
Abstract
Since December 2019, the world has been exposed to a novel virus from the coronaviruses family, named coronavirus disease 2019 (COVID-19), which has affected the life of millions people around the world. This global pandemic causes a wide spectrum of clinical manifestation in children, adults, and elderly. One side of the spectrum in children is being asymptomatic and the other side is severe inflammatory symptoms. In this article, we describe the clinical manifestations, genetic background, and immune response of children with COVID-19, who are presented with severe multisystem inflammatory syndrome (MIS).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the early twenty-first century, different species of Coronaviruses lead to three outbreaks, including severe acute respiratory syndrome (SARS) in 2002–2003, Middle East respiratory syndrome (MERS) in 2012, and more recently severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1, 2]. Other human coronaviruses that cause respiratory diseases are HKU1, OC43, NL63, and 229E. SARS-CoV-2 is a RNA genotype virus that is most closely related with about 90% nucleotide similarity to a group of SARS-like coronaviruses [3, 4].
The symptoms of first patients from China with novel coronavirus disease (COVID-19) were fever, dry cough, weakness, shortness of breath, and respiratory distress. Other symptoms like fatigue, anorexia, diarrhea, nausea, dizziness, headache, vomiting, abdominal pain, various type of rashes, and conjunctivitis were also reported [5, 6]. Moreover lately, central nervous system involvement and myocarditis associated with SARS-CoV-2 have also been reported [7,8,9,10].
Although children were supposed to be less affected than adults [11], they are also at risk of infections at all ages, while the range of infection varies from asymptomatic to severe phenotypes. The immune system has a main role in fighting COVID-19, while it can also be affected during the infection, leading to severe complications and even death in those with immune dysregulation [12, 13]. Recently, a group of COVID-19 positive test children present with multiple organ inflammatory symptoms that bears a striking resemblance to the inflammatory syndrome symptoms like Kawasaki disease (KD). A study in children and adolescents who had been infected by COVID-19 showed a high proportion in African race. They had presented with gastrointestinal symptom and KD shock syndrome [14].
Multisystem Inflammatory Syndrome
Although the evidences showed up a significant relation between the symptoms of KD and COVID-19, not all symptoms were appeared according to the AHA Kawasaki guideline (fever plus four of five criteria); therefore, the World Health Organization (WHO) called this phenomenon as multisystem inflammatory syndrome (MIS) with the following criteria in “children and adolescents” [15]:
-
1.
Age of less than 10 years old
-
2.
Fever which is lasting 3 days or more
-
3.
At least two accompanying symptoms of (a) rash or bilateral non-purulent conjunctivitis or muco-cutaneous inflammation signs; (b) hypotension or shock; (c) features of cardiovascular dysfunction; (d) coagulopathy; (e) diarrhea, vomiting, or abdominal pain
-
4.
Elevated inflammatory markers such as ESR, CRP, or procalcitonin
-
5.
Exclusion of microbial cause of inflammation
-
6.
Evidence of COVID-19
Multisystem inflammatory syndrome may present at any time, but often occurs 1–6 weeks following an infection [16]. Moreover, these days may overlap with COVID-19 presentation [17]. MIS can appear similar to CD in classic form or may occur in adults with COVID-19. MIS in children (MIS-C) come to pass with evidence of multiple organ failure including neurologic involvement and cardiogenic or vasoplegic shock. Our knowledge of this new incident is increasing day by day. Symptoms that have been seen in pediatric patients up to date are very similar to the symptoms of Kawasaki disease [18].
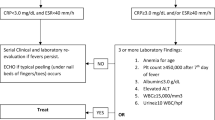
The “guideline for evaluation of a child with suspected MIS-C”, which is proposed by Hennon et al., is presented in Fig. 1 [19]. This guideline describes how we have to approach to a child with inflammatory symptoms.

Guideline for evaluation of a child with suspected MIS-C, proposed by Hennon et al. 2020 [19]; adapted with written permission
Kawasaki Disease
Kawasaki disease is an acute systemic vasculitis. Moreover, it is the most common cause of cardiac complications such as coronary artery aneurysms (CAAs). This disease mostly occurs in ages less than 5 years; notwithstanding, recent studies have shown that heart complications in untreated children may lead to ischemic heart disease and myocarditis in adulthood [20]. Kawasaki disease symptoms include fever, maculopapular rash, erythema of palms and lips, strawberry tongue, erythema of oral and pharyngeal mucosa, periungual peeling of the skin, conjunctivitis, and cervical lymphadenopathy [21, 22]. There are some gastrointestinal manifestations, especially abdominal pain, vomiting, and diarrhea. Musculoskeletal presentations are also expected, which could include arthralgia and arthritis at the first week of disease and also some large weight-bearing joints like the knees and ankles at the second or third week of disease. Genitourinary manifestations as well as respiratory findings are also seen in affected patients. Laboratory tests often show leukocytosis, anemia, elevation of ESR and CRP, elevation in serum transaminases or gammaglutamyl transpeptidase, hyperbilirubinemia and thrombocytosis. Nevertheless, the lack of specific diagnostic tests and biomarkers for KD makes early diagnosis and treatment challenging. Patient’s heart complications which include coronary and peripheral artery aneurysms must be considered because the pericardium, myocardium, endocardium including valves, and the coronary arteries all may be inflamed.
The diagnostic criteria have been modified during years, and they are based on clinical manifestations and the guidelines by the Kawasaki Disease Research Committee guidelines (Japanese guidelines, 2005) [23] and American Heart Association (AHA guideline, 2017) [24].
Kawasaki Disease Features
Although the etiological mechanism of KD is far from being resolved and the seminal factors are also unknown, recent evidence leads us to the role of infectious stimuli, especially viral infections in the patient’s immunity response besides the effect of genetic background in children with KD. These findings will be discussed below.
Genetics
In recent years, the genetic causes of KD have received much attention from researchers to suggest it as a genetic disease. The incidence rate of KD was reported 2.1% and 1.2% higher in children with siblings or parents, who have been diagnosed with KD in 2017 [25]. This incidence rate was higher in Asian, especially in Japanese population [26]. Consequently, genetic studies in KD have been done with the candidate genes or genome-wide association (GWAS) method.
A genetic association study showed that the HLA-E gene polymorphism is involved in disease susceptibility and progression. HLA-E has regulatory functions in both the innate and adaptive immune responses [27]. A recent report suggests the possibility of neutrophil activation and lncRNA transcript profiles involvement in KD and CAA. Therapies that suppress neutrophil activation and gene expression in peripheral blood may prove effective [28]. Another scientific research in KD genome-wide result suggests that HLA-B (HLA-B 54:01) may play a role in pathogenesis of KD, particularly in cases triggering HLA class I and CD8+ T cell-mediated immune response [29]. In genome-wide association studies (GWAS), the research method is to label or type DNA samples from both the normal control group and the disease case group to compare the whole genome of small genomic changes such as single nucleotide polymorphisms (SNPs) to identify involved genes [30,31,32,33].
Immunity
The immune system seems to have a main role in development of KD, particularly in coronary artery dysfunctions. Lee et al. proposed a hypothesis based on the “protein homeostasis system” for the immune pathogenesis of KD in 2012 [34]. Both innate and acquired immune systems are activated after pathogen infection. Meanwhile, the occurrence of KD seems to be more related to the activation of the innate immune system [35]. However, the cell-mediated part of acquired immune system activates macrophages and T cells which leads to the release of lymphokines. Plasma levels of T helper cell type (Th)1 cytokine such as interferon-γ (IFN-γ) and interleukin (IL)-2 and Th2 cytokines such as IL-4 and IL-10 are increased during CK stages [36, 37].
In 2008, Wang et al. showed that the abnormal activation of TLR4 may lead to being one of the initiating factors and caused immune dysfunction in KD patients, which also involved the activation of the nuclear factor κB (NF-κB) and its downstream pathway with the excessive production of pro-inflammatory cytokines such as TNF-α [38]. Furthermore, a study in 2020 revealed that plasma IL-35 that identified from IL-12 cytokine family members increases in patients with KD. IL-35 suppresses CD14+ monocytes and inhibits tumor necrosis factor (TNF)-α and granzyme B secretion by CD14+ monocytes from patients with KD [39]. A recent research identified that TNF and IL-1 would be responsible cytokines key for playing roles in cardiac inflammation and vasculitis in KD patients [40]. In 2020, Sun et al. showed that nuclear factors of activated T cells (NFAT1, NFAT2) and the calcineurin (caN) have been proposed as a significant pathway to be related with KD development; CaN and NFAT are increased at the acute stage of KD, while crucially decreased after treatment with intravenous immunoglobulin (IVIG), along with clinical improvement [6] Moreover, the immune response and clearance of viral infections are directly associated with type I interferon (T1IFN) released amount. A study in mice showed that T cells with a lack of NFAT1 and NFAT2 were unable to clear the acute viral infection [41].
Consistent with bioinformatic analysis data, TLR4, CASP3, and CD40 are the most important genes among all that may be involved in the KD pathways. In addition to participating in apoptosis, CASP3 is reported as a coronary lesions factor in KD. The interaction of CASP3 with the T cell receptor was correlated to the increased transcription of CASP3 and activation of the nuclear factor of the activated T cell signaling pathway, accompanied by the increased level of cytokines such as IL-2 [42].
Besides, it was proven by examining patients that the expression level of CD40 was significantly higher in both acute KD and KD with coronary artery lesion groups, suggesting the possible immunological role of CD40 in the pathogenesis of KD [43].
Infection
Due to the serological and polymerase chain reaction (PCR)-based analyses, possible causal microorganisms of KD have been proposed. In addition to a variety of bacteria and fungi, at least 14 viral species, including coronaviruses, have been reported to be relevant to KD [44]. RNA virus-like inclusion bodies have been detected in the cytoplasm of bronchoepithelial cells of KD patients in the acute phase. In 2011 Rowley et al. reported that KD synthetic antibodies detect antigen. The antigen was localized to intracytoplasmic inclusion bodies (ICI) that has consistent with viral protein and nucleic acid [45]. In another study, Rowley proved that KD antigen can also be detected in ICI in ciliated bronchial epithelium in late-stage KD fatalities in all ages. The high rate of this detection may strengthen the effect of persistent or latent infection following the acute illness hypothesis. A persistent infectious agent could clarify recurrences of KD in at least 1–3% of affected children [46].
Potential Treatment Protocols
Due to the rapid spread and expansion of SARS-CoV-2 infection globally, human society urgently needs to expand ways of treatment and vaccination therapy to overcome the infection. For pediatric patients with COVID-19, who have MIS or KD-like illness (ferritin > 700 ng/mL, CRP > 30 g/dL, or multisystem organ failure or cardiac involvement), the following treatment could be recommended: intravenous immunoglobulin (IVIG) 2 g/kg and aspirin 20–25 mg/kg/dose every 6 h (80–100 mg/kg/day) [47]. Meanwhile, IVIG 2 g/kg as a single infusion with 3-day pulse methylprednisolone could be recommended for those who are at higher risk, such as infants, KD shock syndrome, CRP > 130 g/dL, admission echo Z score > 2.5, or aneurysms. A second dose of IVIG or infliximab (a TNF-alpha inhibitor) could also be considered in the case of first-line treatment failure [19]. It should be mentioned that the first antiviral choice at the moment is Remdesivir. A dose of 5 mg/kg load IV once (max dose 200 mg) on day 1 and then a dose of 2.5 mg/kg (100 mg max dose) IV daily for 9 days are proposed for children [19, 48]. To reduce and control fever, paracetamol or dipyrone is recommended [49]. Antibiotics should be added in patients with secondary bacterial infections base on cultural results. In patients who presented with severe inflammatory symptoms consistent with cytokine storm syndrome (CSS) with or without KD features, the chosen treatment could be anakinra and corticosteroids [50,51,52]. In a very recent study, the administration of IVIG is recommended in COVID-19 patients, with or without MIS features, because it effects on the production of TNF-α and reduces the number of regulatory T cells, which control inflammation and T cell activation. All these therapeutic protocols could affect the cytokine-mediated interstitial and alveolar wall edema in ARDS and consequently on the viral clearance [53].
Sometimes, we encounter a problem in the treatment of KD patients, called IVIG resistance. First time, IVIG resistant was introduced as the persistent fever for 48 h after completion of IVIG treatment [54]. Approximately 10% of KD patients did not respond to IVIG treatment; these patients eventually lead to a higher risk of CALs formation or KD shock syndrome [55]. Several reports and scientific research have suggested various types of biomarkers and genetic genes that may be the reason for being resistance to IVIG. Some of them are clusterin, IL-6, IL-10, IL-17, TNF-α, and regulatory T cells. Other involved risk factors are age, sex, number of illness days, platelet count, ESR, aspartate aminotransferase, hemoglobin concentration, CRP, eosinophil, total bilirubin, lactate dehydrogenase, cholesterol, serum albumin, percentage of neutrophils, and serum sodium level [42]. No alternative treatment has been found yet, but researchers believe that steroids such as prednisolone may reduce the incidence of coronary abnormalities [56]. Infliximab, methotrexate, and plasma exchange have also been reported to be useful in IVIG treatment failure patients [42]. Sometimes standard AAS treatment after IVIG therapy causes an adverse effect called Reye’s syndrome (RS) in KD patients. Reye’s syndrome comes to pass during the treatment of fever with AAS (anabolic-androgenic steroids), and it is usually preceded by a viral infection, specifically influenza and chickenpox in childhood and teenage, and it may lead to possible side effects such as toxic encephalopathy or liver dysfunction [57]. In general, the point that must be considered in the treatment of these patients is that every child would have a different clinical presentation and therapy must be tailored to each case.
Conclusion
COVID-19 could lead to MIS in children, a few days after infection with SARS-CoV-2. However, it should be considered that the viral etiology of KD was previously reported; and it is still unclear if the prevalence of such phenotype after SARS-CoV-2 is more prevalent than other viruses. Anyway, such inflammatory disease should be promptly treated; it is highly recommended that these patients be visited again a week later after treatment and discharge, because of the risk of developing heart failure or vascular disease.
References
Hanaei S, Rezaei N. COVID-19: developing from an outbreak to a pandemic. Arch Med Res. 2020;51:582–4.
Jabbari P, Jabbari F, Ebrahimi S, Rezaei N. COVID-19: a chimera of two pandemics. Disaster Med Public Health Prep. 2020:1–3.
Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–9.
Golshani M, Saghazadeh A, Rezaei N. SARS-CoV-2–a tough opponent for the immune system. Arch Med Res. 2020.
HuiDS IAE, Madani T, Ntoumi F, Koch R, Dar O. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health: the latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020;91:264–6.
Cao Y, Li L, Feng Z, Wan S, Huang P, Sun X, et al. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020;6(1):1–4.
Jahanshahlu L, Rezaei N. Central nervous system involvement in COVID-19. Arch Med Res. 2020.
Lotfi M, Rezaei N. SARS-CoV-2: a comprehensive review from pathogenicity of the virus to clinical consequences. J Med Virol. 2020;92:1864–74.
Saghazadeh A, Rezaei N. Immune-epidemiological parameters of the novel coronavirus - a perspective. Expert Rev Clin Immunol. 2020;16(5):465–70.
Saleki K, Banazadeh M, Saghazadeh A, Rezaei N. The involvement of the central nervous system in patients with COVID-19. Rev Neurosci. 2020;31(4):453–6.
Rezaei N. COVID-19 affects healthy pediatricians more than pediatric patients. Infect Control Hosp Epidemiol. 2020;1.
Saghazadeh A, Rezaei N. Immune-epidemiological parameters of the novel coronavirus–a perspective. Expert Rev Clin Immunol. 2020:1–6.
Yazdanpanah F, Hamblin MR, Rezaei N. The immune system and COVID-19: friend or foe? Life Sci. 2020;256:117900.
Toubiana J, Poirault C, Corsia A, Bajolle F, Fourgeaud J, Angoulvant F, et al. Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: prospective observational study. Bmj. 2020:369.
WHO. Multisystem inflammatory syndrome in children and adolescents with COVID-19. 2020.
Kijima E, Yamamura H, Okada M, Nagasawa M. Kawasaki disease and invasive pneumococcal infection. SN Compr Clin Med. 2020:1–2.
DeBiasi RL, Song X, Delaney M, Bell M, Smith K, Pershad J, et al. Severe COVID-19 in children and young adults in the Washington, DC metropolitan region. J Pediatrics. 2020.
Bahrami A, Vafapour M, Moazzami B, Rezaei N. Hyperinflammatory shock related to COVID-19 in a patient presenting with multisystem inflammatory syndrome in children: first case from Iran. J Paediatr Child Health. 2020.
Hennon TR, Penque MD, Abdul-Aziz R, Alibrahim OS, McGreevy MB, Prout AJ, et al. COVID-19 associated multisystem inflammatory syndrome in children (MIS-C) guidelines; a Western New York approach. Prog Pediatr Cardiol. 2020;57:101232.
Huang Y-H, Chen K-D, Lo M-H, Cai X-Y, Chang L-S, Kuo Y-H, et al. Decreased DNA methyltransferases expression is associated with coronary artery lesion formation in Kawasaki disease. Int J Med Sci. 2019;16(4):576–82.
McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135(17):e927–e99.
Singh S, Jindal AK, Pilania RK. Diagnosis of Kawasaki disease. Int J Rheum Dis. 2018;21(1):36–44.
Ayusawa M, Sonobe T, Uemura S, Ogawa S, Nakamura Y, Kiyosawa N, et al. Revision of diagnostic guidelines for Kawasaki disease (the 5th revised edition). Pediatr Int. 2005;47(2):232–4.
Saguil A, Fargo MV, Grogan SP. Diagnosis and management of Kawasaki disease. Am Fam Physician. 2015;91(6):365–71.
Anthopoulos LG. The Smart City in Practice. Understanding smart cities: a tool for smart government or an industrial trick?: Springer; 2017. p. 47–185.
Singh S, Vignesh P, Burgner D. The epidemiology of Kawasaki disease: a global update. Arch Dis Child. 2015;100(11):1084–8.
Lin YJ, Wan L, Wu JY, Sheu JC, Lin CW, Lan YC, et al. HLA–E gene polymorphism associated with susceptibility to Kawasaki disease and formation of coronary artery aneurysms. Arthritis Rheum. 2009;60(2):604–10.
Tai-Ming K, Jeng-Sheng C, Chen S-P, Yi-Min L, Chia-Jung C, Fuu-Jen T, et al. Genome-wide transcriptome analysis to further understand neutrophil activation and lncRNA transcript profiles in Kawasaki disease. Sci Rep (Nature Publisher Group). 2019;9(1).
Kwon Y-C, Sim BK, Yu JJ, Yun SW, Yoon KL, Lee K-Y, et al. HLA-B* 54: 01 is associated with susceptibility to Kawasaki disease. Circulation. 2019;12(1):e002365.
Lv Y-W, Wang J, Sun L, Zhang J-M, Cao L, Ding Y-Y, et al. Understanding the pathogenesis of Kawasaki disease by network and pathway analysis. Comput Math Methods Med. 2013;2013.
Assari R, Aghighi Y, Ziaee V, Sadr M, Rahmani F, Rezaei A, et al. Pro-inflammatory cytokine single nucleotide polymorphisms in Kawasaki disease. Int J Rheum Dis. 2018;21(5):1120–6.
Assari R, Aghighi Y, Ziaee V, Sadr M, Rezaei A, Rahmani F, et al. Interleukin-4 cytokine single nucleotide polymorphisms in Kawasaki disease: a case-control study and a review of knowledge. Int J Rheum Dis. 2018;21(1):266–70.
Rahmani F, Ziaee V, Assari R, Sadr M, Rezaei A, Sadr Z, et al. Interleukin 10 and transforming growth factor beta polymorphisms as risk factors for Kawasaki disease: a case-control study and meta-analysis. Avicenna J Med Biotechnol. 2019;11(4):325–33.
Lee K-Y, Rhim J-W, Kang J-H. Kawasaki disease: laboratory findings and an immunopathogenesis on the premise of a. Yonsei Med J. 2012;53(2):262–75.
Hara T, Nakashima Y, Sakai Y, Nishio H, Motomura Y, Yamasaki S. Kawasaki disease: a matter of innate immunity. Clin Exp Immunol. 2016;186(2):134–43.
Ferdosian F, Dastgheib SA, Morovati-Sharifabad M, Lookzadeh MH, Noorishadkam M, Mirjalili SR, et al. Cumulative evidence for association between IL-10 polymorphisms and Kawasaki disease susceptibility: a systematic review and meta-analysis. Fetal Pediatr Pathol. 2019:1–13.
Lee SB, Kim YH, Hyun MC, Kim YH, Kim HS, Lee YH. T-helper cytokine profiles in patients with Kawasaki disease. Korean Circ J. 2015;45(6):516–21.
Wang G, Li C, Zu Y, Yang W. Changes and significance for regulatory factors for signal pathways of Toll-like receptors in immunological pathogenesis of Kawasaki disease. Zhonghua er ke za zhi= Chinese journal of pediatrics. 2008;46(1):49–54.
Kandel N, Chungong S, Omaar A, Xing J. Health security capacities in the context of COVID-19 outbreak: an analysis of international health regulations annual report data from 182 countries. Lancet. 2020;395:1047–53.
Stock AT, Jama HA, Hansen JA, Wicks IP. TNF and IL-1 play essential but temporally distinct roles in driving cardiac inflammation in a murine model of Kawasaki disease. J Immunol. 2019;202(11):3151–60.
Xu T, Keller A, Martinez GJ. NFAT1 and NFAT2 differentially regulate CTL differentiation upon acute viral infection. Front Immunol. 2019;10:184.
Tang B, Lo HH, Lei C, Hsiao W-LW, Guo X, Bai J, et al. Adjuvant herbal therapy for targeting susceptibility genes to Kawasaki disease: an overview of epidemiology, pathogenesis, diagnosis and pharmacological treatment of Kawasaki disease. Phytomedicine. 2020;153208.
Wang G, Li C, Zu Y. Change of MyD88-independent signal transduction of Toll-like receptor 4 in immunological pathogenesis of Kawasaki disease. Zhonghua er ke za zhi= Chinese journal of pediatrics. 2007;45(11):818.
Nakamura A, Ikeda K, Hamaoka K. Aetiological significance of infectious stimuli in Kawasaki disease. Front Pediatr. 2019;7.
Rowley AH. Kawasaki disease: novel insights into etiology and genetic susceptibility. Annu Rev Med. 2011;62:69–77.
Rowley AH, Baker SC, Shulman ST, Garcia FL, Fox LM, Kos IM, et al. RNA-containing cytoplasmic inclusion bodies in ciliated bronchial epithelium months to years after acute Kawasaki disease. PLoS One. 2008;3(2):e1582.
Shulman ST. Intravenous immunoglobulin for the treatment of Kawasaki disease. Pediatr Ann. 2017;46(1):e25–e8.
Lotfi M, Hamblin MR, Rezaei N. COVID-19: transmission, prevention, and potential therapeutic opportunities. Clin Chim Acta. 2020;508:254–66.
Carlotti APCP, Carvalho WB, Johnston C, Rodriguez IS, Delgado AF. COVID-19 diagnostic and management protocol for pediatric patients. Clinics. 2020;75.
Rajasekaran S, Kruse K, Kovey K, Davis AT, Hassan NE, Ndika AN, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children. Pediatr Crit Care Med. 2014;15(5):401–8.
Jahanshahlu L, Rezaei N. Monoclonal antibody as a potential anti-COVID-19. Biomed Pharmacother. 2020;129:110337.
Saghazadeh A, Rezaei N. Towards treatment planning of COVID-19: rationale and hypothesis for the use of multiple immunosuppressive agents: anti-antibodies, immunoglobulins, and corticosteroids. Int Immunopharmacol. 2020;84:106560.
Prete M, Favoino E, Catacchio G, Racanelli V, Perosa F. SARS-CoV-2 infection complicated by inflammatory syndrome. Could high-dose human immunoglobulin for intravenous use (IVIG) be beneficial? Autoimmunity Rev. 2020.
Singer LT, Nelson S, Short E, Min MO, Lewis B, Russ S, et al. Prenatal cocaine exposure: drug and environmental effects at 9 years. J Pediatrics. 2008;153(1):105–11. e1.
Gamez-Gonzalez LB, Moribe-Quintero I, Cisneros-Castolo M, Varela-Ortiz J, Muñoz-Ramírez M, Garrido-García M, et al. Kawasaki disease shock syndrome: unique and severe subtype of Kawasaki disease. Pediatr Int. 2018;60(9):781–90.
Miyata K, Kaneko T, Morikawa Y, Sakakibara H, Matsushima T, Misawa M, et al. Efficacy and safety of intravenous immunoglobulin plus prednisolone therapy in patients with Kawasaki disease (post RAISE): a multicentre, prospective cohort study. Lancet Child Adolesc Health. 2018;2(12):855–62.
Chornomydz I, Boyarchuk O, Chornomydz A. Reye (Ray’s) syndrome: a problem everyone should remember. Georgian Med News. 2017;272:110–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Covid-19
Rights and permissions
About this article

Cite this article
Sarzaeim, M., Rezaei, N. Kawasaki Disease and Multisystem Inflammatory Syndrome in Children with COVID-19. SN Compr. Clin. Med. 2, 2096–2101 (2020). https://doi.org/10.1007/s42399-020-00558-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42399-020-00558-9