of tens of simultaneous reactions where products and reactants are dependent on one another. The equilibrium condition is achieved when the forward and the reverse rates of each reaction equalize, and the reaction process stops. At this condition, chemical equilibrium is achieved where the net concentration of the components in the system is constant over time. In other words, we say that the system is in a minimum free Gibbs energy state at standard conditions. For a system at constant temperature and pressure, we can summarize the following results:
-
1.
If \(\varDelta G < 0\) , the process occurs spontaneously.
-
2.
If \(\varDelta G = 0\) , the system is at equilibrium.
-
3.
If \(\varDelta G > 0\) , the process is not spontaneous as written but occurs spontaneously in the reverse direction.
The term \(\varDelta G\) is defined as the change in the free Gibbs energy of the reaction and is denoted by:
where R is the gas constant, T is the temperature, Q is the reaction quotient, and K is the equilibrium ratio. The terms Q and K will be discussed in detail in the section that follows.
4.2 How do we characterize a reaction?
Every reaction is essentially a reversible kinetic reaction. Thus, it is conceptually sound to model every reaction as a kinetic reaction. However, it might be extensive to gather experimental data about the rates of every possible reactions path in a system at different temperature conditions. Some systems might even prove challenging to establish its path and measure the rates in a laboratory setup due to the volatile nature of the components and their critical properties. Thus, we tend to put constraints in place that localize parts of the reaction system as we assume equilibrium conditions for some reactions. In reality, equilibrium is not achieved simultaneously, and the reversible reactions do not stop at equilibrium but rather the concentration change is constant.
Let us assume a generic chemical reaction given by:
At equilibrium, we define a quantity \(\mathbf {K_{eq}}\) called the equilibrium ratio, in which equilibrium is achieved when the rates of the forward and the reverse reactions are equal. In the chemical reaction shown above, \(\mathbf {K_{eq}}\) is written as:
The ratio represents the concentration of the products to that of the reactants at equilibrium. This expression is conventionally called the law of mass action [109]. We illustrate schematically the significance of the equilibrium ratio of Eq. 6 for 3 different scenarios:
-
1.
\(\mathbf {K_{eq}>> 1}\) with \(C_0=0 \ mols\), \(A_0=0.5\ mols\) and \(B_0=0.5\ mols\).
-
2.
\(\mathbf {K_{eq}<< 1}\) with \(C_0=1\ mols\), \(A_0=0\ mols\) and \(B_0=0\ mols\).
-
3.
\(\mathbf {K_{eq} = 1}\) with \(C_0=0.5\ mols\), \(A_0=0.25\ mols\) and \(B_0=0.25\ mols\).
The terms \(A_0\), \(B_o\) and \(C_o\) denote the initial concentration of each component in the 3 scenarios before any reaction occurs.

The concentration of the reactants and the products at initial and equilibrium conditions: a At high equilibrium ratios, the reactants are completely converted to C as the reaction moves forward b At low equilibrium ratios, the product is completely dissociated into A and B as the reaction moves reversibly c When the equilibrium ratio is close to unity, the reactions and the products tend to reach equilibrium at equal concentrations
The illustration in Fig. 7 shows how the concentrations of the components change for the different cases of \(\mathbf {K_{eq}}\). The ratio of change between the components is generally dependent on the stoichiometric ratios of the reactions, and the initial number of moles of the reactants. At initial conditions, the reactants are introduced and the reaction path (driven by the equilibrium ratio) favors either a forward or a backward reaction. For example, in case (a) of Fig. 7, the reaction is driven forward due to \(K_{eq}>>1\), and hence, A and B will fully react to form C with the backward reaction being of little significance. In case (c), where \(K_{eq}=1\), both products and reactants are present at equilibrium conditions and the ratios of the components depend on the stoichiometry of the reaction.
When a reaction follows the law of mass action, we characterize it as an equilibrium reaction. These reactions are reversible and fast, as they occur spontaneously on a small time scale. This means that we can express them algebraically to relate \(\mathbf {K_{eq}}\) with the reaction quotient \(\mathbf {Q_{eq}}\) as follows:
where \(\mathbf {Q_{eq}}\) depends on the initial concentrations of the components in the mixture.
On the other hand, when the reaction has a strong dependency on time and is relatively slow and irreversible, we characterize it as a kinetic reaction. The term kinetic refers to the rate at which a chemical process occurs. If we rewrite the same generic reaction presented earlier in Eq. 6 as an irreversible reaction:
we can then write the reaction rate as follows:
The rate constant is denoted by k which is a reaction property that can be only determined experimentally and is highly dependent on temperature. Since a limited amount of rate measurements at different temperatures can done experimentally, we usually extend and interpolate the range for the rate constant using Arrhenius equation:
where \(E_a\) is the activation energy (J/mol), R is the gas constant (J/k.mol), T is the absolute temperature, and A is the frequency factor (a constant to indicate the number of collisions with correct orientation that leads to products).
We can finally write the rate law with respect to A as:
where x and y are the reactant order determined from the experiments. For simplicity, we can generally assume a reaction to be of first order (x = y = 0), but this treatment ultimately depends on the process being modeled.
To summarize, reaction characterization is a complex process that requires rigorous experimental validation of the reaction mechanism. Since all chemical reactions can be, in essence, modeled as kinetic reactions, we selectively localize some of the reactions by assuming an equilibrium condition to further simplify the reaction network.
4.3 Kinetic or equilibrium?
It is up to the modeler to classify a set of reactions into kinetic and equilibrium reactions, as long as the integrity of the system is preserved. In theory, every reaction can be modeled as a kinetic reaction. However, it is almost impossible to do so because of the lack of experimental data to represent the kinetic rates of the reaction system. In this case, it is practical to assume some reactions are instantaneous and thus treat them as equilibrium reactions.
If we consider the reversible reaction of decomposition of \(\hbox {N}_{2}\hbox {O}_{4}\)(g) and dimerization of 2 NO(g), we write it as follows:
In this reaction, equilibrium is not achieved instantaneously and concentrations stop changing at equilibrium but reactions do not stop. If we have a reaction vessel starting with \(100\%\) \(\hbox {N}_{2}\hbox {O}_{4}\)(g) that undergoes a full conversion to 2NO(g), this indicates that the kinetic rate of the dimerization reaction is zero. To test this out, we vary the kinetic reaction rate k for the decomposition reactions between 0.01 and 10 and we plot the concentration of the reactant and the product in Fig. 8. When the kinetic rate is high, the kinetic reactions approach the equilibrium point and thus can be treated as an equilibrium reaction.

Component concentration for the product (left) and the reactant (right) for different values of the kinetic reaction rate K
5 Flow and transport in porous media
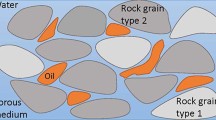
It is essential to understand the process of fluid flow in the subsurface geological structures, especially when multiple fluids are present as in the case of hydrocarbon reservoirs, see Fig. 9. This allows for proper modeling of the fluid path and preserving the integrity of the physical laws in critical processes such as underground water management, chemical weathering and oil extraction from deep reservoirs. Typically, transport is controlled by the phenomena of advection, dispersion and diffusion if no reactions are assumed [1, 3].

A schematic illustration of the flow of fluid in porous media. The path of the fluid is tortuous
Advection is the process where solutes move with the bulk fluid phase at the same velocity. Imagine you are next to a stream or a river, and you gently place a piece of paper on the surface of the water. The paper will start moving with the bulk phase (water) at the same velocity of the flow. Conversely, dispersion and diffusion are concentration driven and depict the extent of spreading of different components.
The generic continuity equation describing the concentration of a component with time for fluid flow in porous media, while accounting for the advective, diffusive and dispersive forces is represented by:
where \(\frac{\partial C_{i}}{\partial t}\) is the accumulation term written as \(C_{i}= \phi \rho _{j} S_{j}X_{ij}\), \(n_{p}\) is the number of fluid phases, \(\rho _{j}\) is the density of phase j that depends on pressure, temperature and molar fraction, \(\phi\) is the porosity , \(X_{ij}\) is the mole fraction fluid species i in the fluid phase j, \(u_{j}\) is the Darcy velocity for phase j, \(\varvec{D_{ij}}\) is the dispersivity tensor for fluid species i in phase j, and \(q^{W}_{i}\) is the source/sink term of species i.
The Darcy velocity \(u_{j}\) is given by Darcy’s law:
where \(k_{rj}\) is the relative permeability of phase j, \(p_{j}\) is the pressure of phase j, g is the acceleration of gravity, and K is the rock permeability. Typically, the Kozeny–Carman equation is used to dynamically represent the permeability in the continuum scale:
where \(K_{0}\) and \(\phi _{0}\) represent the initial permeability and porosity in the system, respectively, before any chemical reaction takes place. In case, the formation porosity is assumed to be constant throughout a certain physical process, and then, \(K=K_{0}\)
6 Coupling reactions with transport
The coupling of kinetic and equilibrium reactions with the mass balance equation of the fluid flow can result in a highly complex system of equations especially when compositional systems are modeled [4,5,6, 71, 123]. Since the reactive transport area is quite mature in the hydrology community, recent efforts in the work of [43,44,45, 62, 75, 95, 108] have been focused on utilizing the advances in the hydrogeological field to design a comprehensive framework for reactive flow in porous media for hydrocarbon reservoirs, see Fig. 10. In this section, we present the different variable formulations that are used to represent the continuity and reaction equations and highlight the key differences in each approach.

A schematic illustration of the pore space showing the reaction sites of the flowing fluids with the ions present in the porous media
6.1 Natural variable formulation
The natural variable formulation was first developed by [31] allowing the implementation of compositional modeling in reservoir simulations with fully implicit schemes. In this formulation, [31] consider \(n_c\) hydrocarbon components plus the water phase which adds up to \(n_c+1\) components, requiring similar number of mass balance equations to be solved. The primary variables are the pressure, phase compositions and saturations, while phase behavior relationships are used as local constraints. Many modifications and alterations to the natural variable formulation have been proposed to fit different physical applications. In the following, we present different forms of the natural variable formulation for reactive transport with multiphase flow problems. The presentation and the analysis of the transport equations are based on the assumption that Darcy’s flow is valid throughout the domain.
6.1.1 Governing equations of transport and chemistry
A component, in thermodynamics, is defined as a chemically independent constituent of a system. If we have a system that constitutes \(n_s\) solid species and \(n_f\) fluid species, then we can write the total number of components in the system as \(n_c=n_s+n_f\). Incorporating the reactive term to the mass balance relation in Eq. 14 results in:
where \(N_i\) is the accumulation term of component i, \(L_i\) is the flux term accounting for the advective, diffusive and dispersive forces and is written as (\(\nabla \cdot \sum _{j=1}^{n_{p}}(\rho _{j}X_{ij} u_{j} -\rho _{j}\phi S_{j} \varvec{D_{ij}}\nabla X_{ij}) + q^{W}_{i}\)), \(v_{i,k}\) is the stoichiometric coefficient of species i in kinetic reaction k, and \(v_{i,q}\) is the stoichiometric coefficient of component i in equilibrium reaction q. The kinetic and equilibrium reaction rates are denoted by \(r_k\) and \(r_q,\) respectively, and \(n_k\) and \(n_q\) are the number of kinetic and equilibrium reactions, respectively. The total number of the reactions in the system can be then expressed by \(n_r=n_k+n_q\).
The accumulation term represents the change of mass of the fluid and solid species with time and is written as,
where \(n_{jf}\) is the number of fluid phases, \(\rho _{j}\) is the density of phase j that depends on pressure, temperature and molar fractions, \(\phi\) is the porosity, \(X_{ij}\) is the mole fraction of fluid species i in the fluid phase j, \(\rho _s\) is the density of the solid phase, and \(X_{is}\) is the mole fraction of the solid phase s.
The natural variable formulation expresses the unknowns in the system in terms of the set
To further simplify the system, we assume that the solid phase does not flow and phase partitioning only occurs when fluid components are present. This leads to a simplified set of equations based on Eq. 17:
where \(C_s\) is the concentration of the solid components s, \(n_f\) is the number of fluid components, and \(n_s\) is the number of solid components ( recall \(n_c=n_f+n_s)\).
Subsequently, the natural variable set of unknowns is then rewritten as,
The governing equation represented in Eq. 17 is the basis of all the simulators discussed earlier in the introduction. The main differences only lie in the treatment of the kinetic and equilibrium reactions. If all the reactions in the system are kinetic reactions, the rate of the reaction is then expressed in terms of the natural variable set and can be solved with the flux term or replaced with a rate law equation similar to the ones discussed in section 4.2. On the other hand, equilibrium reactions are more complicated due to their fast nature and are usually expressed by algebraic equations. This means that each equilibrium reaction will add additional unknowns to the system, and computational complexity overall [74]. In the hydrologic simulators, the number of equilibrium reactions is reduced by formulating the problem in terms of components. A component in this case is defined as the combination of species that are not affected by the equilibrium reactions in the system and is different from the definition of a component in reservoir simulations [80]. This makes such a concept difficult to apply in multiphase flow context, and hence, an alternative approach is needed to both simplify the system and preserve the characteristics of the reactions.
6.1.2 Element-based formulation
Coupling geochemical reactions with transport equations for hydrocarbon production applications poses complexities on the solved system due to the differences in the time scales of the transport and chemistry [110]. Such differences would typically result in a stiff system of equations, and hence, an effective strategy needs to be adopted to solve or simplify such systems. The work of [23, 106] discussed how different time scales could affect the quantification of the deposition process in the porous media at the pore, core and field level using analytical and numerical modeling techniques. The results showed the influence of the Damkohler number (Da), a dimensionless quantity that relates the chemical reaction time scale to the transport phenomena rate occurring in a system, on the properties of the system. If \(D_a>1\), reaction is dominant and clogging effects are exacerbated and evident in the porous media as the variations in the concentration field are significant. However, and for \(D_a<1\), convective forces are dominant and the porous medium properties change smoothly. At a field level, the prediction of simulations can be improved by using relevant core-scale transport properties of reactive flow for each rock-type and reactive flow regime [106].
As mentioned before, one could linearly transform the transport equations to eliminate the dependency on equilibrium reactions (and hence the fast nature of the reactions) using an element-based mass balance equations [20, 99]. The governing equations 20 and 21 can be further simplified and written in a concise matrix format:
where \(\mathbf {N}\) is the vector of total component concentration, \(\mathbf {N}= (N_{1},..., N_{nf}, C1,..., C_{n_{s}})^T\); \(\mathbf {L}\) is the flux vector,
\(\mathbf {L}=(L_{1}, . . . , L_{nf}, 0, . . .,0)^T\); \(\mathbf {R}\) is the reaction term vector, \(\mathbf {R}= \left( {\begin{array}{c}r_{k}\\ r_{q}\end{array}}\right)\) and \(\mathbf {S}\) is the stoichiometric matrix that holds the coefficients of the components in each reaction with dimensions \(n_{t}\times n_{r}\), \(\mathbf {S}= \begin{bmatrix} S_{k}&{}S_{q}\\ \end{bmatrix}\) where the stoichiometric submatrix \(S_k\) corresponds to kinetic reactions and \(S_q\) to equilibrium reactions.
The total number of reactions in the system, \(n_r\), consists of kinetic \(n_k\) and equilibrium \(n_q\) reactions of which we can safely assume that they are linearly independent and balanced (\(n_r=n_k+n_q\)) [43]. By definition, an element is an atom or a compound that does not partition into smaller entities by chemical reactions [44]. Let us take a matrix \(\mathbf {E}_{n_e \times n_c}\) that holds the stoichiometric values for each element in each component. When the set of reactions is assumed to be linearly dependent, then the number of components is equal to the number of reactions and the number of elements in the system (\(n_c=n_e+n_r\)). Thus, if we multiply \(\mathbf {E}\) by the component stoichiometric coefficient matrix \(\mathbf {S},\) since both matrices are nonzero matrices and do not have a full rank, we get:
Let us illustrate the relationship above with an example of a set of independent balanced reactions below (the reaction can be either kinetic or equilibrium) :
The elements in the system are C, H and O forming five components: \(\hbox {H}_{2}\), \(\hbox {O}_{2}\), \(\hbox {H}_{2}\hbox {O}\), \(\hbox {CO}_{2}\) and \(\hbox {C}_{6}\hbox {H}_{12}\hbox {O}_{6}\). The element stoichiometric and reaction stoichiometric matrices are:
Since \(\mathbf {ES=0}\), we can multiply equation 23 by \(\mathbf {E}\) to obtain:
Thus, we managed to eliminate the reaction term from the mass balance equations (whether they are kinetic or equilibrium) which adds an advantage to the element-based formulation. We end up with a system of conservation equations for each element involved in the reactions. In other words, we solve for Eq. 27 plus \(n_r\) reaction relations instead of solving Eq. 23 directly.
6.1.3 Reduction of reaction rates
The approach, in theory, is similar to the element-based formulation reduction procedure, as we have to linearly transform the system to eliminate the equilibrium rates but through multiplying the element stoichiometric matrix, \(\mathbf {E}\) with the canonical or reduced row echelon form of the stoichiometric coefficient matrix, denoted now as \(\mathbf {\bar{S}}\). The applied mathematical manipulations to obtain the canonical matrix depend on the kinetic and equilibrium reactions in the system, and the choice of primary and secondary components. Different forms of \(\mathbf {\bar{S}}\) for specific reaction sets have been discussed in the work of [45, 80].
The product of \(\mathbf {E\bar{S}}\) would then result in \(\begin{bmatrix} \mathbf {I} &{} &{} \mathbf {0} \\ \end{bmatrix}\) where \(\mathbf {I}\) is an identity matrix. If we rewrite Eq. 23 in terms of \(\mathbf {\bar{S}}\) and multiply by the matrix \(\mathbf {E},\) we get:
The resultant system signifies that each kinetic reaction rate only appears in one equation due to the linear combination of the component conservation equations. But since the kinetic and equilibrium reaction relations are provided separately from the mass balance equations, one should have the knowledge to model different types of chemical reactions and their associated rates. Next, we present a generic way to treat chemical reactions despite the fact that such treatment is usually simplified depending on the problem at hand.
6.1.4 Generic reaction implementation
In order to implement the reactions into a Jacobian, we have to obtain the derivatives of the reaction term with the set of variables \(\mathbf {X}\). It is essential to denote that the reaction term \(r_k\) depends on the concentration/activity of the fluid species i in phase j and the concentration/activity of the mineral solid phase s (\(r_k(C_{ij},C_s\))). The reaction is then characterized in terms of both fluid and solid concentrations/activities.
Concentration-based reactions
If we take the derivatives of the reaction term with the respect to the set of unknowns \(\mathbf {X}\), we obtain the generic derivatives for kinetic and equilibrium relations, respectively:
In order to derive the generic reaction relations with respect to the concentration, one must assume a form of the reaction rate, see Table 1, for example.
Let us assume that we have kinetic reaction that follows the law mass of action. In this case, Eq. 29 must be used to obtain the derivatives and the reaction rate would be in the form of:
where \(K_k\) is the reaction rate constant, m and n are the stoichiometric coefficients of the element, and the derivatives of \(r_k\) with respect to \(\mathbf {X}\) for Eq. 29 are :
We also relate \(C_{ij}\) to set \(\mathbf {X}\) by:
The derivatives of the fluid concentration with respect to the unknown variable set will then be:
where \(\psi\) and \(\kappa\) are target phase and species derivative indices and \(\delta\) is the Kronecker delta function.
Similarly, we calculate the derivatives of \(C_s\) with respect to the variable set. Since we have a solid phase, only the derivative of \(C_s\) with itself is nonzero,
where \(\beta\) is a mineral component index.
If Eqs. 32-33 and 35-37 are plugged back to Eq. 29, the resultant expression can then form the basis of the Jacobian representing the reactions in the system. If the equilibrium reactions are present in the system, a similar derivation procedure is adapted through Eq. 30 to obtain the required derivatives for each equilibrium reaction rate, \(Q_q\).
Activity-based reactions
In some problems, the solubility of components in the gas phase is better represented using an activity approach, specifically in precipitation/dissolution processes. Thus, the generic reaction equations must be expressed in terms of activity coefficients. We denote the activity coefficients of fluid species i in phase j and the solid species s by \(a_{ij}\) and \(a_s,\) respectively. The generic derivatives for kinetic and equilibrium relations, respectively, are written as:
Since the activities of the solid phase are set to unity, then the derivative of \(a_s\) with respect to \(\mathbf{X}\) is 0. The kinetic rate is represented in terms of the activity of the components in the fluid phase. An example rate law for such processes is written as:
where \(A_k\) is the reactive surface area used by reaction k, \(K_k\) is the rate constant, and \(K_{eq,k}\) is the equilibrium constant.
The activity relations can be expressed in two distinct forms based on the state of the fluid, as fugacity is used when the components are dissolved in the gaseous phase, while molality is used when the aqueous phase is the solvent. The fugacity is defined as an empirical quantity to account for the fluid deviation from ideality and is measured by the effective pressure of a gas to express its tendency to escape or expand. The dimensionless activity relations then become,
where \(f_{ij}\) is the fugacity of species i in phase \(j=g\), \(P^0\) is standard pressure, \(X_{ij}\) is the mole fraction of solute species i in phase \(j=w\), \(X_{jj}\) is the molar fraction of water in the aqueous phase (\(j=w\)), and \(\gamma _{ij}\) is the activity coefficient for species i in the aqueous phase \(j=w\).n. The constant value 55.508 represents the number of mole of water per kilogram, and the activity coefficient \(\gamma _{ij}\) is calculated from empirically correlated models similar to [57, 85].
Based on the presented definition of activity in Eqs. 42 and 43 and the rate equation in 41 , we can then obtain the derivatives of these expressions with respect to the variable set \(\mathbf {X}\) in a similar fashion to Eqs. 32-33 and 35-37. The derivatives are substituted in Eqs. 39 and 40 to obtain the final form of the Jacobian equations.
6.1.5 Summary of equations
In summary, we have \(2n_p +n_s +n_p n_f\) variables requiring \(n_e + n_r +(n_p-1)n_f +2n_p\) equations. We recall that \(n_r\) is the number of chemical reaction relations \(n_r= n_k +n_q\), \(n_p\) is the number of phases, \(n_f\) is the number of fluid components and \(n_s\) is the number of solid components where \(n_c=n_s+n_f\) . When a system is fully characterized, this implies that \(n_c=n_f+n_s=n_e+n_r\), and thus, the total number of equations reduces to be equal to the total number of unknowns. We would like to also point out that the system is closed with a set of constraints as follows:
In order to simplify the final system, it is possible to split the governing equations, reaction relations and local constraints into primary and secondary equations as we show in the following section.
6.1.6 System reduction: selection of primary and secondary equations
We first solve for the primary unknowns, which are then substituted into the secondary set to compute the secondary variables. This procedure does not alter the total number of the governing equations, but rather selectively categorize the equations into different sets to increase the performance of the simulator at the liner solver stage.
In order to decompose our governing equations into two categories, [44] suggested two approaches: reactions coupled and local reactions decoupled. In the reaction coupled approach, we consider our mass balance equation including the reaction term (both kinetic and equilibrium) to be our primary system, while the local constraints are treated as secondary. The primary variables in this selection would be the pressure (P), phase saturation (\(S_j\)) and some phase composition (\(X_{ij}\)), while the rest of (\(X_{ij}\)) and the solid-phase composition (\(X_{is}\)) are the secondary variables. Similarly, for the second approach when local reactions are decoupled, the primary and secondary variables selection is the same except for the number of the primary equations. The equilibrium reactions are separated from the primary set and treated as secondary equations. In some cases, further reductions can be performed by assuming localized chemical reactions, but this solely depends on the reaction system under study. Table 2 summarizes the equation selection criteria discussed earlier.
6.1.7 Solution procedure
The Jacobian is constructed based on the selection of the primary and secondary equations and is solved in a fully implicit scheme. Although the fully implicit approach guarantees a stable solution, the strong coupling of different physical phenomena in the system results in a stiff matrix. Moreover, this approach might be memory intensive and pose a computational complexity in the simulator. This elevates the need to develop unique pre-conditioners for the system based on the modeled physical processes and the numerical discretization scheme that is used [81].
In a reactive transport system, the primary equations are solved at each Newton step which necessitates the computation of the derivatives of the governing reaction with respect to the variable set at each iteration. Thus, decoupling might prove handy in simplifying the procedure of obtaining such derivatives and speeding up the computation time. After the primary variables are calculated, we move to perform thermodynamic analysis of the system to study the phase stability and determine whether phase disappearance/reappearance will occur at the next Newton iteration. Finally, the secondary variables are then computed. The procedure presented in algorithm 1 summarizes the solution steps.

After we have fully presented the natural variable formulation and the different approaches in representing the governing equations and the reactions relations of a system, we introduce a molar variables basis for the system formulation. In the following, we will discuss two different molar variables formulations and highlight the key differences of each approach.
6.2 Molar variable formulation
The molar variable formulation was first introduced by [97] and fully tested for compositional reservoir simulations by [62]. First, we define a new porosity term, \(\widehat{\phi },\) that refers to the reactive porosity defined as the volume occupied by the solids that do not participate in chemical reactions and related to \(\phi\) by:
6.2.1 Overall-composition formulation
Now, we would like to express the governing equations in terms of overall-composition variables using the following set:
where \(z_c\) is the overall-composition fraction, \(X_{ij}\) is phase composition, and \(\nu _j\) is the phase molar fraction defined as:
The fluid saturation \(S_j\) is expressed as:
We relate the overall composition of each component to the molar fraction of the fluid by:
We also define \(\rho _T\) as the total component density:
The above relationships allow us to propose and alternative form of \(N_i\) in Eq. 18 as follows,
This form allows us to write the governing equations similarly to Eq. 28 and then reduce the \(n_c\) component conservation equations into \(n_e\) element conservation equations using the matrix \(\mathbf {E}\),
The reactions are treated independently similar to the procedure outlined in Sect. 6.1.4. Table 3 summarizes the primary and secondary variables of the system.
The total number of variables in the system is \(n_c n_p +n_c+n_p+1\) closed by the element conservation equations, reaction equations and local constrains which will be discussed next. Note that the number of variables is equal to the number of governing equations. The local constrains are defined as follows:
(1) Phase Equilibrium
(2) Overall mole fraction constraints
(3) Phase molar constraints
(4) Molar phase fraction and overall-composition constraints
Note that not all \(z_i\) values are primary but rather those related to \(n_e+n_k-1\) equations. If we further decouple the chemical reactions to include the equilibrium reactions in the secondary equations, we end up with a set \(\mathbf {R}\) that includes the residuals of the local constraints, phase behavior and equilibrium reactions equations.
The solution procedure is quite different than the one discussed before for natural variables. Here, we assume that the primary variables are known (P and part of \(z_i\)), and the goal is to determine \(\nu _j\), \(X_{ij}\) and the rest of \(z_i\) from the secondary set of equations. The procedure involves two steps; the first step is to determine the number of existing phases in a given cell. This follows with flash calculations to determine the composition of the existing phases (\(X_{ij}\)) as the second step.
After the secondary variables are obtained from the flash calculations, we update the residual of the primary equations to check for convergence. If the results are unsatisfactory, then we are required to calculate the partial derivative of the secondary variables with respect to the primary variables:
This derivative is computed using the results from the last converged step , where then the Jacobian of the primary equations is reconstructed and solved for the primary variables. This process is repeated until Newton iterations converge. Algorithm 2 shows the steps of the process.

The main advantage of this formulation is that the number of equations and variables is the same in any given cell. However, phase computations are required for each cell at every step, and thus increasing the number of flash calculation iterations.
6.2.2 Element molar formulation
In this formulation, we take the elements in the system as the basis of the governing equations as opposed to components. The number of variables is similar to the overall-composition formulation with additional \(n_e\) variables. The following variable set is used:
where \(z_e\) is the ratio of moles of an element to the total moles of an element.
The element molar formulation can be easily expressed using a set of relations to perform component-to-element transformation from \(z_i\) to \(z_e\):
where \(\rho _T^E\) is the total element density, equivalent to the total moles of elements divided by pore volume. The total element density is expressed as:
where \(E_{ei}\) is the number of element e in component i. We apply these changes to the accumulation term of Eq. 54 to write the governing equations in terms of the element molar formulation by dividing with \(\rho _T^E\):
The solution procedure is quite similar to the overall-composition formulation. Here, we assume that the primary variables are known (P, \(z_e\) and part of \(z_i\)), denoted as the primary set \(\varPsi _{e,p}\). Table 4 summarizes the primary and secondary variables of the system.
The goal is to determine \(\nu _j\), \(X_{ij}\) and the rest of \(z_i\) from the secondary set of equations. In this formulation, the last \(z_e\) value is not solved for using the primary equations but rather computed from the constraint:
When we formulate the secondary equation set \(\mathbf {R}\), we add an additional constraint to represent the component-to-element transformation in the system. Since phase behavior computations are performed on components, we need to constantly combine the elements to form components when needed.

In molar formulations, phase behavior computations are straightforward if chemical reactions are not involved. Typically, the pressure and the molar composition \(z_i\) are known throughout the calculations, and hence, a two-stage procedure is easily implemented. First, we perform a stability test to determine the number of phases in a grid block [49, 78], followed by traditional flash or negative flash calculations to determine the molar fraction of each component in each phase. However, when chemical reactions are implemented, the molar composition of the mixtures is not totally known. In particular, \(n_e+n_k -1\) components of \(z_i\) are known, which might limit phase behavior procedures in existing reservoir simulators. The chemical reactions pose an additional constraint on the parameters represented by chemical equilibrium, a term that describes the interactions of multicomponents in multiple phase. Adding this constraint to the existing phase equilibrium requirement presents critical challenges to phase behavior computations. In brief, chemical reactions need a special combined thermodynamic and chemical equilibrium approach to ensure phase computations are accurate and representative of the physical system being investigated.
6.3 Which formulation is better: molar or natural?
Traditionally, natural variables have been widely used in commercial reservoir simulators due to the ease of implementation and the simple phase behavior algorithms. However, as the more complex physics are required to be modeled, molar formulation started to be implemented for specific simulation scenarios. Each formulation has its own advantages and drawbacks, which we summarize in four main points.
6.3.1 Jacobian construction
Typically, the derivatives of the governing equations are required to construct the Jacobian matrix and solve for the primary unknowns. When using natural formulation, computing the derivatives is a straightforward process since the governing equations are naturally expressed in the terms of the set of the unknowns (hence the name of the formulation). However, in the molar formulation, the expression of the derivatives is much more complicated as we have seen in Eq.59. In a reservoir simulator, such approach would increase the processing time of setting up the Jacobian matrix and thus reduces its desirability.
6.3.2 Variable substitution
The size of the Jacobian matrix is decided by the number of equations and unknowns in the system. Thus, the Jacobian has to dynamically represent the system when any changes occur. When natural variables are used, the primary variables set has to represent the phases present in the system. As the simulation progresses, phase appearance/disappearance is experienced in some cells, and thus, the number of unknowns varies accordingly. Such behavior adds unwanted complexities in a reservoir simulation when performing phase behavior calculations, especially when parallel processors are utilized. The partitioning of the Jacobian into different computer cores is not straightforward as the size of the system varies with each time step. On the other hand, molar formulation does not suffer from phase appearance/disappearance issues since overall variables are used. This means that the number of unknowns is the same in each grid at any given time step. The phase behavior is decoupled from the primary set and is solved separately.
6.3.3 Flash calculations
In the natural variable formulation, the phase behavior is solved together with the governing equations to determine the phase equilibrium conditions. However, in the molar formulation, the flash calculations are performed separately and referred to as exact flash. This treatment ensures that accurate depiction of the phase state of the system is achieved as the time progresses in a reservoir simulation. However, this approach has to be further investigated when chemical reactions are involved.
6.3.4 Computational cost
The phase behavior computations in the molar formulation increase the complexity of the system and subsequently increase the cost. However, this should not be the sole factor in picking a method over the other. Factors such as the time-stepping size and Newton iterations vary with each formulation based on the system being studied. Furthermore, decoupling chemical equilibrium reactions in the natural formulations will reduce the computation cost further and this should be taken into consideration when assessing the performance of both formulations. The advantages and the disadvantages of the natural and molar variable formulation are summarized in Table 5.
Earlier, we presented the advantages and disadvantages of utilizing molar and natural formulations for coupled reaction systems. Ultimately, the choice of the method will be based on the physics of the problem and the desired efficiency of the simulation code. Next, we present two computational cases from the literature that relies on the previously discussed formulations, and we briefly comment on the results of each numerical case.
7 Review of applied numerical cases
7.1 In situ upgrading of shale oil
In the first case, we show an in situ upgrading of oil shale model by [43]. The process is highly dependent on temperature which directly affects the rates of the kinetic reactions as compositional flow is experienced in the reservoir. [43] modeled the problem using a component-based formulation that we presented in Sect. 6.1.1 using natural variables. In the model, the kinetic chemical reactions are strongly coupled with the mass and energy balance since they are highly dependent on temperature, while average values are used to represent the porosity and permeability changes. Furthermore, the decomposition of kerogen into multiple mixtures yields four different phases (gas, water, oil and solid prechar) in which the solid phase is assumed to not occupy any pore space. The decomposition reaction of the kerogen adapted from [64] and reported in [112] is as follows,
where the symbol IC indicates lumped components. All the reactions are assumed to follow the elementary rate law in Eq.31, and the rate constant \(K_k\) is given by Arrhenius law in Eq. 11.
Moreover, since most in situ upgrading processes occur above the boiling point of water, the problem is reduced to a gas–oil equilibrium analysis. However, if the water production is significant, three phase flash calculations are required to thermodynamically represent the system. The unknowns to be computed in the system are pressure, temperature, mole fractions of each fluid component and solid concentrations, while localized reactions are strongly coupled. The model of [43] was used to validate the results of the Mahogany Demonstration Project-South (MDP-S) pilot in situ upgrading project [47] where the pore space of the matrix is initially assumed to be saturated with brine or gases. Heaters are spaced around the producers to facilitate the decomposition of kerogen where oil starts accumulating in the pore space and gets produced by the well, see Fig. 11.

The heater spacing around the producers is shown in a while the gridding of the domain is depicted in b (reported in [43])
The results of the simulation are presented in Fig. 12 where high initial gas production is noticed due to the heating effect to accelerate the decomposition of the kerogen into smaller carbon chains. The plots of the cumulative production show a great agreement with the field results. The importance of using a reactive transport approach in this problem is related to the decomposition of the kerogen into smaller hydrocarbons that need to be tracked, while the bulk fluid phase flows in the reservoir. Since the water phase is not of great importance in the in situ upgrading, the component-based formulation (presented in Sect. 6.1.1) is used to monitor different alkanes in the system as the temperature changes and the kerogen is consumed.

The plots show oil and gas production rates (left) and the cumulative oil and total production (right) for the simulation of in situ upgrading of oil shale in the MDPS-S project [43]. The numerical and field production data are in good agreement, providing the possibility of modeling and designing in situ shale upgrading projects using the component-based approach
7.2 Coupling high-order linearization schemes with molar variables for reactive transport modeling
In the previous sections, we have extensively detailed the different formulations used to set up a reactive transport problem mathematically and discussed the pros and cons of each approach. The selection of the formulation that optimizes the problem is essential as the main objective of this procedure is to drive the residuals of the secondary equations to zero at each Newton iteration accurately and efficiently. However, this process is quite expensive especially when complicated derivatives are computed as in the molar formulation.
To overcome this issue, [66] developed a multilinear state-dependent operators based on an operator-based linearization (OBL) technique to simplify the computation of the Jacobian derivatives. The technique is based on dividing the grid into smaller subdomains, encompassing one or more grid cells, and calculating the physical properties at specific points in the subdomains. This results in pre-computed properties stored in specific operators which are used to compute the derivatives inside the Jacobian. The derivatives are easily computed at any location in the grid through interpolating the values at the populated points, see Fig. 13

The domain of the reservoir grid. The black points represent the locations at which the OBL operators are computed. The red points A and B are interpolated based on the pre-computed operators at the surrounding black points, thus easily determining the value of the derivatives at the red points
In OBL, the governing equation (Eq. 63) is discretized and rewritten in terms of state-based (physical properties of fluid and rock) and space-based (properties transformed in space) operators:
where \(\omega\) is a state-dependent parameter, \(\xi\) is a space-dependent parameter, and \(\omega\) and \(\omega _n\) are nonlinear unknowns at the current and the previous time step, respectively.. The operators for a simple equilibrium reaction case are given by,
where \(c_r\) is defined as the rock compressibility, \(T^{ab}\) is the geometric part of transmissibility (which involves permeability and the geometry of the control volume) , and \(e_{ic}\) is the amount of element i in component c.
The values of the operators \(\alpha\) and \(\beta\) can be computed beforehand and prepared in tables at different compositions for a specified pressure range. It is important to note that the summation of \(z_i\) of all elements equals to unity, and thus, one can produce tables of the operator values at a given resolution as a pre-processing stage. It is also possible to integrate an adaptive OBL scheme within the process of the simulation adding to the efficiency of the reservoir simulator. This technique is quite useful in reducing the complications of the phase behavior calculations and has been rigorously tested for complex geothermal and multicomponent flow and transport problems with buoyancy [65], and for reactive flow of species in reservoirs [62]. The working flow of the OBL approach is shown in Fig. 14.

Flow diagram
The work of [62] presented a 1-D case of calcite dissolution using element formulation and molar variables. The dissolution and precipitation reactions were given by,
The test was implemented using the OBL approach, and the equilibrium constants for the equilibrium reactions were scaled up to show the effects of the reactions on the overall system. The initial elements present in the reservoir are water and some other ions, while \(\hbox {CO}_{2}\) is injected continuously. The simulation results highlight the effect of \(\hbox {CO}_{2}\) on the behavior of ions in the reservoir, and the variations in their concentrations after 2000 days of simulation, as shown in Fig. 15.

Element (left) and component (right) for the reaction system presented by [62]
The injection of \(\hbox {CO}_{2}\) starts at distance 0 m causing the vaporization of water which in turn accelerates the calcite deposition. This causes an increase in the reactive porosity of the system and in turn impacts the permeability of the reservoir. The production (precipitation) and dissolution of ions will contribute to the transport of fluids in the system as more and more \(\hbox {CO}_{2}\) is injected. The molar formulation was used in the latest case opposed to the approach of [44] to easily couple it with the OBL scheme and significantly improve the run-time of the simulation. This also shows that the molar framework is capable of handling multiple equilibrium reactions when integrated with high linearization schemes such as OBL.
8 Closing arguments and the way forward
The continuous increase for the need of hydrocarbons in the marketplace has pushed for constant improvements on the performance of the existing petroleum reservoir simulators physically and numerically. The area of reactive transport where chemical reactions greatly affect the flow of different fluids in the reservoir and the characterization of rock–fluid interactions through precipitation and dissolution processes are a new attraction for current research in the petroleum applications of reactive transport, which contributes to improving the quality and accuracy of numerical reservoir simulations.
In this work, we described the physical significance of geochemical equilibrium and the reaction systems on the surrounding environment in porous media. A detailed procedure was outlined on how to mathematically implement the reactions in the framework of an existing reservoir simulator for both natural and molar variables formulations. A brief comparison was drawn on the performance of the two formulations and was supported with a review of two numerical cases from the literature. Ultimately, the choice of the formulation is dictated by the physical problem to be modeled where a trade-off has to be made between accuracy and efficiency.
In the future, we aim to integrate these reaction implementation techniques with a mimetic finite difference (MFD) discretization scheme [5] and a parameterized set of governing equations using the OBL method while using unstructured grids and full tensor permeability [2]. Upon the completion of such a simulator, it would be able to simulate field problems of hydrocarbon recovery with realistic scenarios such as heterogeneous 3-D aquifer and artificial groundwater recharge, CO2 storage and sequestration and in situ upgrading of shale oil.
References
Abd A, Alyafie N (2019) A parametric investigation on the effect of rock and fluid properties in upscaling of spontaneous imbibition. Reservoir Management in Carbonates, Third EAGE WIPIC Workshop
Abd AS, Abushaikha A (2019) A review of numerical modelling techniques for reactive transport in subsurface reservoirs and application in mimetic finite difference discretization schemes. SPE Europec featured at 81st EAGE Conference and Exhibition
Abd AS, Elhafyan E, Siddiqui AR, Alnoush W, Blunt MJ, Alyafei N (2019) A review of the phenomenon of counter-current spontaneous imbibition: analysis and data interpretation. J Petrol Sci Eng 180:456–470
Abushaikha A, Terekhov K (2018) Hybrid-mixed mimetic method for reservoir simulation with full tensor permeability. ECMOR XVI-16th European Conference on the Mathematics of Oil Recovery
Abushaikha AS, Terekhov KM (2020) A fully implicit mimetic finite difference scheme for general purpose subsurface reservoir simulation with full tensor permeability. J Comput Phys 406:109194
Abushaikha AS, Voskov DV, Tchelepi HA (2017) Fully implicit mixed-hybrid finite-element discretization for general purpose subsurface reservoir simulation. J Comput Phys 346:514–538
Acharya RC, Valocchi AJ, Werth CJ, Willingham TW (2007) Pore-scale simulation of dispersion and reaction along a transverse mixing zone in two-dimensional porous media. Water Resour Res 43(10)
Alhashmi Z, Blunt M, Bijeljic B (2015) Predictions of dynamic changes in reaction rates as a consequence of incomplete mixing using pore scale reactive transport modeling on images of porous media. J Cont Hydrol 179:171–181
Anbeek C (1992) Surface roughness of minerals and implications for dissolution studies. Geochim Cosmochim Acta 56(4):1461–1469
Andersen GR (1991) Physical mechanisms controlling the strength and deformation behavior of frozen sand. PhD Thesis, Massachusetts Institute of Technology
Andre L, Audigane P, Azaroual M, Menjoz A (2007) Numerical modeling of fluid-rock chemical interactions at the supercritical CO2-liquid interface during \(CO_2\) injection into a carbonate reservoir, the dogger aquifer (paris basin, france). Energy Convers Manag 48(6):1782–1797
Angeli M, Soldal M, Skurtveit E, Aker E (2009) Experimental percolation of supercritical \(CO_2\) through a caprock. Energy Proced 1(1):3351–3358
Annunziatellis A, Beaubien S, Bigi S, Ciotoli G, Coltella M, Lombardi S (2008) Gas migration along fault systems and through the vadose zone in the latera caldera (central Italy): implications for \(CO_2\) geological storage. Int J Greenhouse Gas Control 2(3):353–372
Apps JA, Zheng L, Zhang Y, Xu T, Birkholzer JT (2010) Evaluation of potential changes in groundwater quality in response to \(CO_2\) leakage from deep geologic storage. Trans Porous Media 82(1):215–246
Arbogast T, Bryant S, Dawson C, Saaf F, Wang C, Wheeler M (1996) Computational methods for multiphase flow and reactive transport problems arising in subsurface contaminant remediation. J Comput Appl Math 74(1–2):19–32
Baker J (1995) Continental-scale magmatic carbon dioxide seepage recorded by dawsonite in the bowen-gunnedah-sydney basin system, eastern australia. SEPM J Sedim Res 65A
Beaulieu E, Goddéris Y, Labat D, Roelandt C, Calmels D, Gaillardet J (2011) Modeling of water-rock interaction in the mackenzie basin: competition between sulfuric and carbonic acids. Chem Geol 289(1–2):114–123
Beckingham L, Peters C, Um W, Jones K, Lindquist W (2013) 2D and 3D imaging resolution trade-offs in quantifying pore throats for prediction of permeability. Adv Water Resour 62:1–12
Beisman JJ, Maxwell RM, Navarre-Sitchler AK, Steefel CI, Molins S (2015) Parcrunchflow: an efficient, parallel reactive transport simulation tool for physically and chemically heterogeneous saturated subsurface environments. Comput Geosci 19(2):403–422
Bethke C (2007) Geochemical and biogeochemical reaction modeling, 2nd edn. Cambridge University Press, Cambridge
Brown BD, Rolston DE (1980) Transport and transformation of methyl bromide in soils. Soil Sci 130(2):68–75
Busch A, Alles S, Gensterblum Y, Prinz D, Dewhurst D, Raven M, Stanjek H, Krooss B (2008) Carbon dioxide storage potential of shales. Int J Greenhouse Gas Control 2(3):297–308
Békri S, Thovert J-F, Adler P (1997) Dissolution and deposition in fractures. Eng Geol 48(3–4):283–308
Cao J, Kitanidis PK (1998) Pore-scale dilution of conservative solutes: an example. Water Resour Res 34(8):1941–1949
Carroll S, Carey JW, Dzombak D, Huerta NJ, Li L, Richard T, Um W, Walsh SD, Zhang L (2016) Review: role of chemistry, mechanics, and transport on well integrity in \(CO_2\) storage environments. Int J Greenhouse Gas Control 49:149–160
Chapman B, James R, Jung R, Washington H (1982) Modelling the transport of reacting chemical contaminants in natural streams. Mar Freshwater Res 33(4):617
Chapman BM (1982) Numerical simulation of the transport and speciation of nonconservative chemical reactants in rivers. Water Resour Res 18(1):155–167
Charlton SR, Parkhurst DL (2011) Modules based on the geochemical model phreeqc for use in scripting and programming languages. Comput Geosci 37(10):1653–1663
Cirpka OA, Olsson A, Ju Q, Rahman MA, Grathwohl P (2006) Determination of transverse dispersion coefficients from reactive plume lengths. Ground Water 44(2):212–221
Class H, Ebigbo A, Helmig R, Dahle HK, Nordbotten JM, Celia MA, Audigane P, Darcis M, Ennis-King J, Yea Fan (2009) A benchmark study on problems related to CO2 storage in geologic formations. Comput Geosci 13(4):409–434
Coats KH (1980) An equation of state compositional model. Soc Petrol Eng J 20(05):363–376
Credoz A, Bildstein O, Jullien M, Raynal J, Pétronin J-C, Lillo M, Pozo C, Geniaut G (2009) Experimental and modeling study of geochemical reactivity between clayey caprocks and \(CO_2\) in geological storage conditions. Energy Proced 1(1):3445–3452
Cubillas P, Köhler S, Prieto M, Causserand C, Oelkers EH (2005) How do mineral coatings affect dissolution rates? an experimental study of coupled caco3 dissolution-cdco3 precipitation. Geochim Cosmochim Acta 69(23):5459–5476
Daccord G (1987) Chemical dissolution of a porous medium by a reactive fluid. Phys Rev Lett 58(5):479–482
Daccord G, Lenormand R, Liétard O (1993) Chemical dissolution of a porous medium by a reactive fluid–I. Model for the “wormholing’’ phenomenon. Chem Eng Sci 48(1):169–178
Darcis M, Class H, Flemisch B, Helmig R (2011) Sequential model coupling for feasibility studies of \(CO_2\) storage in deep saline aquifers. Oil Gas Sci Technol Revue d’IFP Energies nouvelles 66(1):93–103
Detwiler RL (2008) Experimental observations of deformation caused by mineral dissolution in variable-aperture fractures. J Geophys Res Solid Earth 113(B8)
Dewanckele J, De Kock T, Boone M, Cnudde V, Brabant L, Boone M, Fronteau G, Van Hoorebeke L, Jacobs P (2012) 4D imaging and quantification of pore structure modifications inside natural building stones by means of high resolution x-ray ct. Sci Total Environ 416:436–448
Dijk P, Berkowitz B (1998) Precipitation and dissolution of reactive solutes in fractures. Water Resour Res 34(3):457–470
Duan Z, Møller N, Weare JH (1992) An equation of state for the \(CH_4\)–\(CO_2\)–\(H_{2}O\) system: I. Pure systems from 0 to 1000 c and 0 to 8000 bar. Geochim Cosmochim Acta 56(7):2605–2617
Emmanuel S, Berkowitz B (2005) Mixing-induced precipitation and porosity evolution in porous media. Adv Water Resour 28(4):337–344
Fahs M, Carrayrou J, Younes A, Ackerer P (2008) On the efficiency of the direct substitution approach for reactive transport problems in porous media. Water Air Soil Pollut 193(1–4):299–308
Fan Y, Durlofsky L, Tchelepi HA (2010) Numerical simulation of the in-situ upgrading of oil shale. SPE J 15(02):368–381
Fan Y, Durlofsky LJ, Tchelepi HA (2012) A fully-coupled flow-reactive-transport formulation based on element conservation, with application to \(CO_2\) storage simulations. Adv Water Resour 42:47–61
Farshidi SF, Fan Y, Durlofsky LJ, Tchelepi HA (2013) Chemical reaction modeling in a compositional reservoir-simulation framework. SPE Reserv Simul Symp
Fischer M, Botz R, Schmidt M, Rockenbauch K, Garbe-Schönberg D, Glodny J, Gerling P, Littke R (2006) Origins of CO2 in permian carbonate reservoir rocks (zechstein, ca2) of the nw-german basin (lower saxony). Chem Geol 227(3–4):184–213
Fowler TD, Vinegar HJ (2009) Oil shale icp - colorado field pilots. SPE Western Regional Meeting
Gaus I, Azaroual M, Czernichowski-Lauriol I (2005) Reactive transport modelling of the impact of CO2 injection on the clayey cap rock at sleipner (north sea). Chem Geol 217(3–4):319–337
Gautam R, Seider WD (1979) Computation of phase and chemical equilibrium: Part II. Phase-splitting. AIChE J 25(6):999–1006
Gilfillan SM, Ballentine CJ, Holland G, Blagburn D, Lollar BS, Stevens S, Schoell M, Cassidy M (2008) The noble gas geochemistry of natural CO2 gas reservoirs from the colorado plateau and rocky mountain provinces, USA. Geochim Cosmochim Acta 72(4):1174–1198
Gramling CM, Harvey CF, Meigs LC (2002) Reactive transport in porous media: a comparison of model prediction with laboratory visualization. Environ Sci Technol 36(11):2508–2514
Group CM (2008) GEM user manual
Group CM (2008) STARS user manual
Guadagnini A, Sanchez-Vila X, Saaltink MW, Bussini M, Berkowitz B (2009) Application of a mixing-ratios based formulation to model mixing-driven dissolution experiments. Adv Water Resour 32(5):756–766
Gunter W, Bachu S, Law D-S, Marwaha V, Drysdale D, Macdonald D, McCann T (1996) Technical and economic feasibility of CO2 disposal in aquifers within the alberta sedimentary basin, Canada. Energy Convers Manage 37(6–8):1135–1142
Hammond GE, Lichtner PC, Mills RT (2014) Evaluating the performance of parallel subsurface simulators: an illustrative example with pflotran. Water Resour Res 50(1):208–228
Helgeson HC (1968) Evaluation of irreversible reactions in geochemical processes involving minerals and aqueous solutions—I. Thermodynamic relations. Geochim Cosmochim Acta 32(8):853–877
Helgeson HC, Garrels RM, MacKenzie FT (1969) Evaluation of irreversible reactions in geochemical processes involving minerals and aqueous solutions—I. Applications. Geochim Cosmochim Acta 33(4):455–481
Hochstetler DL, Rolle M, Chiogna G, Haberer CM, Grathwohl P, Kitanidis PK (2013) Effects of compound-specific transverse mixing on steady-state reactive plumes: insights from pore-scale simulations and darcy-scale experiments. Adv Water Resour 54:1–10
Izgec O (2009) Reactive flow in vuggy carbonates: methods and models applied to matrix acidizing of carbonates. PhD Thesis, Texas A&M University
Jiang X (2011) A review of physical modelling and numerical simulation of long-term geological storage of CO2. Appl Energy 88(11):3557–3566
Kala K, Voskov D (2018) Parameterization of element balance formulation in reactive compositional flow and transport. ECMOR XVI: 16th European Conference on the Mathematics of Oil Recovery
Kaszuba JP, Janecky DR, Snow MG (2005) Experimental evaluation of mixed fluid reactions between supercritical carbon dioxide and Nacl brine: relevance to the integrity of a geologic carbon repository. Chem Geol 217(3–4):277–293
Katz GE, Berkowitz B, Guadagnini A, Saaltink MW (2011) Experimental and modeling investigation of multicomponent reactive transport in porous media. J Contam Hydrol 120–121:27–44
Khait M, Voskov D (2018) Operator-based linearization for efficient modeling of geothermal processes. Geothermics 74:7–18
Khait M, Voskov DV (2017) Operator-based linearization for general purpose reservoir simulation. J Petrol Sci Eng 157:990–998
Kharaka Y, Cole D, Hovorka S, Gunter W, Knauss K, Freifeld B (2006) Gas-water-rock interactions in frio formation following CO2 injection: implications for the storage of greenhouse gases in sedimentary basins. Geology 34(7):577
Knodel P, Sheahan T, Germaine J (1992) Computer automation of conventional triaxial equipment. Geotech Test J 15(4):311
Lagneau V (2000) Influence des processus géochimiques sur le transport en milieu poreux: application au colmatage de barrières de confinement potentielles dans un stockage en formation géologique. PhD Thesis. Ecole des Mines de Paris
Lasaga AC (1984) Chemical kinetics of water-rock interactions. J Geophys Res Solid Earth 89(B6):4009–4025
Li L, Khait M, Voskov D, Abushaikhaa A (2020) Parallel framework for complex reservoir simulation with advanced discretization and linearization schemes. SPE EAGE EUROPEC
Li W, Einstein HH, Germaine JT (2019) An experimental study of matrix dissolution and wormhole formation using gypsum core flood tests: 1. Permeability evolution and wormhole geometry analysis. J Geophys Res Solid Earth 124(11):11055–11073
Lichtner PC (1985) Continuum model for simultaneous chemical reactions and mass transport in hydrothermal systems. Geochim Cosmochim Acta 49(3):779–800
Lichtner PC (1993) Scaling properties of the time-space kinetic mass transport equations and the local equilibrium limit. Am J Sci 293(4):257–296
Lucia A, Henley H, Thomas E (2015) Multiphase equilibrium flash with salt precipitation in systems with multiple salts. Chem Eng Res Des 93:662–674
Manual SG (2008) Eclipse technical description
Mayer K, Benner S, Frind E, Thornton S, Lerner D (2001) Reactive transport modeling of processes controlling the distribution and natural attenuation of phenolic compounds in a deep sandstone aquifer. J Contam Hydrol 53(3–4):341–368
Michelsen ML (1982) The isothermal flash problem. Part I. Stability. Fluid Phase Equilib 9(1):1–19
Mito S, Xue Z, Ohsumi T (2008) Case study of geochemical reactions at the nagaoka CO2 injection site, japan. Int J Greenhouse Gas Control 2(3):309–318
Molins S, Carrera J, Ayora C, Saaltink MW (2004) A formulation for decoupling components in reactive transport problems. Water Resour Res 40(10)
Nardean S, Ferronato M, Abushaikha A (2020) A novel block non-symmetric preconditioner for mixed-hybrid finite-element-based flow simulations. arXiv preprint arXiv:2009.13916
Navarre-Sitchler A, Steefel CI, Sak PB, Brantley SL (2011) A reactive-transport model for weathering rind formation on basalt. Geochim Cosmochim Acta 75(23):7644–7667
T. U. of Texas at Austin (2000) UTCHEM-9.0: A three-dimensional chemical flood simulator. Reservoir Engineering Research Program, Center for Petroleum and Geosystems Engineering, Technical Documentation
Oostrom M, Mehmani Y, Romero-Gomez P, Tang Y, Liu H, Yoon H, Kang Q, Joekar-Niasar V, Balhoff MT, Dewers T (2014) Pore-scale and continuum simulations of solute transport micromodel benchmark experiments. Comput Geosci 20(4):857–879
Pitzer KS (2018) Activity coefficients in electrolyte solutions
Pokrovsky OS, Schott J (1999) Processes at the magnesium-bearing carbonates/solution interface. II. Kinetics and mechanism of magnesite dissolution. Geochim Cosmochim Acta 63(6):881–897
Porta GM, Chaynikov S, Thovert J-F, Riva M, Guadagnini A, Adler PM (2013) Numerical investigation of pore and continuum scale formulations of bimolecular reactive transport in porous media. Adv Water Resour 62:243–253
Rashidi M, Peurrung L, Tompson A, Kulp T (1996) Experimental analysis of pore-scale flow and transport in porous media. Adv Water Resour 19(3):163–180
Rolle M, Chiogna G, Hochstetler DL, Kitanidis PK (2013) On the importance of diffusion and compound-specific mixing for groundwater transport: an investigation from pore to field scale. J Contam Hydrol 153:51–68
Saaltink MW, Carrera J, Ayora C (2001) On the behavior of approaches to simulate reactive transport. J Contam Hydrol 48(3–4):213–235
Saito H, Nobuoka D, Azuma H, Xue Z, Tanase D (2006) Time-lapse crosswell seismic tomography for monitoring injected CO2 in an onshore aquifer, Nagaoka, Japan. Explor Geophys 37(1):30–36
Schutze C, Sauer U, Beyer K, Lamert H, Brauer K, Strauch G, Flechsig C, Kampf H, Dietrich P (2012) Natural analogues: a potential approach for developing reliable monitoring methods to understand subsurface CO2 migration processes. Environ Earth Sci 67(2):411–423
Singurindy O (2004) Dedolomitization and flow in fractures. Geophys Res Lett 31(24)
Smith MM, Sholokhova Y, Hao Y, Carroll SA (2013) CO2-induced dissolution of low permeability carbonates. Part I: Characterization and experiments. Adv Water Resour 62:370–387
Soulaine C, Roman S, Kovscek A, Tchelepi HA (2018) Pore-scale modelling of multiphase reactive flow: application to mineral dissolution with production of CO2. J Fluid Mech 855:616–645
Spycher N, Pruess K (2005) CO2–H2O mixtures in the geological sequestration of CO2. II. Partitioning in chloride brines at 12–100c and up to 600 bar. Geochim Cosmochim Acta 69(13):3309–3320
Sriyanong P (2013) Element based formulations for coupled flow, transport and chemical reactions. MSc Thesis, Department of Energy Resources Engineering, Stanford University
Steefel C, Depaolo D, Lichtner P (2005) Reactive transport modeling: An essential tool and a new research approach for the earth sciences. Earth Planet Sci Lett 240(3–4):539–558
Steefel CI, Lasaga AC (1994) A coupled model for transport of multiple chemical species and kinetic precipitation/dissolution reactions with application to reactive flow in single phase hydrothermal systems. Am J Sci 294(5)
Steefel CI, Yabusaki SB, Mayer KU (2015) Reactive transport benchmarks for subsurface environmental simulation. Comput Geosci 19(3):439–443
Su D, Mayer KU, MacQuarrie KTB (2020) Min3p-hpc: A high-performance unstructured grid code for subsurface flow and reactive transport simulation. Math Geosci
Suits LD, Sheahan TC, Abdulhadi NO, Germaine JT, Whittle AJ (2011) Thick-walled cylinder testing of clays for the study of wellbore instability. Geotech Test J 34(6):103611
Tartakovsky AM, Redden G, Lichtner PC, Scheibe TD, Meakin P (2008) Mixing-induced precipitation: Experimental study and multiscale numerical analysis. Water Resour Res 44(6)
Tomadakis MM, Sotirchos SV (1993) Transport properties of random arrays of freely overlapping cylinders with various orientation distributions. J Chem Phys 98(1):616–626
van der Lee J, De Windt L, Lagneau V, Goblet P (2003) Module-oriented modeling of reactive transport with hytec. Comput Geosci 29(3):265–275
Varloteaux C, Békri S, Adler PM (2013) Pore network modelling to determine the transport properties in presence of a reactive fluid: from pore to reservoir scale. Adv Water Resour 53:87–100
Vilcáez J, Morad S, Shikazono N (2017) Pore-scale simulation of transport properties of carbonate rocks using fib-sem 3D microstructure: implications for field scale solute transport simulations. J Natl Gas Sci Eng 42:13–22
Voskov DV, Henley H, Lucia A (2017) Fully compositional multi-scale reservoir simulation of various CO2 sequestration mechanisms. Comput Chem Eng 96:183–195
Waage P, Gulberg CM (1986) Studies concerning affinity. J Chem Educ 63(12):1044
Wartha E-M, Bösenhofer M, Harasek v (2020) Characteristic chemical time scales for reactive flow modeling. Combust Sci Technol pp 1–26
Watson M, Boreham C, Tingate P (2004) Carbon dioxide and carbonate cements in the otway basin: implications for geological storage of carbon dioxide. APPEA J 44(1):703
Wellington SL, Berchenko IE, De Rouffignac EP, Fowler TD, Ryan RC, Shahin GT, Stegemeier GL, Vinegar HJ, Zhang E (2005) In situ thermal processing of an oil shale formation to produce a desired product. US Patent
White AF, Brantley SL (1995) Chemical weathering rates of silicate minerals. De Gruyter, Inc.
White AF, Brantley SL (2003) The effect of time on the weathering of silicate minerals: why do weathering rates differ in the laboratory and field? Chem Geol 202(3–4):479–506
Willingham TW, Werth CJ, Valocchi AJ (2008) Evaluation of the effects of porous media structure on mixing-controlled reactions using pore-scale modeling and micromodel experiments. Environ Sci Technol 42(9):3185–3193
Wollenweber J, Alles S, Busch A, Krooss B, Stanjek H, Littke R (2010) Experimental investigation of the CO2 sealing efficiency of caprocks. Int J Greenhouse Gas Control 4(2):231–241
Wollenweber J, Alles SA, Kronimus A, Busch A, Stanjek H, Krooss BM (2009) Caprock and overburden processes in geological CO2 storage: an experimental study on sealing efficiency and mineral alterations. Energy Proced 1(1):3469–3476
Worden RH (2006) Dawsonite cement in the triassic lam formation, shabwa basin, yemen: a natural analogue for a potential mineral product of subsurface CO2 storage for greenhouse gas reduction. Mar Pet Geol 23(1):61–77
Xu R, Li R, Ma J, He D, Jiang P (2017) Effect of mineral dissolution/precipitation and CO2 exsolution on CO2 transport in geological carbon storage. Acc Chem Res 50(9):2056–2066
Xu T, Apps JA, Pruess K (2003) Reactive geochemical transport simulation to study mineral trapping for CO2 disposal in deep arenaceous formations. J Geophys Res Solid Earth 108(B2)
Xu T, Pruess K (1998) Coupled modeling of non-isothermal multiphase ow, solute transport and reactive chemistry in porous and fractured media: 1. Model development and validation. Lawrence Berkeley National Laboratory Report LBNL-42050
Yeh G-T, Fang Y, Zhang F, Sun J, Li Y, Li M-H, Siegel MD (2009) Numerical modeling of coupled fluid flow and thermal and reactive biogeochemical transport in porous and fractured media. Comput Geosci 14(1):149–170
Zhang N, Abushaikha AS (2019) Fully implicit reservoir simulation using mimetic finite difference method in fractured carbonate reservoirs. SPE Reservoir Characterisation and Simulation Conference and Exhibition
Acknowledgements
This publication was supported by the National Priorities Research Program grant NPRP11S-1210-170079 from Qatar National Research Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Declarations
The authors have no conflicts of interest to declare that are relevant to the content of this article
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abd, A.S., Abushaikha, A.S. Reactive transport in porous media: a review of recent mathematical efforts in modeling geochemical reactions in petroleum subsurface reservoirs. SN Appl. Sci. 3, 401 (2021). https://doi.org/10.1007/s42452-021-04396-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-021-04396-9