
Abstract
Background
Genetic diversity of mutations in the CYP21A2 gene is the main cause of the monogenic congenital adrenal hyperplasia (CAH) disorder. On chromosome 6p21.3, the CYP21A2 gene is partially overlapped by the TNXB gene, the two residing in tandem with their highly homologous corresponding pseudogenes (CYP21A1P and TNXA), which leads to recurrent homologous recombination.
Methods and results
In the present study, the genetic status of an ethnic Greek-Cypriot family, with a female neonate that was originally classified as male and manifested the salt-wasting (SW) form, is presented. Genetic defects in the CYP21A2 and TNXB genes were investigated by Sanger sequencing multiplex ligation-dependent probe amplification (MLPA) and a real-time PCR assay. The neonate carried in compound heterozygosity the TNXA/TNXB chimeric gene complex (termed CAH-X CH-1) that results in a contiguous CYP21A2 and TNXB deletion and in her second allele the pathogenic IVS2-13A/C > G (c.655A/C > G) in CYP21A2.
Conclusions
The classic SW-CAH due to 21-hydroxylase (21-OH) deficiency may result from various complex etiological mechanisms and, as such, can involve the formation of monoallelic TNXA/TNXB chimeras found in trans with other CYP21A2 pathogenic variants. This is a rare case of CAH due to 21-hydroxylase deficiency, which elucidates the role of the complex RCCX CNV structure in the development of the disease. Identification of the correct CAH genotypes for a given phenotype is of considerable value in assisting clinicians in prenatal diagnosis, appropriate treatment, and genetic counseling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders that affect cortisol biosynthesis and cause defective steroidogenesis [1]. The most prevalent type of CAH is 21-hydroxylase (21-OH) (90–95% of cases), followed by the next most common type of 11β-hydroxylase (11β-OH) (∼ 5% of cases) and other infrequent types [2]. The disorder has a wide spectrum of clinical phenotypes, and severity depends on the patients’ underlying CYP21A2 genotype [1, 3, 4]. Currently, based on the inherited genetic background, the disorder is classified into the “classic” and “non-classic” (NC late onset) CAH forms [1, 2]. In “classic CAH,” during the course of fetal development, the adrenal cannot sufficiently produce cortisol and overproduces androgens, leading to varying degrees of prenatal virilization of the external genitalia in affected girls [5]. The multiallelic and tandem RCCX copy number variation (CNV) locus is found on chromosome 6p23.1 within the major histocompatibility complex (MHC) class III region. The RCCX structure is characteristically defined by the copy number of a DNA segment containing a series of genes, namely, serine/threonine kinase 19 (STK19), complement 4 (C4), steroid 21-OH (CYP21A2), and tenascin-X (TNX). The most frequent RCCX haplotype (69%) comprises two segments that are situated close to each other, with the first one containing the pseudogenes STK19-C4A-CYP21A1P-TNXA and the second containing the genes STK19B-C4B-CYP21A2-TNXB [6]. The majority of deleterious variants, including about 75% of those reported in 21-OH deficiency cases, are transferred to the CYP21A2 gene by small conversions from the CYP21A1P pseudogene during meiosis [6,7,8]. It is estimated that the remaining 20–30% of the common CYP21A2 pathogenic variants in CAH cases involve large gene deletions or amplifications of the CYP21A2 gene and the other contiguous genes. The formation of these mutants is the result of misalignments due to unequal crossing over during meiosis [6, 9,10,11]. To date, nine different haplotypes of chimeric CYP21A1P/CYP21A2 genes (CH-1 to CH-9) in 21-OH deficiency have been detected [6, 12, 13]. Likewise, so far, three other chimeric recombination events between the TNXB/TNXA have been reported, which result in the deletion of the CYP21A2 gene, thus creating a CAH disease-causing allele [14, 15]. These three types of TNXB/TNXA chimeras disrupt both the CYP21A2 and the TNXB genes and, according to the type of disruption, have been termed CAH-X CH-1, CH-2, and CH-3 [15]. Approximately 10% of patients with CAH have CAH-X syndrome due to the presence of a monoallelic nonfunctional TNXB/TNXA chimeric gene in combination with another CYP21A2 pathogenic mutation in the second allele [2]. The clinical phenotype of CAH-X includes joint hypermobility, arthralgias, joint dislocations, hernias, and cardiac malformations [2, 15]. The TNXB gene encodes for the large extracellular matrix glycoprotein tenascin-X (TNX) and has a significant structural function in the assembly of collagen deposition by dermal fibroblasts and the connective tissues of the heart and skeletal muscle [16]. Ehlers-Danlos syndrome (EDS), characterized by two autosomal recessive mutations in the TNXB gene, leads to complete deficiency of tenascin-X: patients typically have the classical hypermobile type of EDS, with a characteristically large range of joint movements [17, 18]. Interestingly, there have also been reports of heterozygous TNXB gene mutations causing tenascin-X haploinsufficiency, which also results in the signs and symptoms of hypermobile-type EDS [14, 19, 20].
In recent years, numerous studies have established a robust correlation between the genotype and the phenotype of CAH of a large number of CYP21A2 defects [1,2,3,4, 21,22,23,24,25,26,27]. In this study, we present the genetic features of the disease in a family of Cypriot descent with the salt-wasting (SW) classic form as a result of the TNXA/TNXB chimera, resulting in deletions of CYP21A2 and TNXB in one allele [7, 15] and a severe case of CYP21A2 mutation in the second allele.
Case description
Patients and bioethics approval
Written and oral informed consent was obtained from both parents of the infant under investigation screened for mutations in the CYP21A2 gene. The study was approved by the Cyprus National Ethics Committee, and all methods performed were in accordance with the relevant guidelines and regulations.
History
A female neonate of non-consanguineous and healthy ethnic Greek-Cypriot parents was born at 40 weeks of gestation with normal delivery and an APGAR score of 10. On initial examination, the neonate was assigned as male based on the phenotypic appearance of the genitalia with an external genital appearance of Prader 5. After the first 2 weeks of life, the neonate was diagnosed with the salt-wasting (SW) form of CAH and reassigned as female based on biochemical findings of SW (failure to thrive, hyponatremia, hyperkalemia, high plasma renin activity (PRA), significantly high 17-OHP > 75.7 nmol/L, and high ACTH). Following the diagnosis of SW-CAH, the infant was started on replacement treatment with glucocorticoids and salt supplementation (hydrocortisone and fludrocortisone), with excellent response regarding linear growth and mental development. Salt supplementation was stopped at the age of 4 months, at which time the infant underwent surgery to correct the appearance of her genitalia where a vaginal opening was formed.
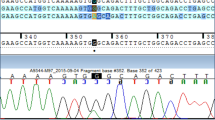
A possible genetic diagnosis with the most severe SW classic form of CAH was speculated, and genetic investigation for mutations in the CYP21A2 gene based on a cascade strategy, as formerly described, was undertaken for the infant and the parents [4, 28, 29]. The neonate was the first child born to this family, and no other close relative was reported with similar clinical issues. SW-CAH secondary to 21-OH deficiency in the infant was justified by the obtained Sanger sequencing and MLPA (MRC, Holland/SALSA MLPA Probemix P050 CAH) analyses (Fig. 1 pedigree and sequences, MLPA). The neonate was identified as carrying in compound heterozygosity the IVS2-13A/C > G (c.655A/C > G)/TNXA/TNXB chimera (termed CAH-X CH-1) and additionally proved to be a female and remained as such as evidenced by the MLPA analyses. The 30-year-old mother was identified as carrying in heterozygosity the pathogenic IVS2-13A/C > G, while the 28-year-old father was concurrently found to also carry in heterozygosity the TNXA/TNXB chimera (Fig. 1 pedigree and sequences, MLPA). The TNXA/TNXB chimera results from an infrequent breakpoint of the TNXB gene, leading to complete deletion of C4B and CYP21A2 [15, 30].

A Pedigree of the investigated family with congenital adrenal hyperplasia. Gray shading indicates the presence of the TNXA/TNXB CH-1 chimeric allele, and hatched lines indicate the presence of the IVS2-13A/C > G mutation. B Sanger sequencing electropherograms indicating the severe IVS2-13A/C > G mutation identified in heterozygosity in the mother and in hemizygosity in the neonate female patient. The position of the IVS2-13A/C > G mutation is shown by red arrows. C MLPA electropherograms (left) and ratio charts (right) showing the CYP21A2 gene deletion extending to exon 35 of TNXB. The father and the neonate patient carried in heterozygosity the TNXA/TNXB CH-1 chimeric gene. Red arrows indicate the reduction of specific sites located on the CYP21A2 and TNXB genes
This rare chimera is characterized by a 120-bp deletion in exon 35 of TNXB and results in TNXB haploinsufficiency and disrupted TGF-β signaling; when found in the homozygous state, it leads to a contiguous gene syndrome consisting of CAH and Ehlers-Danlos syndrome (EDS) [7, 31]. It has been reported that heterozygous TNXB mutations may be associated with the mild joint hypermobility form of EDS [1, 15] and that up to 10% of classic SW-CAH patients harboring in compound heterozygosity a TNXB/CYP21A2 genotype tend to demonstrate an extended phenotype termed CAH-X syndrome [1, 31, 32] (Fig. 2). These patients have also been identified with clinical features of EDS, such as joint hypermobility, chronic arthralgia, hernias, and cardiac defects [7, 15]. The genetic investigation was extended by employing another test in addition to those suggested by the CAH Best Practice Guidelines [33]. Our investigation included the CAH Real Fast CNV Assay (real-time PCR, Vienna Lab) so as to re-confirm the large CYP21A2 deletion that was detected both in the neonate and in the paternal samples. As demonstrated in Fig. 3, the large CYP21A2 was likewise detected both in the neonate and in the paternal samples via the use of this method as well.

Schematic representation of the RCCX locus on chromosome 6p21.3 in which the TNXA and TNXB genes undergo unequal crossover resulting in a TNXA/TNXB CH-1 chimera

CAH Real Fast CNV Assay (real-time PCR, Vienna Lab) showing the large CYP21A2 deletion that was detected both in the patient and in the paternal samples and which confirms the monoallelic TNXA/TNXB CH-1 chimera that was identified both in the father and in the neonate
Discussion
CAH-X syndrome is estimated to be found in approximately 7–9% of CAH patients [7, 14, 34] and is classified into three types (CAH-X CH-1, CAH-X CH-2, and CAH-X CH-3) as a result of chimeric recombination events between the TNXB/TNXA during meiosis, resulting in the deletion of the CYP21A2 gene [15]. The CAH-X CH-1 type is the most frequent of the three and retains a 120-bp deletion at the boundary of exon 35 and intron 35 of TNXB. CAH-X CH-2 and CAH-X CH-3 types are less frequently found in CAH patients, with CAH-X CH-2 being characterized by the variant c.12174C > G (p.Cys4058Trp), which derives from the TNXA pseudogene [7], and CAH-X CH-3, being characterized by a cluster of three mutations (p.Arg4073His, p.Asp4172Asn, and p.Ser4175Asn) also derived from the TNXA pseudogene [35]. CAH-X CH-1 does not produce structural changes in the TNX protein but, rather, reduces the TNX expression, this being in contrast to the altered production of TNX proteins from both CAH-X CH-2 and CAH-X CH-3 [35]. An in-depth molecular genetic approach for the correct diagnosis of the severe forms of CAH, including CAH-X syndrome, is absolutely essential and indeed makes a difference to the everyday practice of an experienced endocrinologist. Lately, in addition to Sanger analysis, which still remains the gold standard for the detection of CYP21A2 gene mutations, the PCR-based techniques of MLPA and real-time PCR assays are widely used for the detection of the severe deletions or duplications. Therefore, in the endocrinologist’s everyday practice, genetic information obtained by using these techniques aids towards taking the most appropriate decisions in cases of prenatal and preimplantation genetic diagnosis, hormonal treatment, and genetic counseling.
The female neonate with the classic SW-CAH of the present study presented the highest degree of virilization, with an external genital appearance of Prader 5, and, when salt supplementation was stopped at the age of 4 months, she underwent surgery for the correction of the appearance of her genitalia where a vaginal opening was formed.
Similarly to the neonate of the present study, CAH patients that carry in compound heterozygosity the splice IVS2-13A/C > G with the TNXA/TNXB (CAH-X CH-1) chimera generally have the most severe SW phenotype [36,37,38]. Several reports in European, Mediterranean, Middle Eastern, and Chinese populations confirmed a high frequency of the pathogenic IVS2-13A/C > G and found it to comprise 20 to 25% of the CYP21A2 mutant alleles and to translate to the most severe SW forms of the disease when inherited in homozygosity [1, 4, 27, 39,40,41,42,43]. Since the first CAH-X syndrome report by Merke et al. [14], it has been established that only 10% of CAH-X patients also have EDS manifestations [15, 44]. Several other studies that followed have reported an incidence of IVS2-13A/C > G combined with the chimeric TNXA/TNXB (CAH-X CH-1) genotype in patients with 21-OH deficiency and with no EDS manifestations [32, 44]. Similarly, the neonate of the present case with a compound heterozygous IVS2-13A/C > G/CAH-X CH-1 genotype presented only SW 21-hyroxylase deficiency and no EDS-related clinical manifestations. More specifically, our case has normal skin when compared to certain patients with biallelic CAH-X genotype and who typically demonstrate skin laxity and abnormal wound healing [45, 46]. The father of the neonate who was a carrier of the chimeric TNXA/TNXB (CAH-X CH-1) genotype also did not present any clinical manifestations of EDS.
In conclusion, we herein report an infrequent case of CAH illustrating that its clinical phenotype depends on the severity of the underlying variants, while the role of the complex RCCX CNV structure in the development of the disease is explained. Our report moreover constitutes an example of the complexities encountered in patients with classic CAH and adds to the understanding of the spectrum of CAH phenotype and the TNX-related disorders, including the CAH-X syndrome.
References
Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, Falhammar H, Fluck CE, Guasti L, Huebner A, Kortmann BBM, Krone N, Merke DP, Miller WL, Nordenstrom A, Reisch N, Sandberg DE, Stikkelbroeck N, Touraine P, Utari A, Wudy SA, White PC (2022) Congenital adrenal hyperplasia-current insights in pathophysiology, diagnostics, and management. Endocr Rev 43(1):91–159. https://doi.org/10.1210/endrev/bnab016
Merke DP, Auchus RJ (2020) Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med 383(13):1248–1261. https://doi.org/10.1056/NEJMra1909786
Neocleous V, Fanis P, Toumba M, Stylianou C, Picolos M, Andreou E, Kyriakou A, Iasonides M, Nicolaou S, Kyriakides TC, Tanteles GA, Skordis N, Phylactou LA (2019) The spectrum of genetic defects in congenital adrenal hyperplasia in the population of Cyprus: a retrospective analysis. Horm Metab Res 51(9):586–594. https://doi.org/10.1055/a-0957-3297
Neocleous V, Fanis P, Phylactou LA, Skordis N (2018) Genotype is associated to the degree of virilization in patients with classic congenital adrenal hyperplasia. Front Endocrinol (Lausanne) 9:733. https://doi.org/10.3389/fendo.2018.00733
Wedell A, Thilen A, Ritzen EM, Stengler B, Luthman H (1994) Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: implications for genetic diagnosis and association with disease manifestation. J Clin Endocrinol Metab 78(5):1145–1152. https://doi.org/10.1210/jcem.78.5.8175971
Carrozza C, Foca L, De Paolis E, Concolino P (2021) Genes and pseudogenes: complexity of the RCCX locus and disease. Front Endocrinol (Lausanne) 12:709758. https://doi.org/10.3389/fendo.2021.709758
Morissette R, Chen W, Perritt AF, Dreiling JL, Arai AE, Sachdev V, Hannoush H, Mallappa A, Xu Z, McDonnell NB, Quezado M, Merke DP (2015) Broadening the spectrum of Ehlers Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab 100(8):E1143-1152. https://doi.org/10.1210/jc.2015-2232
Concolino P, Costella A (2018) Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency: a comprehensive focus on 233 pathogenic variants of CYP21A2 gene. Mol Diagn Ther 22(3):261–280. https://doi.org/10.1007/s40291-018-0319-y
Higashi Y, Tanae A, Inoue H, Fujii-Kuriyama Y (1988) Evidence for frequent gene conversion in the steroid 21-hydroxylase P-450(C21) gene: implications for steroid 21-hydroxylase deficiency. Am J Hum Genet 42(1):17–25
Tusie-Luna MT, White PC (1995) Gene conversions and unequal crossovers between CYP21 (steroid 21-hydroxylase gene) and CYP21P involve different mechanisms. Proc Natl Acad Sci USA 92(23):10796–10800. https://doi.org/10.1073/pnas.92.23.10796
Pignatelli D, Carvalho BL, Palmeiro A, Barros A, Guerreiro SG, Macut D (2019) The complexities in genotyping of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Front Endocrinol (Lausanne) 10:432. https://doi.org/10.3389/fendo.2019.00432
Chen W, Xu Z, Sullivan A, Finkielstain GP, Van Ryzin C, Merke DP, McDonnell NB (2012) Junction site analysis of chimeric CYP21A1P/CYP21A2 genes in 21-hydroxylase deficiency. Clin Chem 58(2):421–430. https://doi.org/10.1373/clinchem.2011.174037
Concolino P (2020) A rare CYP21A2 haplotype clarifies the phenotype-genotype discrepancy in an Italian patient with Non Classical Congenital Adrenal Hyperplasia (NC-CAH). Mol Biol Rep 47(4):3049–3052. https://doi.org/10.1007/s11033-020-05379-6
Merke DP, Chen W, Morissette R, Xu Z, Van Ryzin C, Sachdev V, Hannoush H, Shanbhag SM, Acevedo AT, Nishitani M, Arai AE, McDonnell NB (2013) Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab 98(2):E379-387. https://doi.org/10.1210/jc.2012-3148
Miller WL, Merke DP (2018) Tenascin-X, Congenital adrenal hyperplasia, and the CAH-X syndrome. Horm Res Paediatr 89(5):352–361. https://doi.org/10.1159/000481911
Bristow J, Tee MK, Gitelman SE, Mellon SH, Miller WL (1993) Tenascin-X: a novel extracellular matrix protein encoded by the human XB gene overlapping P450c21B. J Cell Biol 122(1):265–278. https://doi.org/10.1083/jcb.122.1.265
Miller WL (2020) Tenascin-X-Discovery and early research. Front Immunol 11:612497. https://doi.org/10.3389/fimmu.2020.612497
Lindor NM, Bristow J (2005) Tenascin-X deficiency in autosomal recessive Ehlers-Danlos syndrome. Am J Med Genet A 135(1):75–80. https://doi.org/10.1002/ajmg.a.30671
Byers PH, Murray ML (2014) Ehlers-Danlos syndrome: a showcase of conditions that lead to understanding matrix biology. Matrix Biol 33:10–15. https://doi.org/10.1016/j.matbio.2013.07.005
Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, Kucharekova M, Boezeman JB, Schalkwijk J (2003) Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet 73(1):214–217. https://doi.org/10.1086/376564
Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, Lesser M, New MI, White PC (1992) Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest 90(2):584–595. https://doi.org/10.1172/JCI115897
Carrera P, Bordone L, Azzani T, Brunelli V, Garancini MP, Chiumello G, Ferrari M (1996) Point mutations in Italian patients with classic, non-classic, and cryptic forms of steroid 21-hydroxylase deficiency. Hum Genet 98(6):662–665. https://doi.org/10.1007/s004390050280
Stikkelbroeck NM, Hoefsloot LH, de Wijs IJ, Otten BJ, Hermus AR, Sistermans EA (2003) CYP21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: six novel mutations and a specific cluster of four mutations. J Clin Endocrinol Metab 88(8):3852–3859. https://doi.org/10.1210/jc.2002-021681
Loidi L, Quinteiro C, Parajes S, Barreiro J, Leston DG, Cabezas-Agricola JM, Sueiro AM, Araujo-Vilar D, Catro-Feijoo L, Costas J, Pombo M, Dominguez F (2006) High variability in CYP21A2 mutated alleles in Spanish 21-hydroxylase deficiency patients, six novel mutations and a founder effect. Clin Endocrinol (Oxf) 64(3):330–336. https://doi.org/10.1111/j.1365-2265.2006.02465.x
Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Najmabadi H, Saffari F, New MI (2007) Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab 90(4):414–421. https://doi.org/10.1016/j.ymgme.2006.12.005
Krone N, Rose IT, Willis DS, Hodson J, Wild SH, Doherty EJ, Hahner S, Parajes S, Stimson RH, Han TS, Carroll PV, Conway GS, Walker BR, MacDonald F, Ross RJ, Arlt W, United Kingdom Congenital adrenal Hyperplasia Adult Study E (2013) Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: analysis of the United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE) cohort. J Clin Endocrinol Metab 98(2):E346-354. https://doi.org/10.1210/jc.2012-3343
New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, Sun L, Zaidi M, Wilson RC, Yuen T (2013) Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci USA 110(7):2611–2616. https://doi.org/10.1073/pnas.1300057110
Phedonos AA, Shammas C, Skordis N, Kyriakides TC, Neocleous V, Phylactou LA (2013) High carrier frequency of 21-hydroxylase deficiency in Cyprus. Clin Genet 84(6):585–588. https://doi.org/10.1111/cge.12153
Neocleous V, Fanis P, Toumba M, Phedonos AAP, Picolos M, Andreou E, Kyriakides TC, Tanteles GA, Shammas C, Phylactou LA, Skordis N (2017) Variations in the 3’UTR of the CYP21A2 gene in heterozygous females with hyperandrogenaemia. Int J Endocrinol 2017:8984365. https://doi.org/10.1155/2017/8984365
Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, Zipf WB, Rennebohm RM, Yung YuC (2000) Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. J Exp Med 191(12):2183–2196
Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, Miller WL, Bristow J (1997) Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet 17(1):104–108. https://doi.org/10.1038/ng0997-104
Gao Y, Lu L, Yu B, Mao J, Wang X, Nie M, Wu X (2020) The prevalence of the chimeric TNXA/TNXB gene and clinical symptoms of Ehlers-Danlos syndrome with 21-hydroxylase deficiency. J Clin Endocrinol Metab 105(7):2288. https://doi.org/10.1210/clinem/dgaa199
Baumgartner-Parzer S, Witsch-Baumgartner M, Hoeppner W (2020) EMQN best practice guidelines for molecular genetic testing and reporting of 21-hydroxylase deficiency. Eur J Hum Genet 28(10):1341–1367. https://doi.org/10.1038/s41431-020-0653-5
Lao Q, Brookner B, Merke DP (2019) High-throughput screening for CYP21A1P-TNXA/TNXB chimeric genes responsible for Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J Mol Diagn 21(5):924–931. https://doi.org/10.1016/j.jmoldx.2019.06.001
Chen W, Perritt AF, Morissette R, Dreiling JL, Bohn MF, Mallappa A, Xu Z, Quezado M, Merke DP (2016) Ehlers-Danlos syndrome caused by biallelic TNXB variants in patients with congenital adrenal hyperplasiA. Hum Mutat 37(9):893–897. https://doi.org/10.1002/humu.23028
Lee HH (2005) Chimeric CYP21P/CYP21 and TNXA/TNXB genes in the RCCX module. Mol Genet Metab 84(1):4–8. https://doi.org/10.1016/j.ymgme.2004.09.009
Concolino P, Mello E, Minucci A, Giardina E, Zuppi C, Toscano V, Capoluongo E (2009) A new CYP21A1P/CYP21A2 chimeric gene identified in an Italian woman suffering from classical congenital adrenal hyperplasia form. Bmc Med Genet 10:72. https://doi.org/10.1186/1471-2350-10-72
L’Allemand D, Tardy V, Gruters A, Schnabel D, Krude H, Morel Y (2000) How a patient homozygous for a 30-kb deletion of the C4-CYP 21 genomic region can have a nonclassic form of 21-hydroxylase deficiency. J Clin Endocrinol Metab 85(12):4562–4567. https://doi.org/10.1210/jcem.85.12.7018
Finkielstain GP, Chen W, Mehta SP, Fujimura FK, Hanna RM, Van Ryzin C, McDonnell NB, Merke DP (2011) Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 96(1):E161-172. https://doi.org/10.1210/jc.2010-0319
Tolba A, Mandour I, Musa N, Elmougy F, Hafez M, Abdelatty S, Ibrahim A, Soliman H, Labib B, Elshiwy Y, Ramzy T, Elsharkawy M (2022) Copy number variations in genetic diagnosis of congenital adrenal hyperplasia children. Front Genet 13:785570. https://doi.org/10.3389/fgene.2022.785570
Xu C, Jia W, Cheng X, Ying H, Chen J, Xu J, Guan Q, Zhou X, Zheng D, Li G, Zhao J (2019) Genotype-phenotype correlation study and mutational and hormonal analysis in a Chinese cohort with 21-hydroxylase deficiency. Mol Genet Genomic Med 7(6):e671. https://doi.org/10.1002/mgg3.671
Tardy V, Menassa R, Sulmont V, Lienhardt-Roussie A, Lecointre C, Brauner R, David M, Morel Y (2010) Phenotype-genotype correlations of 13 rare CYP21A2 mutations detected in 46 patients affected with 21-hydroxylase deficiency and in one carrier. J Clin Endocrinol Metab 95(3):1288–1300. https://doi.org/10.1210/jc.2009-1202
Vakili R, Baradaran-Heravi A, Barid-Fatehi B, Gholamin M, Ghaemi N, Abbaszadegan MR (2005) Molecular analysis of the CYP21 gene and prenatal diagnosis in families with 21-hydroxylase deficiency in northeastern Iran. Horm Res 63(3):119–124. https://doi.org/10.1159/000084570
Marino R, Garrido NP, Ramirez P, Notaristefano G, Moresco A, Touzon MS, Vaiani E, Finkielstain G, Obregon MG, Balbi V, Soria I, Belgorosky A (2021) Ehlers-Danlos syndrome: molecular and clinical characterization of TNXA/TNXB chimeras in congenital adrenal hyperplasia. J Clin Endocrinol Metab 106(7):e2789–e2802. https://doi.org/10.1210/clinem/dgab033
Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL, Bristow J (2001) A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med 345(16):1167–1175. https://doi.org/10.1056/NEJMoa002939
Demirdas S, Dulfer E, Robert L, Kempers M, van Beek D, Micha D, van Engelen BG, Hamel B, Schalkwijk J, Loeys B, Maugeri A, Voermans NC (2017) Recognizing the tenascin-X deficient type of Ehlers-Danlos syndrome: a cross-sectional study in 17 patients. Clin Genet 91(3):411–425. https://doi.org/10.1111/cge.12853
Funding
This work was supported by the A.G. Leventis Foundation. All four authors (PF, VN, LAP, and NS) of this publication are members of the European Reference Network on Rare Endocrine Conditions: Project ID N0 739543 (https://endo-ern.eu/about/reference-centres/).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
The study was approved by the Cyprus National Ethics Committee and all methods performed were in accordance with the relevant guidelines and regulations.
Informed consent
Written informed consent was obtained from both parents of the infant under investigation.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fanis, P., Skordis, N., Phylactou, L.A. et al. Salt-wasting congenital adrenal hyperplasia phenotype as a result of the TNXA/TNXB chimera 1 (CAH-X CH-1) and the pathogenic IVS2-13A/C > G in CYP21A2 gene. Hormones 22, 71–77 (2023). https://doi.org/10.1007/s42000-022-00410-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42000-022-00410-w