
Abstract
Background
Despite the extensive use of bevacizumab in a range of oncology indications, the US FDA revoked its approval for breast cancers, and multiple negative trials in several solid malignancies have been reported, so the need for predictive biomarkers has increased. The development of predictive biomarkers for anti-angiogenic bevacizumab therapy has long been pursued but without success.
Introduction
Heat shock protein (HSP)-27 expression has recently been identified as a predictive biomarker for bevacizumab in treating metastatic melanoma. This study aimed to evaluate the cost effectiveness of HSP27 biomarker testing before administration of bevacizumab.
Methods
A partitioned survival analysis model with three mutually exclusive health states (progression-free survival, progressed disease, and death) was developed using a Norwegian health system perspective. The proportion of patients in each state was calculated using the area under the Kaplan–Meier curve for progression-free and overall survival derived from trials of bevacizumab and dacarbazine. Three strategies were compared: (1) test-treat with HSP27 biomarker and bevacizumab, (2) treat-all with dacarbazine without HSP27 testing, (3) treat-all with bevacizumab without HSP27 testing. Quality-adjusted life-years (QALYs) and costs were calculated for each strategy and discounted at 4%. A lifetime horizon was applied. Uncertainty analyses were performed. Expected value of perfect information (EVPI) was estimated to assess the potential value of further research to generate more evidence.
Results
Although the test-treat strategy was cost effective compared with treat-all with dacarbazine, it was not cost effective compared with treat-all with bevacizumab without HSP27 testing. However, EVPI results showed very minimal or no value in conducting further research efforts to reduce uncertainties around current information.
Conclusion
The results of this study suggested that testing for HSP27 expression before administering bevacizumab is not cost effective compared with treat-all with bevacizumab without testing. It indicates that HSP27 expression is not cost effective as a potential predictive biomarker for bevacizumab. This may not necessarily mean that HSP27 is a bad biomarker for bevacizumab, but it may mean that bevacizumab is much better than dacarbazine regardless of HSP27 expression, so patient stratification according to HSP27 status is meaningless. Or, indeed, it may imply that HSP27 is not sufficiently good at identifying the right patients for bevacizumab.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Testing for heat shock protein (HSP)-27 expression before administering bevacizumab in patients with metastatic melanoma was not cost effective when compared with treat-all with bevacizumab without testing. It may imply that HSP27 expression is not a good enough biomarker for bevacizumab in identifying patients likely to be responsive or unresponsive to the treatment. |
Further research to identify a predictive biomarker for bevacizumab is yet to be achieved; however, the expected value of perfect information (EVPI) results showed either very low or no value would be obtained from research efforts to generate more evidence around the HSP27 biomarker for bevacizumab. |
EVPI results clearly showed that investing in further study, such as a phase III trial, is not justified given the number of patients with metastatic melanoma in Norway. |
1 Introduction
Cutaneous malignant melanoma is common in fair-skinned populations in many countries [1,2,3,4]. Worldwide, 132,000 melanoma skin cancers occur each year [5]. Incidence and mortality continue to rise across the world [4, 6,7,8]. Norway has among the highest incidence of melanoma in the world [9]. In Norway, the 5-year relative survival is 90% for patients with localized melanoma but only 16% for those with distant melanoma [10].
The routinely available treatment options for metastatic melanoma were high-dose interleukin-2 and dacarbazine, with a low response rate of around 10% [11,12,13]. Chemotherapy has long been the main treatment option for metastatic tumours, even though it is marginally effective, with dacarbazine—the only US FDA-approved drug—the standard drug for most melanoma cases [9]. However, dacarbazine has shown low response rates with no life-extending effect [9]. Recently, new targeted drugs have been developed; in particular, the introduction of BRAF and MEK inhibitors has improved the progression-free survival (PFS) and overall survival (OS) of patients with advanced melanoma [14,15,16,17,18,19,20]. Immunotherapy has also improved the outlook for those with metastatic disease. Thus, biomarker-guided therapies have demonstrated considerable efficacy in the treatment of metastatic melanoma [21, 22]. Currently, the presence of a specific BRAF mutation is the biomarker recommended for routine clinical practice to administer the corresponding targeted therapies (vemurafenib, dabrafenib, trametinib, cobimetinib) in advanced melanoma [21, 23,24,25]. In addition, immune checkpoint inhibition for metastatic melanoma has created significant optimism in recent years [15, 26], but no predictive biomarkers have been validated for immunotherapy.
Given bevacizumab’s mechanism of action as a vascular endothelial growth factor (VEGF) inhibitor, certain patient populations might be less likely to benefit from the drug as indicated by measured VEGF levels. Thus, development of predictive biomarkers for bevacizumab has long been pursued but without success. However, recently, a study identified heat shock protein (HSP)-27 as a potential predictive biomarker for bevacizumab in treating metastatic melanoma (ClinicalTrials.govt ID: NCT00139360) [27]. HSP27 is associated with poor prognosis and treatment resistance in many cancers [28]. Schuster et al. [27] suggested that strong HSP27 tissue expression in melanoma metastasis can predict response to bevacizumab. Based on this recent study, our analysis aimed to assess the cost effectiveness of HSP27 testing before the administration of bevacizumab in patients with metastatic melanoma. Given the early stage of companion biomarker discovery, we also aim to inform decisions about investing in further research to generate more evidence. To the best of our knowledge, no economic evaluations of biomarker testing before the administration of bevacizumab for melanoma have been conducted.
2 Methods
2.1 Overview
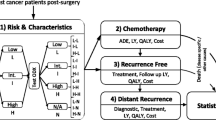
A partitioned survival analysis model, similar to previous economic evaluations of treatments of advanced or metastatic cancers, including the cost effectiveness of a BRAF inhibitor (dabrafenib) and bevacizumab [28, 29], was developed using Microsoft Excel. A hypothetical cohort of 10,000 patients with metastatic melanoma was modelled. The model had three mutually exclusive health states: alive with no progression (PFS), alive with progression (progressed disease [PD]), or dead (Fig. 1). The proportion of patients in each health state over time was calculated using the Kaplan–Meier (KM) survival curves for PFS and OS. Partitioned survival analysis assumes that, at any discrete time point, the difference between the proportion of patients in OS and the proportion of patients in PFS determines the proportion of patients who are alive with PD.

The diagram of health states included in the model
A Norwegian health system perspective was employed, which considered direct costs in treating metastatic melanoma. The model has a monthly cycle and a lifetime horizon to capture all consequences in health benefits and costs. Costs and health outcomes were discounted at 4% annually as recommended by the Norwegian Ministry of Finance and guidelines for health economic evaluation in the health sector [30]. The primary measure of cost effectiveness was the incremental cost per quality-adjusted life-year (QALY) gained.
2.2 Strategies compared
We compared and assessed three strategies in this study. The intervention strategy of interest was testing the HSP27 biomarker status of patients before the administration of bevacizumab (hereafter, referred to as the test-treat strategy). This intervention strategy was compared against two comparator strategies: first, treating all patients with dacarbazine without HSP27 biomarker testing (hereafter, treat-all with dacarbazine strategy), and, second, treating all patients with bevacizumab without HSP27 biomarker testing (hereafter, treat-all with bevacizumab strategy).
Under the test-treat strategy, patients who truly tested positive for HSP27 expression received bevacizumab (online supplementary material 1), and HSP27-negative patients received dacarbazine. However, for patients who falsely tested positive, the health effect of dacarbazine was assumed even though they were treated with bevacizumab. Also, we assumed that dacarbazine was efficacious in patients who truly or falsely tested negative because they were not treated with bevacizumab. Based on the findings of Schuster et al. [27], patients with HSP27 tissue expression with a staining index ≥4 were considered HSP27 biomarker positive, and those below index 4 were considered HSP27 negative.
2.3 Survival estimates for partitioned survival analysis modelling
The survival analysis for bevacizumab used PFS and OS KM data from the phase II study (35 patients) [31], which identified a potential predictive biomarker to guide administration of bevacizumab in treating patients with metastatic melanoma [27]. We obtained the individual patient dataset (IPD) from the clinical research group, which reported the clinical data beyond the published trial period. In this newly obtained IPD, only one patient was censored, and this IPD was used in simulating survival for the bevacizumab arms. Therefore, given that only one patient was censored by the end of the trial period of bevacizumab, use of the actual KM curves was preferred as it was not necessary to make assumptions regarding the parametric distribution of survival times. The OS and PFS survival curves for patients treated with bevacizumab are presented in supplementary material 1.
Dacarbazine survival was based on the PFS and OS KM curves from the dacarbazine arm of a phase III study (675 patients) [32, 33]. This trial was selected based on patient characteristics such as age, Eastern Cooperative Oncology Group (ECOG) performance status, and sex, as they were broadly similar to those in the bevacizumab study (supplementary material 2). No head-to-head trial and no pooled analysis (i.e. meta-analysis) provided a treatment effect for bevacizumab compared with dacarbazine for patients with metastatic melanoma. The KM OS and PFS survival curves were extracted using Digitizelt [34], and individual-level time-to-event data were reconstructed using an algorithm developed in R by Guyot et al. [35]. Parametric survival distributions were fitted to the individual patient data reconstructed from the published KM curves using Stata (StataCorp., 2015. Stata Statistical Software: Release 14. College Station, TX, USA). When fitting parametric survival distributions to KM curves, the exponential, Weibull, log-logistic, log-normal, and gamma distributions were considered. The log-normal distribution was selected for OS and the generalized gamma distribution was selected for PFS based on visual inspection, the Akaike information criterion (AIC) and the Bayesian information criterion (BIC). The pertinent parameters used in extrapolating survival are provided in supplementary material 3. The log-normal distribution provided the lowest AIC and BIC values for OS and the generalised gamma distribution for PFS. AIC and BIC results are provided in supplementary material 4.
2.4 Costs
Costs were calculated from the perspective of the Norwegian healthcare system. Direct costs included drug costs (drug acquisition and administration), HSP27 biomarker testing costs and monitoring costs during and after drug administration [30, 31, 36,37,38,39]. Costs other than healthcare costs were not included. The costs for testing HSP27 biomarker status assumed the units of 200 µg/ml of HSP27 antibody and converted from $US to Norwegian krone (NOK) using the exchange rate of $US1 = NOK7.72, year 2019 values [36]. The drug costs depended on the acquisition price, the dosage, and the treatment duration. The estimated cost of dacarbazine assumed that 850 mg/m2 body surface was administered on day 1 and then once every 3 weeks by intravenous infusion. Dacarbazine could be administered for up to 24 months while in the PFS state and then no dacarbazine given afterwards. After the 24 months of treatment with dacarbazine, monitoring costs were included for patients continuing in PFS and PD states.
Patients received intravenous bevacizumab 10 mg/kg as an infusion on day 1 of a 2-week cycle until progression or for up to 12 cycles (24 weeks). Only monitoring costs were included for patients in PFS who had finished treatment after 6 months. Monitoring costs were considered in PFS and PD states. Treatment was assumed to cease on progression. Cost calculations were made with respect to a monthly cycle length of 30.42 days. An average body weight of 80 kg was assumed. The dosages used in this model follow the information in the summary of product characteristics or trial protocol. Details of the costs are shown in Table 1.
2.5 Health outcome (quality-adjusted life-years)
The primary health outcome of interest in this analysis was QALYs gained. However, utility data on HSP27 testing and bevacizumab in metastatic melanoma were not available, so we used utility values from another study, which at least collected utility data on dacarbazine. The health state utility values were based on data collected in the BREAK-3 trial of dabrafenib versus dacarbazine using the EuroQoL-5 Dimensions, 3 Levels instrument [40]. The health state utility of patients receiving dacarbazine was 0.750. Patients treated with bevacizumab were assumed to have the same health state utility as those receiving dabrafenib (0.767). The health state utility of all patients following progression was 0.677.
2.6 Cost-effectiveness threshold
Whether or not the test-treat strategy is cost effective depends on how much a payer is willing to pay for additional health outcomes gained (QALYs or life-years [LYs]). When the intervention strategy is both cheaper and more effective than comparator strategies, it is a dominant strategy and clearly recommended as the optimal strategy to implement. However, if the intervention strategy is more effective and more expensive than comparator strategies, decisions should be made according to the cost-effectiveness threshold (CET) set by healthcare payers. Norway does not have a specific CET, but the Ministry of Health have argued that NOK275,000 per additional QALY gained is the best estimate of the opportunity cost of healthcare in Norway [41]. While it is suggested that a higher CET per QALY should be accepted for more serious conditions [41], and NOK500,000 per QALY has been used for some disease conditions, NOK275,000 per QALY was used in this study in the absence of an explicit definition of what constitutes a serious condition.
2.7 Receiver operating characteristic analysis
Receiver operating characteristic (ROC) analysis was conducted using the clinical trial data [27] to estimate the optimal threshold of true-positive fraction and false-positive fraction of HSP27 biomarker testing. ROC analysis is a simple but useful tool to evaluate the accuracy of a diagnostic test [42]. The ROC curve shows the trade-off between sensitivity and specificity; any increase in sensitivity will lead to a decrease in specificity. The closer the curve follows the left-hand border of the ROC curve, the more accurate the test. Meanwhile, the closer the curve comes to the 45° diagonal of the ROC curve, the less accurate the test. ROC curve analysis was used to test the performance of a test in identifying eligible patients for the treatment of interest. In Fig. 2, the true-positive rate (sensitivity) is plotted against the false-positive rate (1-specificity) for a series of cut-off points of a parameter. It predicted the optimal cut-off threshold of HSP27 biomarker testing performance at the sensitivity of 81.8%. Following this ROC analysis, a sensitivity of 81.8% and specificity of 41.7% were incorporated in the cost-effectiveness model. Given the low specificity, we also considered a higher index, but when an HSP27 staining index >4 was applied (which is staining index 6 or 9 in this case), it was worse than the random selection (45° diagonal of the ROC space). For example, under HSP27 index 6, both sensitivity and specificity improved to over 90% but were located below the 45° diagonal of the ROC curve (Fig. 2). The estimated cut-off thresholds according to different levels of the HSP27 staining indices are reported in supplementary material 5. Thus, based on the ROC analysis results (Fig. 2), the best cut-off threshold for determining HSP27 biomarker positivity or negativity is at staining index 4.

Receiver operating characteristics (ROC) curve
2.8 Uncertainty analysis
2.8.1 Sensitivity analysis: handling parameter uncertainty
We conducted deterministic sensitivity analysis (DSA) to identify key drivers in the model while holding all other variables at their baseline values. We also performed probabilistic sensitivity analysis (PSA) to assess the uncertainty around the base-case incremental cost-effectiveness ratio (ICER) by varying all relevant parameters simultaneously [43]. When available, we used the bounds of 95% confidence intervals (CIs) as high and low estimates in the sensitivity analysis. When the bounds of 95% CIs were unavailable, we used a range of ± 30%. Survival estimates were based on a beta distribution. Distributions of cost inputs used for PSA are detailed in Table 1. Monte Carlo simulation was used to assess the effect of simultaneous variation of all relevant parameters [44]. Additionally, we performed scenario analyses for sensitivity and specificity of the HSP27 expression testing to examine their impact on the cost-effectiveness results.
2.8.2 Expected value of perfect information (EVPI): handling decision-making uncertainty with current evidence
Healthcare decisions made based on existing information incur the costs of uncertainty. If an incorrect decision is made based on existing information, opportunity costs will be incurred in terms of healthcare resources and health benefits. The expected costs of uncertainty can be interpreted as EVPI because perfect information can remove the possibility of making incorrect decisions [45]. The opportunity costs of making incorrect decisions can be estimated using the value-of-information techniques. The EVPI estimates the upper bound of the value of conducting further research.
3 Results
3.1 Base-case results
The base-case ICER per QALY for the test-treat strategy (bevacizumab plus HSP27 testing) compared with treat-all patients with dacarbazine without HSP27 testing was NOK21,069, being cost effective. However, the test-treat strategy was not cost effective when compared with treat-all with bevacizumab without HSP27 testing (Table 2) because it cost less and produced fewer QALYs (Table 2). To be cost effective in this situation, the ICER needs to be above the CET, otherwise, the cost saving is not compensating adequately for the loss of benefit. In other words, we should be able to save costs per QALY at a rate above the CET, otherwise, it is not worth giving up the QALYs and we would rather keep them. The base-case ICER results were clearly lower than the Norwegian CET (NOK275,000).
3.2 Sensitivity analysis results
3.2.1 Tornado diagram
The DSA results are presented in a tornado diagram (supplementary material 6). The key drivers in the model were the bevacizumab costs and the proportion of HSP27-positive patients. However, they did not ultimately change the cost-effectiveness decision.
3.2.2 Probabilistic sensitivity analysis
PSA was performed to assess the effect of parameter variation across all relevant parameters on the base-case ICER when all parameters simultaneously varied. We ran 1000 simulations with QALYs gained as effectiveness measures. The total costs and QALYs gained for individuals tested for HSP27 and treated with bevacizumab were NOK94,740 and 7.80 QALYs, whereas those for patients simply treated with dacarbazine were NOK1450 and 2.91 QALYs, respectively (Table 3). Therefore, the ICER per QALY was NOK19,079.
However, the test-treat strategy was not cost effective when compared with the treat-all with bevacizumab strategy. It saved costs but produced fewer QALYs as observed in the base-case results. Likewise, the cost savings per QALY need to be above the CET in order for the intervention strategy to be cost effective. However, the ICER per QALY was NOK13,796, far below the Norwegian CET.
The scatterplot of the incremental costs and incremental QALYs from these simulations are presented in Figs. 3 and 4. All the iterations were contained in the north-east quadrant of Fig. 3, which means that the test-treat strategy of bevacizumab and HSP27 biomarker testing is costlier and more effective than the strategy of treating all patients with dacarbazine without HSP27 testing. However, when the test-treat strategy was compared with the treat-all with bevacizumab strategy, a majority of the 1000 simulations were located in the south-west quadrant of the scatterplot, suggesting that the intervention strategy is less costly but also less effective (Fig. 4). The PSA results confirmed that, although the test-treat strategy was cost effective compared with treat-all with dacarbazine, it was not cost effective compared with treat-all with bevacizumab without HSP27 testing, and base-case results were robust to changes in all variables.

Probabilistic sensitivity analysis (PSA) scatterplot for test-treat strategy compared with treat-all with dacarbazine

Probabilistic sensitivity analysis (PSA) scatterplot for test-treat strategy compared with treat-all with bevacizumab
3.2.3 Scenario analysis on the sensitivity and specificity of HSP27 expression testing
Scenario analysis was performed to examine the impact of the sensitivity and specificity of HSP27 testing on the cost-effectiveness results. It did not change the results in the different scenarios of the sensitivity and specificity of HSP27 testing under the different staining index of HSP27 expression. The cost-effectiveness results according to different combination scenarios of sensitivity and specificity of HSP27 expression testing are provided in Table 4.
3.3 EVPI analysis results
EVPI estimated the expected value of a decision made with current information against perfect information (Eq. 1).
Equation 1. Expected value of perfect information
where π refers to unknown parameters, NB the net benefit, t the treatment, and NB(t, π) the net benefit of treatment if parameters take the value π.
A willingness-to-pay threshold of NOK275,000 was assumed in the EVPI analysis. The EVPI was estimated at NOK5910 for the test-treat strategy versus treat-all with bevacizumab, whereas the EVPI was estimated at zero value for the comparison of the test-treat strategy and treat-all with dacarbazine (Table 5). The EVPI for the test-treat strategy against the treat-all with dacarbazine strategy implies that further research to reduce the uncertainties around current information would not be warranted. Likewise, the EVPI of NOK5910 for the test-treat strategy against the treat-all with bevacizumab implies further research might not be worthwhile either, given the small number of new cases of metastatic melanoma in Norway (annual average of 173 cases [10]). The upper bound of the population EVPI of the comparative analysis between the test-treat and the treat-all bevacizumab strategy was only NOK1,022,430 per annum. In other words, to justify further investment in research efforts of data collection such as conducting a phase III trial for HSP27 testing and bevacizumab, the research costs need to be lower than this upper bound, which is very unlikely for Norway. EVPI results are provided in Table 5. The EVPI graph depicted in Fig. 5 shows the change of EVPI depending on different thresholds. The spike is when we have maximum uncertainty where CET equals the ICER.

Expected value of perfect information (EVPI) graph. EVPI expected value of perfect information, NOK Norwegian Krone, QALY quality-adjusted life year
4 Discussion
The cost effectiveness of administering bevacizumab to those testing positive for HSP27 was compared with two alternative strategies (treating all patients with bevacizumab without HSP27 testing and treating all patients with dacarbazine without HSP27 testing) using a partitioned survival model. From the Norwegian health system perspective, a strategy of HSP27 biomarker testing was not cost effective. Treating all patients with bevacizumab is the best of the three strategies. This is the first study analysing the cost effectiveness of HSP27 biomarker testing before the administration of bevacizumab. No cost-effectiveness analyses of potential biomarkers (newly discovered yet unregistered for routine clinical use) in metastatic melanoma have been conducted.
Previous studies have assessed the cost effectiveness of BRAF and MEK inhibitors in metastatic melanoma. Delea et al. [29] evaluated the cost effectiveness of BRAF inhibitors but did not assess the impact of BRAF testing separately. Tarhini et al. [46] assessed the sequence of different targeted therapy options in melanoma but did not consider biomarker status. Curl et al. [47] also estimated the cost effectiveness of treatments for patients with BRAF-mutated metastatic melanoma. Likewise, Bohensky et al. [48] conducted a cost effectiveness analysis for the treatment of BRAF wild-type advanced melanoma in Australia, but all patients entering the model had BRAF wild-type melanoma. However, none of these studies analysed the cost effectiveness of biomarker testing before the provision of corresponding targeted therapies. However, Oh et al. [49] analysed the cost effectiveness of targeted therapy depending on the biomarker status (programmed death ligand-1 [PD-L1]-positive and -negative patients) and found that PD-L1 biomarker status contributed the most uncertainty to their model.
This study has several limitations. First, the survival data for dacarbazine were derived from one clinical study not through meta-analysis. We found no meta-analysis of the effect of dacarbazine, so we chose a study based on the patient characteristics in a clinical trial among other studies considered for dacarbazine monotherapy for patients with metastatic melanoma [50, 51]. Although the survival data for HSP27 testing and bevacizumab were derived from a small single-arm study, these were the only clinical data available on HSP27 expression and bevacizumab for patients with metastatic melanoma. Second, bevacizumab does not have marketing authorization for metastatic melanoma. However, the objective of this study was to determine the value of HSP27 testing in terms of cost effectiveness and EVPI. Third, given the early stage of development of the HSP27 biomarker for bevacizumab, we had to make some assumptions with regard to HSP27 testing. Fourth, it is a naïve indirect comparison and we did not match patients between dacarbazine and bevacizumab. This may lead to some potential bias in the results. For example, patients in the dacarbazine trial were younger than those in the bevacizumab trial; however, ECOG performance status was better for patients in the bevacizumab trial than for those in the dacarbazine trial. Also, the prognosis of female patients with metastatic melanoma is known to be better than that of male patients. However, both trials showed the same proportion of male and female participants. Fifth, utility values were not available for patients with metastatic melanoma treated with bevacizumab. However, utility values for patients treated with dacarbazine were available, and the model applied utility values for a targeted therapy for patients treated with bevacizumab.
5 Conclusion
The cost-effectiveness results showed that testing HSP27 biomarker status before the administration of bevacizumab was not cost effective. This finding may imply that this HSP27 biomarker is not good enough at identifying the right patients for treatment, as shown in the results of the ROC curve analysis or that bevacizumab is much better than dacarbazine in terms of health outcomes regardless of whether eligible patients are identified. The EVPI suggests that no further research is required to generate more evidence for assessing the test-treat strategy against treat-all with dacarbazine; however, it suggests some health gains to reduce the uncertainties around the comparative analysis of the test-treat and treat-all with bevacizumab strategies. Depending on the budget required to conduct further studies such as clinical trials, the decisions regarding additional research efforts can be reasonably determined by Norwegian stakeholders by considering the expected gain in health and the upper bound of the monetary value of perfect information as suggested by EVPI.
Data Availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Arnold M, de Vries E, Whiteman DC, Jemal A, Bray F, Parkin DM, et al. Global burden of cutaneous melanoma attributable to ultraviolet radiation in 2012. Int J Cancer. 2018;143(6):1305–14. https://doi.org/10.1002/ijc.31527(Epub 2018 May 7).
Giblin AV, Thomas JM. Incidence, mortality and survival in cutaneous melanoma. J Plast Reconstr Aesthet Surg. 2007;60(1):32–40. https://doi.org/10.1016/j.bjps.2006.05.008(Epub Jul 7).
Godar DE. Worldwide increasing incidences of cutaneous malignant melanoma. J Skin Cancer. 2011;2011:858425. https://doi.org/10.1155/2011/858425.
Whiteman DC, Green AC, Olsen CM. The growing burden of invasive melanoma: projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J Investig Dermatol. 2016;136(6):1161–71. https://doi.org/10.1016/j.jid.2016.01.035Epub Feb 20.
World Health Organization. How common is skin cancer? 2018. https://www.who.int/uv/faq/skincancer/en/index1.html. Accessed 21 Aug 2018.
Erdmann F, Lortet-Tieulent J, Schuz J, Zeeb H, Greinert R, Breitbart EW, et al. International trends in the incidence of malignant melanoma 1953–2008—are recent generations at higher or lower risk? Int J Cancer. 2013;132(2):385–400. https://doi.org/10.1002/ijc.27616.
Guy GP Jr, Thomas CC, Thompson T, Watson M, Massetti GM, Richardson LC. Vital signs: melanoma incidence and mortality trends and projections—United States, 1982–2030. MMWR Morb Mortal Wkly Rep. 2015;64(21):591–6.
Linos E, Swetter SM, Cockburn MG, Colditz GA, Clarke CA. Increasing burden of melanoma in the United States. J Investig Dermatol. 2009;129(7):1666–744. https://doi.org/10.1038/jid.2008.423.
Geisler J, Bachmann IM, Nyakas M, Helsing P, Fjosne HE, Maehle LO, et al. Malignant melanoma–diagnosis, treatment and follow-up in Norway. Tidsskrift for den Norske laegeforening: tidsskrift for praktisk medicin, ny raekke. 2013;133(20):2154–9. https://doi.org/10.4045/tidsskr.12.1416.
Cancer Registry of Norway. Cancer in Norway 2016—cancer incidence, mortality, survival and prevalence in Norway. Oslo: Cancer Registry of Norway; 2017.
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–16. https://doi.org/10.1200/jco.1999.17.7.2105.
Busch C, Geisler J, Lillehaug JR, Lonning PE. MGMT expression levels predict disease stabilisation, progression-free and overall survival in patients with advanced melanomas treated with DTIC. Eur J Cancer (Oxford, England: 1990). 2010;46(11):2127–33. https://doi.org/10.1016/j.ejca.2010.04.023.
Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18(1):158–66. https://doi.org/10.1200/jco.2000.18.1.158.
Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet (London, England). 2017;390(10105):1853–62. https://doi.org/10.1016/s0140-6736(17)31601-x.
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–32. https://doi.org/10.1056/NEJMoa1503093.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. https://doi.org/10.1056/NEJMoa1103782.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (London, England). 2012;380(9839):358–65. https://doi.org/10.1016/s0140-6736(12)60868-x.
Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76. https://doi.org/10.1056/NEJMoa1408868.
Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–9. https://doi.org/10.1056/NEJMoa1412690.
Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–88. https://doi.org/10.1056/NEJMoa1406037.
Atkinson V. Medical management of malignant melanoma. Aust Prescr. 2015;38(3):74.
Dummer R, Hauschild A, Lindenblatt N, Pentheroudakis G, Keilholz U. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(suppl_5):v126–v132132.
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30. https://doi.org/10.1056/NEJMoa1412082.
Barone A, Hazarika M, Theoret MR, Mishra-Kalyani P, Chen H, He K, et al. FDA approval summary: pembrolizumab for the treatment of patients with unresectable or metastatic melanoma. Clin Cancer Res. 2017;23(19):5661–5. https://doi.org/10.1158/1078-0432.ccr-16-0664.
Simeone E, Ascierto PA. Anti-PD-1 and PD-L1 antibodies in metastatic melanoma. Melanoma Manag. 2017;4(4):175–8. https://doi.org/10.2217/mmt-2017-0018.
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–177. https://doi.org/10.1056/NEJMoa1414428Epub 2015 Apr 20.
Schuster C, Akslen LA, Straume O. Expression of heat shock protein 27 in melanoma metastases is associated with overall response to bevacizumab monotherapy: analyses of predictive markers in a clinical phase II study. PLoS ONE. 2016;11(5):e0155242. https://doi.org/10.1371/journal.pone.0155242.
Zoubeidi A, Gleave M. Small heat shock proteins in cancer therapy and prognosis. Int J Biochem Cell Biol. 2012;44(10):1646–56. https://doi.org/10.1016/j.biocel.2012.04.010.
Delea TE, Amdahl J, Wang A, Amonkar MM, Thabane M. Cost effectiveness of dabrafenib as a first-line treatment in patients with BRAF V600 mutation-positive unresectable or metastatic melanoma in Canada. PharmacoEconomics. 2015;33(4):367–80. https://doi.org/10.1007/s40273-014-0241-z.
Norwegian Directorate of Health (Helsedirektoratet). Økonomisk evaluering av helsetiltak. [Updated 01.11.2012]. https://helsedirektoratet.no/retningslinjer/veileder-i-okonomisk-evaluering-av-helsetiltak. Accessed 23 Oct 2018.
Schuster C, Eikesdal HP, Puntervoll H, Geisler J, Geisler S, Heinrich D, et al. Clinical efficacy and safety of bevacizumab monotherapy in patients with metastatic melanoma: predictive importance of induced early hypertension. PLoS ONE. 2012;7(6):e38364. https://doi.org/10.1371/journal.pone.0038364.
Roche. Melanoma (BRAF V600 mutation positive, unresectable metastatic)—vemurafenib: manufacturer submission. 2012. https://www.nice.org.uk/guidance/ta269/documents/melanoma-braf-v600-mutation-positive-unresectable-metastatic-vemurafenib-roche-products4. Accessed 1 Feb 2019.
McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–32. https://doi.org/10.1016/S470-2045(14)70012-9(Epub 2014 Feb 7).
Bormann I. Digitizer software. 2015. https://www.digitizeit.de/. Accessed 1 Feb 2019.
Guyot P, Ades AE, Ouwens MJ, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan–Meier survival curves. BMC Med Res Methodol. 2012;12:9. https://doi.org/10.1186/1471-2288-12-9.
Santa Cruz Biotechnology. HSP27 antibody (C-20): sc-1048, https://www.scbt.com/scbt/product/hsp-27-antibody-c-20. Accessed 18 Mar 2018.
Health Products Regulatory Authority. Summary of product charateristics 2016. https://www.hpra.ie/img/uploaded/swedocuments/LicenseSPC_PA1760-001-001_11052016123106.pdf. Accessed 28 Oct 2018.
Pike ETE, Sæterdal I, Jimenez E, Odgaard-Jensen J, Harboe I, Klemp M. A health technology assessment of the new drugs for inoperable or metastatic malignant melanoma patients. Report from Kunnskapssenteret no. 22-2015. Oslo: Norwegian Knowledge Centre for the Health Services. 2015.
Vogler S, Vitry A. Cancer drugs in 16 European countries, Australia, and New Zealand: a cross-country price comparison study. Lancet Oncol. 2016;17(1):39–47.
National Institute for Health and Care Excellence (NICE). Dabrafenib for treating unresectable or metastatic BRAF V600 mutation‑positive melanoma. Technology appraisal guidance (TA321). https://www.nice.org.uk/guidance/ta321. Accessed 4 Mar 2019.
Royal Health and Care Department. Values in the patient's health service: message about prioritization: Meld. St. 34 (2015–2016). 2015. https://www.regjeringen.no/contentassets/439a420e01914a18b21f351143ccc6af/no/pdfs/stm201520160034000dddpdfs.pdf. Accessed 21 Oct 2018.
Metz CE. Basic principles of ROC analysis. Semin Nucl Med. 1978;8(4):283–98.
Briggs A, Sculpher M, Buxton M. Uncertainty in the economic evaluation of health care technologies: the role of sensitivity analysis. Health Econ. 1994;3(2):95–104.
Doubilet P, Begg CB, Weinstein MC, Braun P, McNeil BJ. Probabilistic sensitivity analysis using Monte Carlo simulation. A practical approach. Med Decis Making Int J Soc Med Decis Making. 1985;5(2):157–77. https://doi.org/10.1177/0272989x8500500205.
Claxton K, Ginnelly L, Sculpher M, Philips Z, Palmer S. A pilot study on the use of decision theory and value of information analysis as part of the NHS Health Technology Assessment programme. Health Technol Assess (Winchester, England). 2004;8(31):1–103.
Tarhini A, Benedict A, McDermott D, Rao S, Ambavane A, Gupte-Singh K, et al. Sequential treatment approaches in the management of BRAF wild-type advanced melanoma: a cost-effectiveness analysis. Immunotherapy. 2018;10(14):1241–52. https://doi.org/10.2217/imt-2018-0085.
Curl P, Vujic I, van 't Veer LJ, Ortiz-Urda S, Kahn JG. Cost-effectiveness of treatment strategies for BRAF-mutated metastatic melanoma. PLoS ONE. 2014;9(9):e107255. https://doi.org/10.1371/journal.pone.0107255.
Bohensky MA, Pasupathi K, Gorelik A, Kim H, Harrison JP, Liew D. A cost-effectiveness analysis of nivolumab compared with ipilimumab for the treatment of BRAF wild-type advanced melanoma in Australia. Value Health J Int Soc Pharmacoecon Outcomes Res. 2016;19(8):1009–155. https://doi.org/10.1016/j.jval.2016.05.013.
Oh A, Tran DM, McDowell LC, Keyvani D, Barcelon JA, Merino O, et al. Cost-effectiveness of nivolumab-ipilimumab combination therapy compared with monotherapy for first-line treatment of metastatic melanoma in the United States. J Managed Care Spec Pharm. 2017;23(6):653–64. https://doi.org/10.18553/jmcp.2017.23.6.653.
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30.
Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65.
Acknowledgements
The authors are grateful to Cornelia Schuster for her input in relation to the clinical trial data, Alec Miners for model validation, and Jack Williams for his help with an R algorithm.
Author information
Authors and Affiliations
Contributions
Study concept and design: MKS, JC. Data analysis: MKS. Model development: MKS. Manuscript writing, original draft: MKS. Manuscript review and editing: JC, OS, LAA.
Corresponding author
Ethics declarations
Funding
Mikyung Kelly Seo receives a PhD studentship from the Centre for Cancer Biomarkers (CCBIO), Centre of Excellence at the Faculty of Medicine and Dentistry, University of Bergen. No sources of funding were used to conduct this study or prepare this manuscript.
Conflict of interest
MKS, OS, LAA, and JC have no conflicts of interest that are directly relevant to the content of this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Seo, M.K., Straume, O., Akslen, L.A. et al. HSP27 Expression as a Novel Predictive Biomarker for Bevacizumab: is it Cost Effective?. PharmacoEconomics Open 4, 529–539 (2020). https://doi.org/10.1007/s41669-019-00193-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41669-019-00193-8