
Abstract
The toxin–antitoxin (TA) system is composed of a stable toxin and an unstable antitoxin that neutralizes the toxin. Being perhaps the most studied among the different TA types, type II TA systems are widely distributed and often exist in multiple copies within chromosomes of eubacteria and archaea. Exhibiting diverse molecular activities such as RNases, kinases, and acetyltransferases, type II TA systems have been confirmed to be involved in diverse biological processes including plasmid maintenance, phage inhibition, persistence, stress response, and biofilm formation. In this review, we summarize the current state of the research in the type II TA field, emphasizing the activation mechanism, structure–function relationship, and biological functions of type II TA systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
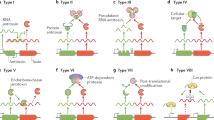
Toxin–antitoxin (TA) systems are small genetical modules encoding two components: a stable toxin protein and its cognate labile antitoxin (Harms et al. 2018). They were firstly discovered in the 1980s as plasmid-borne “addiction modules” that played a role in plasmid maintenance (Gerdes et al. 1986). Subsequently, homologues of TA systems were also discovered in chromosomes of bacteria and archaea, involving in diverse physiological processes including phage abortive infection (Dy et al. 2014), persistence (Moyed and Bertrand 1983), biofilm formation (Wang et al. 2011), and virulence (Zhu et al. 2009). TA systems were generally divided into seven types based on the nature of the antitoxins and its mode of action on toxins (Fig. 1). Antitoxins of type I and type III TA systems are small RNAs that neutralize their cognate toxins by either inhibiting translation of toxins (type I) or directly interfering with the toxin activities (type III). For the other types of TA systems, antitoxins are exclusive proteins but display different mechanisms of antagonizing the toxin activities. For type II TA systems, antitoxins can form stable complexes with the cognate toxins to inhibit the catalytic activities of the toxins. Antitoxins of type IV TA systems can disturb the activities of toxins by protecting their molecular targets. Three single instances of TA modules with regulatory principles different from type I–IV TA systems have been described as type V, type VI, and type VII TA modules, respectively (Marimon et al. 2016; Page and Peti 2016; Wang et al. 2019). The type V antitoxin GhoS specifically cleaves the mRNA of its cognate toxin GhoT to block its translation (Wang et al. 2012), while the type VI antitoxin SocA acts as an adaptor protein to direct the toxin SocB to proteolysis, leading to SocB inactivation (Aakre et al. 2013). The type VII antitoxin TomB enhances the spontaneous oxidation of a conserved cysteine residue of its cognate toxin Hha, resulting in the destabilization and inactivation of Hha (Marimon et al. 2016).

Modes of toxin neutralization by cognate antitoxins in the four major types of TA systems (Type I–IV). Toxins and antitoxins are shown in purple and green, respectively. Genetic loci are shown in colored arrows and RNA transcripts are drawn as curly lines. A Type I antitoxins encode antisense RNAs pairing with the toxin mRNA and result in translation inhibition of the cognate toxins. B Type II antitoxins are proteins which form stable complexes with the cognate toxins, leading the inhibition of the toxin activities. C Type III antitoxins are RNAs (not antisense RNAs) which directly bind to cognate toxins to neutralize the toxicity. D Type IV antitoxins are proteins that bind to the toxin targets (rather than the toxins) to prevent toxic effects
Among the different TA types, type II TA systems are mostly studied and undergo frequent horizontal gene transfers (Ramisetty and Santhosh 2016; Rocker and Meinhart 2016). They are widely distributed and often exist in multiple copies within chromosomes of eubacteria and archaea. Toxins of type II TA systems have diverse molecular activities such as RNases, kinases, and acetyltransferases and play important roles in plasmid maintenance, phage inhibition, persistence, stress response, and biofilm formation. In this review, we summarize the current state of the research in the type II TA field, emphasizing the activation mechanism, structure–function relationship, and biological functions of type II TA systems. Our current knowledge on type II TA systems is just a tip of the iceberg. More understanding are needed of the molecular mechanisms of diverse type II TA systems which would facilitate their application in biotechnology industry and clinical settings.
DISTRIBUTION OF THE TYPE II TA SYSTEMS
Evolutionary analysis indicated that type II TA systems are prone to move between microbial genomes through horizontal gene transfer (HGT), which may account for the surprisingly wide distribution and great numbers of type II TA systems in chromosomes of archaea and eubacteria (Ramisetty and Santhosh 2016) (Table 1). The first genomic-scale survey of the type II TA system was performed by Pandey et al. in 2005 and demonstrated that although type II TA systems were abundant on bacterial chromosomes, the number and the location of TA loci varied greatly between unrelated bacteria. Of the 126 investigated prokaryotic genomes, a large fraction (~50%) had 1–5 TA loci whereas no TA loci were detected in ~25% of the organisms (Pandey and Gerdes 2005). A prediction algorithm RASTA-Bacteria was later developed by Sevin et al. to computationally identify the type II TAs and at least 65 TA systems were found in Mycobacterium bovis genome (Sevin and Barloy-Hubler 2007). Most recently, Xie et al. predicted type II TA systems out of 2786 completely sequenced prokaryotic genomes using the newly developed TA finder algorithm and showed that 66% of species harbored 1–20 type II TA loci in individual strains while 20% of species carried more than 20 loci (Xie et al. 2018). These results indicated that type II TA systems were not evenly distributed in prokaryotic genomes. However, no correlation was found between the number and nature of type II TA loci with the prokaryotic phylum or ecosystem (Sevin and Barloy-Hubler 2007).
ACTIVATION OF THE TYPE II TA SYSTEMS
In type II TA systems, the antitoxin genes are usually located in the upstream of the toxin genes, except for the hipBA, higBA, and mqsRA loci where the gene orders are exchanged. The general feature of the type II TA operon is that two genes are often separated by a short region (e.g., hipA/hipB) or overlap (e.g., mqsR/mqsA). The difference in gene order or intergenic spacing may affect the ratio of the toxin/antitoxin production rates, and thereby regulating the activity of the toxin (Harms et al. 2018). Despite the bactericidal or bacteriostatic effects of the toxin, the antitoxin is present at higher concentrations than the toxin under normal growth conditions, ensuring the bacterial growth is not affected by the toxin. Under the stressed conditions (e.g., nutrient starvation, oxidative stress, antibiotic challenge), the antitoxins are more quickly degraded by protease systems than the toxins (Chan et al. 2016) (Fig. 2). This is because, in many cases, antitoxins are intrinsically disordered proteins or have disordered terminal regions which make them more vulnerable to degradation by intracellular proteases than toxins (Cherny et al. 2005). Interestingly, in Mycobacterium tuberculosis, a SecB-like chaperone was found to be adjacent to the HigBA TA module and protect the antitoxin HigA from degradation and aggregation by interacting with its C-terminal fragment, which is known as a chaperone addiction sequence and makes the antitoxin aggregation-prone (Bordes et al. 2011, 2016; Guillet et al. 2019). Degradation of antitoxins by proteases finally leads to activation of toxins (Fig. 2), which inhibit the bacterial growth by targeting multiple physiological processes including DNA replication, gene transcription or protein translation (Chan et al. 2016).

Activation pathways of the type II TA systems. Genetic loci and promoters of type II TA modules are shown in colored arrows. The antitoxins usually negatively regulate the TA promoter. Upon exposure to environmental stress, te intracellular proteases (e.g., Lon, ClpAP, ClpCP and ClpXP) are activated and degrade antitoxins, resulting in overexpression and release of toxins, which subsequently act on the cellular targets and cause cell growth arrest
As activation of the toxin depends on proteolytic degradation of the antitoxin, it is critical to understand the regulation mechanism of the antitoxin stability. Most of the well-characterized antitoxins were demonstrated to be degraded by the Lon protease (Muthuramalingam et al. 2016), which belongs to the AAA+ protease family that couples the ATP hydrolysis to drive substrate translocation and degradation. There were also reports showing that the two-component protease ClpP, cooperating with the ATP-dependent chaperones ClpA, ClpC or ClpX, carries out degradation of antitoxins in certain type II TA systems (Flynn et al. 2003; Loris and Garcia-Pino 2014; Page and Peti 2016; Smith et al. 1999; Wu et al. 1999). Moreover, multiple proteases can be involved in degradation of one antitoxin, implying that controlling intracellular stability of the antitoxin is a complex process (Flynn et al. 2003; Wu et al. 1999). Notably, for a newly identified TA module GraTA, with its toxin GraT targeting ribosome biogenesis (Tamman et al. 2014), the antitoxin GraA is degraded by neither Lon nor Clp protease but an unidentified ATP-independent protease (Tamman et al. 2015). Oddly, an intrinsically disordered region was found in the toxin GraT rather than the antitoxin, raising an intriguing possibility of the toxin GraT being under proteolytic control (Talavera et al. 2019). This result significantly deviates from the current model that toxins are always more stable than antitoxins, suggesting alternative models should be considered when investigating proteolytic activation of novel families of type II TA systems.
Although antitoxins are generally considered to be intrinsically disordered proteins or possess unstructured terminal regions, there are still many characterized antitoxins forming rigid and stable structures in the absence of their cognate toxins (e.g., MqsA and GraA) (Brown et al. 2011; Talavera et al. 2019). Questions remain how these antitoxins are specifically recognized and degraded by the Lon or ClpP family proteases under stressed conditions. One possible mechanism ensuring the specific degradation of antitoxins is through adaptor proteins that target the substrates for degradation by interacting simultaneously with substrates and proteases (Kirstein et al. 2009). In Staphylococcus aureus, three antitoxins of Axe1, Axe2 and MazE were delivered by the adaptor protein TrfA to the ClpCP protease for degradation (Donegan et al. 2010, 2014). Recently, an adaptor protein for the Lon protease, named HspQ, was characterized to enhance the Lon substrate selectivity (Puri and Karzai 2017), but it was not clear whether this adaptor protein could mediate the Lon-dependent degradation of antitoxins. Another important question still awaiting an answer is whether degradation of antitoxins under different stress conditions is dependent on the elevated expression or allosteric activation of the proteases.
TYPE II TOXINS: CLASSIFICATION AND MODE OF ACTION
Based on structural similarities, the currently characterized toxins of type II TA systems can be classified into nine superfamilies, namely ParE/RelE, MazF, HicA, VapC, HipA, FicT/Doc, AtaT/TacT, Zeta and MbcT (Table 2). The RelE superfamily toxins adopt the RelE-like structural fold (Fig. 3A, CATH ID: 3.30.2310.20 and 3.30.2310.40) which is composed of three α-helices and a five-strand β-sheet (Takagi et al. 2005). Although sharing similar structures, the molecular functions of the RelE superfamily (RelE, ParE, HigA, MqsA, YafQ, YafO, etc.) are heavily diverged. The MazF superfamily toxins share a core structure consisting of a seven-stranded, twisted antiparallel β-sheet surrounded by three α-helices (Fig. 3B, CATH ID: 2.30.30.110) (Kamada et al. 2003). Members of the MazF superfamily include MazF and CcdB but their mode of action can also be very different. The HicA superfamily forms a dsRNA-binding domain-like fold (Fig. 3C, CATH ID: 3.30.920.30) composed of a triple-stranded β-sheet, with two helices packed against one face (Butt et al. 2014). Members of the HicA superfamily mediate ribosome-independent mRNA cleavage, which results in inhibition of bacterial growth (Turnbull and Gerdes 2017). The VapC superfamily generally exhibits a nuclease activity that leads to growth arrest by cleaving tRNAfMet in the anticodon region and contains a typical PIN domain structure (Fig. 3D, CATH ID: 3.40.50.1010) consisting of a small and central five-stranded β-sheet (β1–β5) surrounded by seven α-helices (Dienemann et al. 2011). The HipA superfamily has a structure similar to human CDK2/cyclin A kinase (Fig. 3E), consisting of an N-terminal α/β domain and a C-terminal all α-helical domain (Schumacher et al. 2009). HipA possesses the Ser/Thr kinase activity and phosphorylates the glutamyl-tRNA-synthetase GltX in vivo to promote antibiotic persistence (Germain et al. 2013; Kaspy et al. 2013). The FicT/Doc toxins adopt the Fic domains that are defined by their conserved active-site motif [HXFX(D/E)(A/G)N(G/K)R] consisting of six α-helices with helices α3 and α4 supporting the catalytic loop (Fig. 3F) (Stanger et al. 2016). The currently characterized members of FicT/Doc toxins can function as either adenylylation enzymes that add AMP moieties to target proteins (FicT) or kinases that phosphorylate other proteins (Doc) (Castro-Roa et al. 2013; Harms et al. 2015). Interestingly, FicT toxins can also be linked to effectors of secretion systems as a Fic-domain-containing protein VbhT which was recently identified to be fused with a type IV secretion signal (T4SS) domain BID, and secreted into competing bacterial cells as a bactericidal effector protein (Engel et al. 2012; Harms et al. 2017). The AtaT toxins act as acetyltransferases and adopt a structural fold comprising a central seven-stranded β-sheet bounded by four α-helices (Fig. 3G), with the α3 helix containing the (Q/R-x-x-G-x-G/A) signature motif (Jurenas et al. 2019). The Zeta toxins function as a UDP-N-acetylglucosamine kinases to inhibit cell wall biosynthesis and exhibit an α/β structure (Fig. 3H, CATH ID: 3.40.50.30), with a six β-stranded central β-sheet surrounded by several α-helices (Meinhart et al. 2003). Most recently, type II toxins with ADP-ribosyltransferase activities were characterized (Freire et al. 2019; Piscotta et al. 2019; Skjerning et al. 2019) and the representative toxin MbcT exhibits a β-sandwich fold formed by six β-strands flanked by nine α-helices (Fig. 3I) and triggers bacterial cell death by depleting the intracellular NAD+ pool.

Reprehensive structural folds of the nine superfamilies of the type II toxins. A The structure of the RelE toxin (PDB ID: 3BPQ) from Methanococcus jannaschii (Francuski and Saenger 2009). B The structure of the MazF (PDB ID: 4MDX) toxin from Bacillus subtilis. C The structure of the HicA toxin (PDB ID: 4C26) from Burkholderia pseudomallei (Butt et al. 2014). D The structure of the VapC toxin (PDB ID: 5ECW) from Shigella flexneri (Xu et al. 2016). E The structure of the HipA toxin (PDB ID: 3FBR) from E. coli (Schumacher et al. 2009). F The structure of the FicT toxin (PDB ID: 5JFF) from E. coli (Stanger et al. 2016). G The structure of the AtaT toxin (PDB ID: 6GTP) from E. coli (Jurenas et al. 2019). H The structure of zeta toxin from Streptococcus pyogenes (PDB ID: 1GVN) (Meinhart et al. 2003). I The structure of MbcT toxin from Mycobacterium tuberculosis (PDB ID: 6FKG) (Freire et al. 2019)
Notably, toxins from the same superfamily can have low sequence similarities and exhibit great divergence in their molecular activities. For example, within the ParE/RelE superfamily, although ParE superfamily shares a similar fold with the RelE superfamily, the ParE superfamily is a gyrase poison (Jiang et al. 2002) that leads to the accumulation of DNA breaks whereas the RelE superfamily exhibits ribosome-dependent mRNA endonuclease activity (Pedersen et al. 2003). Functional divergence was also observed within the AtaT/TacT superfamily. While the AtaT toxin inhibits translation initiation by acetylating the amino acid moiety of the initiator tRNAMet (Jurenas et al. 2019), the TacT toxin inhibits translation elongation by targeting aminoacylated elongation tRNA (Cheverton et al. 2016). Most recently, an acetyltransferase toxin ItaT that belongs to the AtaT/TacT superfamily was shown to specifically and exclusively acetylate Ile-tRNAIle thereby blocking translation and inhibiting cell growth (Wilcox et al. 2018). Moreover, toxins from the CcdB/MazF superfamily also display distinct activities and targets. The MazF toxins are ribosome-independent mRNA endoribonucleases (Zhang et al. 2003), whereas the CcdB toxins function as DNA gyrase inhibitors (Dao-Thi et al. 2005). For the FicT/Doc superfamily, while the FicT superfamily toxins adenylylate DNA gyrase and/or topoisomerase IV at their ATP-binding sites (Harms et al. 2015; Lu et al. 2016), the Doc toxins function as protein kinases to phosphorylate the translation elongation factor EF-Tu (Castro-Roa et al. 2013). Since the biological functions of TA systems are closely related to the target selection and specificity of toxins, it would be possible to unveil novel biological roles of type II TA systems if new targets or molecular activities could be identified and characterized for certain toxins within the type II toxin superfamilies.
Although conventional type II toxins are known as small proteins with a single domain, bioinformatics analysis also revealed elongated type II toxins-like proteins containing multiple domains in the bacterial genomes. Two instances of these types of toxins were characterized, either having negative effects on the cell growth or disturbing the peptidoglycan synthesis. For example, the toxin protein PhoH2 consisted of an N-terminal PIN domain with RNase activity and a C-terminal RNA helicase domain (Andrews and Arcus 2015). It was proposed that the coupled RNA helicase domain could unwind RNA and facilitate the RNA cleavage by the PIN domain in a sequence specific manner. Another example is the EzeT multi-domain toxin encoded in genomes of many different bacterial species. EzeT contains a C-terminal domain homologous to the Zeta toxins and an N-terminal cis-acting antitoxin domain. The C-terminal domain of EzeT catalyzes the phosphorylation UDP-N-acetylglucosamine, leading to cell lysis upon overexpression while the N-terminal domain strongly attenuates kinase activity and keeps EzeT in an autoinhibited state (Rocker and Meinhart 2015). Although activation mechanism of EzeT has not been demonstrated by experiments, it was hypothesized partial unfolding of the N-terminal domain mediated by certain chaperones or other protein factors could possibly lead to EzeT activation. These data imply that the organization and architecture of type II TA systems are much more complicated than previous thought and many novel functionalities of type II TA systems are worth deeper investigation.
TYPE II ANTITOXINS: CLASSIFICATION AND MODE OF ACTION
Most of the type II antitoxins have an N-terminal toxin-neutralizing domain and a C-terminal DNA-binding domain. Based on sequence similarities, type II antitoxins have been classified into 20 superfamilies (Leplae et al. 2011). However, the structures of the DNA-binding domains of the type II antitoxins generally fall in only four different categories, namely helix-turn-helix (HTH), ribbon-helix helix (RHH), YefM/Phd-type, and VapB-type (Fig. 4). The HTH-motif is a classical DNA-binding module that can be found in many prokaryotic transcription regulators and it usually binds to the major groove of DNA via the second α-helix (Wintjens and Rooman 1996). Examples of HTH-motif containing antitoxins include PezA (Khoo et al. 2007), HigA (Schureck et al. 2014) and MqsA (Brown et al. 2011). The RHH-motif has the topology of β-strand–α-helix–α-helix and the β-strands mediate dimerization by forming an antiparallel β-sheet, which recognizes specific DNA sequences (Schreiter and Drennan 2007). The RHH-motif contained proteins are ubiquitous in prokaryotic transcription factors that involve in diverse physiological processes. The antitoxins containing RHH-motifs include CcdA (Madl et al. 2006), ParD (Oberer et al. 2007), RelB (Boggild et al. 2012), DinJ (Liang et al. 2014) and FitA (Mattison et al. 2006). The YefM/Phd-type domain has three β-strands and two α-helices, with the three β-strands mediating dimerization by forming a six-stranded antiparallel β-sheet (Garcia-Pino et al. 2016). The bottom surface of the YefM/Phd-type domain has a patch of positive charged residues involved in DNA recognition. Currently, the YefM/Phd-type domains are only found in the YefM and Phd antitoxins. The VapB-type domain dimerizes to form a so-called layered swapped-hairpin β-barrel structure (Bendtsen et al. 2017a), with the first β-hairpins from each antitoxin binding to the DNA major grove. Antitoxins containing the VapB-type domains include VapB (Bendtsen et al. 2017b; Mate et al. 2012), MazE (Bobay et al. 2005; Kamada et al. 2003) and Kis (Kamphuis et al. 2007).

Protein-DNA recognition of representative antitoxins. A The structure of the antitoxin MqsA in complex with its cognate operator sequence (PDB ID: 3O9X) from E. coli (Brown et al. 2011). B The structure of the antitoxin CcdA in complex with its operator sequence (PDB ID: 2H3C) from E. coli (Madl et al. 2006). C The crystal structure of the antitoxin Phd in complex with the DNA sequence (PDB ID: 4ZM0) from Escherichia phage P1 (Garcia-Pino et al. 2016). D The crystal structure of the antitoxin VapB in complex with the DNA sequence (PDB ID: 5L6L) from Caulobacter crescentus (Bendtsen et al. 2017a)
Although antitoxins specifically interact with their cognate toxins, toxins and antitoxins from different superfamilies can associate to form novel TA pairs (Guglielmini and van Melderen, 2011). For example, the Phd antitoxin homologues can neutralize toxins from at least three superfamilies of toxins including Fic/Doc, RelE/ParE and VapC (Arbing et al. 2010). Toxins from the RelE/ParE superfamily can also associate with RelB, Phd, HigA and even antitoxins that do not belong to any well-characterized antitoxin superfamilies (Guglielmini and van Melderen 2011). This “mix and match” phenomenon, which was useful to develop a “guilt by association” algorithm to predict novel toxins associated with known antitoxins and vice versa (Sevin and Barloy-Hubler 2007), suggests that classification of toxins and antitoxins should be considered independently.
In general, the type II antitoxins antagonize the activities of their cognate toxins by blocking or masking the toxin active sites (Blower et al. 2011; Bøggild et al. 2012; Schureck et al. 2014). For example, the Zeta and PezT toxins are inactivated due to the steric hindering of the ATP/GTP binding sites by their cognate antitoxins such as epsilon (Meinhart et al. 2003) and PezA (Khoo et al. 2007). The MazE wraps around the MazF toxin to occlude the enzymatic active site, leading to neutralization of MazF toxic effect in the cell. In the case of the RelE toxin, binding of the RelB antitoxin leads to displacement of a C-terminal α-helix essential for the RelE activity (Li et al. 2009). However, some antitoxins do not act upon their cognate toxins by blocking the active sites. The HipB antitoxin allosterically regulates the activity of the HipA toxin by locking the toxin in an inactive conformation (Schumacher et al. 2009). For the MqsR/MqsA and HigB/HigA TA systems, the corresponding antitoxins bind to sites that are distant from the toxin active sites. It was suggested that both the MqsA and HigA antitoxins neutralize the toxin activities by competing its binding to ribosome or RNA (Brown et al. 2009; Schureck et al. 2014), but the precise mechanism needs further confirming or investigation.
BIOLOGICAL FUNCTIONS OF TYPE II TA SYSTEMS
The plasmid-borne TA systems are generally thought to stabilize the plasmids through a mechanism called post-segregational killing (PSK), which leads to death of the daughter cells that do not inherit the parental plasmids. This is because antitoxins were degraded much faster than toxins so that the plasmid-free daughter cells could not survive the poisoning of the intracellular toxins. For example, Hok/Sok of plasmid R1, CcdA/CcdB of plasmid F, and ParD/ParE of plasmid RK2 were well-characterized TA systems involved in PSK in E. coli (Harms et al. 2018). For the chromosome-borne TA systems, it is believed that they have broader impacts to the host cells, especially participating in stress responses when living in unfavorable circumstances. For instance, TA systems can help the host bacteria to resist phage infection through the mechanism known as phage abortive infection, which leads to the activation of toxins by antitoxin degradation in the phage-infected cells. Two type II TA systems, namely MazE/MazF and RnlA/RnlB, have been shown to significantly block infection of phage P1 and phage T4, respectively (Alawneh et al. 2016; Koga et al. 2011).
Another stress response phenomenon mediated by TA systems is persister formation upon antibiotic challenges, as observed in E. coli and other bacteria. Persister cells are a small portion of cells showing dormant and multidrug-tolerant characteristics when exposed to antibiotics (Lewis 2010). The link between persister formation and type II TA systems was first established in 1983 when Moyed and Bertrand isolated a hipA7 mutant, which bears two substitutions in the HipA toxin (G22S and D291S) and exhibits increased persistence by 1000–10,000-fold versus the wildtype strain in E. coli K-12 upon ampicillin treatment (Moyed and Bertrand 1983). HipA is a serine or threonine kinase that phosphorylates the aminoacyl-tRNA-Glu (tRNAGlu) synthetase GltX, leading to the accumulation of uncharged tRNAGlu. The RelA protein was then activated, triggering the stringent response with increased levels of the (p)ppGpp alarmone (Germain et al. 2013; Kaspy et al. 2013). Stringent response had been demonstrated to be involved in persister formation as mutants devoid of the (p)ppGpp alarmone has significantly decreased persistence upon antibiotic exposure (Germain et al. 2015). Therefore, HipA may stimulate persister formation via triggering the stringent response. Recently, HipA was shown to phosphorylate additional targets such as ribosomal proteins, which could also contribute to persister formation. However, the HipA7 toxin mutant was found to exclusively phosphorylate GltX, suggesting targeting tRNAGlu synthetase alone is sufficient to stimulate high proportions of persisters (Semanjski et al. 2018).
Besides the HipAB TA system, other type II TA systems were also shown to be related to bacterial persistence. Deletion of the mqsRA (Kim and Wood 2010; Luidalepp et al. 2011) or yafQ (Harrison et al. 2009) has been shown to reduce the survival of E. coli under antibiotic exposure, suggesting these TA loci contribute to bacterial persistence. Overexpression of RelE or MazF toxins can drastically promote persister formation of E. coli, suggesting these toxins are involved in persistence in conditions when their cognate antitoxins are degraded (Keren et al. 2004; Tripathi et al. 2014). Moreover, spontaneous mutations to antitoxins YafN or VapB were demonstrated to result in extended lag phase and higher persistence, implying that activation of their cognate toxins YafO or VapC could play important roles in persister formation in the population (Levin-Reisman et al. 2017).
One of the most cited pieces of evidence supporting the role of type II TA modules in persister formation was published by Gerdes group, showing the decreased persistence of ΔTA10 E. coli strain with ten type II toxins as endoribonucleases or toxins/antitoxins (ΔmazF, ΔchpB, ΔrelBE, ΔdinJ/yafQ, ΔyefM/yoeB, ΔhigBA, ΔprlF/yhaV, ΔyafNO, ΔmqsRA, ΔhicAB) deleted (Maisonneuve et al. 2013). However, this result was recently challenged by Ramisetty et al., who independently constructed the ΔTA10 E. coli strain and demonstrated that this mutant did not show significant differences in persistence with chloramphenicol, erythromycin, and kanamycin, compared with the wildtype strain (Ramisetty et al. 2016). Melderen group further constructed a new ΔTA10 E. coli strain and confirmed again that there is no direct link between induction of the ten TA systems and persistence to antibiotics, and the authors also suggested that proper and defined experimental conditions are of major importance when performing persistence assays (Goormaghtigh et al. 2018). These results provide strong evidence that the ten endoribonuclease TA modules do not play an essential role in spontaneous persister formation, however, due to the great numbers and diverse molecular activities of type II TA systems in the bacterial genomes, the role of other type II TA modules played in bacterial persistence needs re-evaluation.
CONCLUSION
As exemplified above, type II TA systems are widespread and involve in a broad range of physiological activities such as post-segregational killing, phage abortive infection, and persistence. After significant efforts of the scientific community, the molecular features and activities of many TA systems were characterized and reported in detail. Nevertheless, the biological function of a large proportion of type II TA systems remains mysterious. Intriguingly, several non-classical toxins, which have multi-domains or fusion domains, increase the complexity of the type II TA systems. Moreover, the extent of functional divergence between the homologous type II TA systems in different bacteria is still elusive. Investigations of the predicted type II TA systems in diverse prokaryotic genomes are likely to have unanticipated findings as novel functionalities and regulatory rewiring can be developed through horizontal gene transfer, which could add to the complexity of type II TA systems.
References
Aakre CD, Phung TN, Huang D, Laub MT (2013) A bacterial toxin inhibits DNA replication elongation through a direct interaction with the beta sliding clamp. Mol Cell 52:617–628
Alawneh AM, Qi D, Yonesaki T, Otsuka Y (2016) An ADP-ribosyltransferase Alt of bacteriophage T4 negatively regulates the Escherichia coli MazF toxin of a toxin-antitoxin module. Mol Microbiol 99:188–198
Andrews ES, Arcus VL (2015) The mycobacterial PhoH2 proteins are type II toxin antitoxins coupled to RNA helicase domains. Tuberculosis (Edinb) 95:385–394
Arbing MA, Handelman SK, Kuzin AP, Verdon G, Wang C, Su M, Rothenbacher FP, Abashidze M, Liu M, Hurley JM, Xiao R, Acton T, Inouye M, Montelione GT, Woychik NA, Hunt JF (2010) Crystal structures of Phd-Doc, HigA, and YeeU establish multiple evolutionary links between microbial growth-regulating toxin-antitoxin systems. Structure 18:996–1010
Bendtsen KL, Xu K, Luckmann M, Winther KS, Shah SA, Pedersen CNS, Brodersen DE (2017a) Toxin inhibition in C. crescentus VapBC1 is mediated by a flexible pseudo-palindromic protein motif and modulated by DNA binding. Nucleic Acids Res 45:2875–2886
Bendtsen KL, Xu KH, Luckmann M, Winther KS, Shah SA, Pedersen CNS, Brodersen DE (2017b) Toxin inhibition in C.crescentus VapBC1 is mediated by a flexible pseudo-palindromic protein motif and modulated by DNA binding. Nucleic Acids Res 45:2875–2886
Bobay BG, Andreeva A, Mueller GA, Cavanagh J, Murzin AG (2005) Revised structure of the AbrB N-terminal domain unifies a diverse superfamily of putative DNA-binding proteins. FEBS Lett 579:5669–5674
Boggild A, Sofos N, Andersen KR, Feddersen A, Easter AD, Passmore LA, Brodersen DE (2012) The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure 20:1641–1648
Bordes P, Cirinesi AM, Ummels R, Sala A, Sakr S, Bitter W, Genevaux P (2011) SecB-like chaperone controls a toxin-antitoxin stress-responsive system in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 108:8438–8443
Bordes P, Sala AJ, Ayala S, Texier P, Slama N, Cirinesi AM, Guillet V, Moureya L, Genevaux P (2016) Chaperone addiction of toxin-antitoxin systems. Nature Commun 7:13339
Brown BL, Grigoriu S, Kim Y, Arruda JM, Davenport A, Wood TK, Peti W, Page R (2009) Three dimensional structure of the MqsR:MqsA complex: a novel TA pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog 5:e1000706
Brown BL, Wood TK, Peti W, Page R (2011) Structure of the Escherichia coli antitoxin MqsA (YgiT/b3021) bound to its gene promoter reveals extensive domain rearrangements and the specificity of transcriptional regulation. J Biol Chem 286:2285–2296
Butt A, Higman VA, Williams C, Crump MP, Hemsley CM, Harmer N, Titball RW (2014) The HicA toxin from Burkholderia pseudomallei has a role in persister cell formation. Biochem J 459:333–344
Castro-Roa D, Garcia-Pino A, De Gieter S, van Nuland NAJ, Loris R, Zenkin N (2013) The Fic protein Doc uses an inverted substrate to phosphorylate and inactivate EF-Tu. Nat Chem Biol 9:811–817
Chan WT, Espinosa M, Yeo CC (2016) Keeping the wolves at bay: antitoxins of prokaryotic type II toxin-antitoxin systems. Front Mol Biosci 3:9
Cherny I, Rockah L, Gazit E (2005) The YoeB toxin is a folded protein that forms a physical complex with the unfolded YefM antitoxin. J Biol Chem 280:30063–30072
Cheverton AM, Gollan B, Przydacz M, Wong CT, Mylona A, Hare SA, Helaine S (2016) A Salmonella toxin promotes persister formation through acetylation of tRNA. Mol Cell 63:86–96
Dao-Thi MH, van Melderen L, De Genst E, Afif H, Buts L, Wyns L, Loris R (2005) Molecular basis of gyrase poisoning by the addiction toxin CcdB. J Mol Biol 348:1091–1102
Dienemann C, Boggild A, Winther KS, Gerdes K, Brodersen DE (2011) Crystal structure of the VapBC toxin-antitoxin complex from Shigella flexneri reveals a hetero-octameric DNA-binding assembly. J Mol Biol 414:713–722
Donegan NP, Marvin JS, Cheung AL (2014) Role of adaptor TrfA and ClpPC in controlling levels of SsrA-tagged proteins and antitoxins in Staphylococcus aureus. J Bacteriol 196:4140–4151
Donegan NP, Thompson ET, Fu Z, Cheung AL (2010) Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J Bacteriol 192:1416–1422
Dy RL, Przybilski R, Semeijn K, Salmond GP, Fineran PC (2014) A widespread bacteriophage abortive infection system functions through a type IV toxin-antitoxin mechanism. Nucleic Acids Res 42:4590–4605
Engel P, Goepfert A, Stanger FV, Harms A, Schmidt A, Schirmer T, Dehio C (2012) Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature 482:107–110
Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA (2003) Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell 11:671–683
Francuski D, Saenger W (2009) Crystal structure of the antitoxin-toxin protein complex RelB-RelE from Methanococcus jannaschii. J Mol Biol 393:898–908
Freire DM, Gutierrez C, Garza-Garcia A, Grabowska AD, Sala AJ, Ariyachaokun K, Panikova T, Beckham KSH, Colom A, Pogenberg V, Cianci M, Tuukkanen A, Boudehen YM, Peixoto A, Botella L, Svergun DI, Schnappinger D, Schneider TR, Genevaux P, de Carvalho LPS, Wilmanns M, Parret AHA, Neyrolles O (2019) An NAD(+) phosphorylase toxin triggers mycobacterium tuberculosis cell death. Mol Cell 73(1282–1291):e8
Garcia-Pino A, De Gieter S, Talavera A, De Greve H, Efremov RG, Loris R (2016) An intrinsically disordered entropic switch determines allostery in Phd-Doc regulation. Nat Chem Biol 12:490–496
Gerdes K, Rasmussen PB, Molin S (1986) Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc Natl Acad Sci USA 83:3116–3120
Germain E, Castro-Roa D, Zenkin N, Gerdes K (2013) Molecular mechanism of bacterial persistence by HipA. Mol Cell 52:248–254
Germain E, Roghanian M, Gerdes K, Maisonneuve E (2015) Stochastic induction of persister cells by HipA through (p)ppGpp-mediated activation of mRNA endonucleases. Proc Natl Acad Sci USA 112:5171–5176
Goormaghtigh F, Fraikin N, Putrins M, Hallaert T, Hauryliuk V, Garcia-Pino A, Sjodin A, Kasvandik S, Udekwu K, Tenson T, Kaldalu N, van Melderen L (2018) Reassessing the role of type II toxin-antitoxin systems in formation of Escherichia coli type II persister cells. MBio 9:e00640
Guglielmini J, van Melderen L (2011) Bacterial toxin-antitoxin systems: translation inhibitors everywhere. Mob Genet Elements 1:283–290
Guillet V, Bordes P, Bon C, Marcoux J, Gervais V, Sala AJ, Dos Reis S, Slama N, Mares-Mejia I, Cirinesi AM, Maveyraud L, Genevaux P, Mourey L (2019) Structural insights into chaperone addiction of toxin-antitoxin systems. Nat Commun 10:782
Harms A, Brodersen DE, Mitarai N, Gerdes K (2018) Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol Cell 70:768–784
Harms A, Liesch M, Korner J, Quebatte M, Engel P, Dehio C (2017) A bacterial toxin-antitoxin module is the origin of inter-bacterial and inter-kingdom effectors of Bartonella. PLoS Genet 13:e1007077
Harms A, Stanger FV, Scheu PD, de Jong IG, Goepfert A, Glatter T, Gerdes K, Schirmer T, Dehio C (2015) Adenylylation of gyrase and topo IV by ficT toxins disrupts bacterial DNA topology. Cell Rep 12:1497–1507
Jiang Y, Pogliano J, Helinski DR, Konieczny I (2002) ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol 44:971–979
Jurenas D, van Melderen L, Garcia-Pino A (2019) Mechanism of regulation and neutralization of the AtaR-AtaT toxin-antitoxin system. Nat Chem Biol 15:285–294
Kamada K, Hanaoka F, Burley SK (2003) Crystal structure of the MazE/MazF complex: molecular bases of antidote-toxin recognition. Mol Cell 11:875–884
Kamphuis MB, Monti MC, van den Heuvel RH, Lopez-Villarejo J, Diaz-Orejas R, Boelens R (2007) Structure and function of bacterial kid-kis and related toxin-antitoxin systems. Protein Pept Lett 14:113–124
Kaspy I, Rotem E, Weiss N, Ronin I, Balaban NQ, Glaser G (2013) HipA-mediated antibiotic persistence via phosphorylation of the glutamyl-tRNA-synthetase. Nat Commun 4:3001
Keren I, Shah D, Spoering A, Kaldalu N, Lewis K (2004) Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186:8172–8180
Khoo SK, Loll B, Chan WT, Shoeman RL, Ngoo L, Yeo CC, Meinhart A (2007) Molecular and structural characterization of the PezAT chromosomal toxin-antitoxin system of the human pathogen Streptococcus pneumoniae. J Biol Chem 282:19606–19618
Kirstein J, Moliere N, Dougan DA, Turgay K (2009) Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Microbiol 7:589–599
Koga M, Otsuka Y, Lemire S, Yonesaki T (2011) Escherichia coli rnlA and rnlB compose a novel toxin-antitoxin system. Genetics 187:123–130
Leplae R, Geeraerts D, Hallez R, Guglielmini J, Dreze P, van Melderen L (2011) Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res 39:5513–5525
Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ (2017) Antibiotic tolerance facilitates the evolution of resistance. Science 355:826–830
Lewis K (2010) Persister cells. Annu Rev Microbiol 64:357–372
Li GY, Zhang Y, Inouye M, Ikura M (2009) Inhibitory mechanism of Escherichia coli RelE-RelB toxin-antitoxin module involves a helix displacement near an mRNA interferase active site. J Biol Chem 284:14628–14636
Liang Y, Gao Z, Wang F, Zhang Y, Dong Y, Liu Q (2014) Structural and functional characterization of Escherichia coli toxin-antitoxin complex DinJ-YafQ. J Biol Chem 289:21191–21202
Loris R, Garcia-Pino A (2014) Disorder- and dynamics-based regulatory mechanisms in toxin-antitoxin modules. Chem Rev 114:6933–6947
Lu CH, Nakayasu ES, Zhang LQ, Luo ZQ (2016) Identification of Fic-1 as an enzyme that inhibits bacterial DNA replication by AMPylating GyrB, promoting filament formation. Sci Signal 9:ra11
Madl T, van Melderen L, Mine N, Respondek M, Oberer M, Keller W, Khatai L, Zangger K (2006) Structural basis for nucleic acid and toxin recognition of the bacterial antitoxin CcdA. J Mol Biol 364:170–185
Maisonneuve E, Castro-Camargo M, Gerdes K (2013) (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154:1140–1150
Marimon O, Teixeira JM, Cordeiro TN, Soo VW, Wood TL, Mayzel M, Amata I, Garcia J, Morera A, Gay M, Vilaseca M, Orekhov VY, Wood TK, Pons M (2016) An oxygen-sensitive toxin-antitoxin system. Nat Commun 7:13634
Mate MJ, Vincentelli R, Foos N, Raoult D, Cambillau C, Ortiz-Lombardia M (2012) Crystal structure of the DNA-bound VapBC2 antitoxin/toxin pair from Rickettsia felis. Nucleic Acids Res 40:3245–3258
Mattison K, Wilbur JS, So M, Brennan RG (2006) Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J Biol Chem 281:37942–37951
Meinhart A, Alonso JC, Strater N, Saenger W (2003) Crystal structure of the plasmid maintenance system epsilon/zeta: functional mechanism of toxin zeta and inactivation by epsilon 2 zeta 2 complex formation. Proc Natl Acad Sci USA 100:1661–1666
Moyed HS, Bertrand KP (1983) hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J Bacteriol 155:768–775
Muthuramalingam M, White JC, Bourne CR (2016) Toxin-antitoxin modules are pliable switches activated by multiple protease pathways. Toxins 8:214
Oberer M, Zangger K, Gruber K, Keller W (2007) The solution structure of ParD, the antidote of the ParDE toxin antitoxin module, provides the structural basis for DNA and toxin binding. Protein Sci 16:1676–1688
Page R, Peti W (2016) Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat Chem Biol 12:208–214
Pandey DP, Gerdes K (2005) Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res 33:966–976
Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M (2003) The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112:131–140
Piscotta FJ, Jeffrey PD, Link AJ (2019) ParST is a widespread toxin-antitoxin module that targets nucleotide metabolism. Proc Natl Acad Sci USA 116:826–834
Puri N, Karzai AW (2017) HspQ functions as a unique specificity-enhancing factor for the AAA plus lon protease. Mol Cell 66:672–683
Ramisetty BC, Santhosh RS (2016) Horizontal gene transfer of chromosomal type II toxin-antitoxin systems of Escherichia coli. FEMS Microbiol Lett 363:fnv238
Ramisetty BCM, Ghosh D, Chowdhury MR, Santhosh RS (2016) What is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front Microbiol 8:458
Rocker A, Meinhart A (2015) A cis-acting antitoxin domain within the chromosomal toxin-antitoxin module EzeT of Escherichia coli quenches toxin activity. Mol Microbiol 97:589–604
Rocker A, Meinhart A (2016) Type II toxin: antitoxin systems. More than small selfish entities? Curr Genet 62:287–290
Schreiter ER, Drennan CL (2007) Ribbon-helix-helix transcription factors: variations on a theme. Nat Rev Microbiol 5:710–720
Schumacher MA, Piro KM, Xu W, Hansen S, Lewis K, Brennan RG (2009) Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323:396–401
Schureck MA, Maehigashi T, Miles SJ, Marquez J, Cho SE, Erdman R, Dunham CM (2014) Structure of the Proteus vulgaris HigB-(HigA)2-HigB toxin-antitoxin complex. J Biol Chem 289:1060–1070
Semanjski M, Germain E, Bratl K, Kiessling A, Gerdes K, Macek B (2018) The kinases HipA and HipA7 phosphorylate different substrate pools in Escherichia coli to promote multidrug tolerance. Sci Signal 11:eaat5750
Sevin EW, Barloy-Hubler F (2007) RASTA-Bacteria: a web-based tool for identifying toxin-antitoxin loci in prokaryotes. Genome Biol 8:R155
Skjerning RB, Senissar M, Winther KS, Gerdes K, Brodersen DE (2019) The RES domain toxins of RES-Xre toxin-antitoxin modules induce cell stasis by degrading NAD. Mol Microbiol 111:221–236
Smith CK, Baker TA, Sauer RT (1999) Lon and Clp family proteases and chaperones share homologous substrate-recognition domains. Proc Natl Acad Sci USA 96:6678–6682
Stanger FV, Harms A, Dehio C, Schirmer T (2016) Crystal structure of the Escherichia coli fic toxin-like protein in complex with its cognate antitoxin. PLoS ONE 11:e0163654
Takagi H, Kakuta Y, Okada T, Yao M, Tanaka I, Kimura M (2005) Crystal structure of archaeal toxin-antitoxin RelE-RelB complex with implications for toxin activity and antitoxin effects. Nat Struct Mol Biol 12:327–331
Talavera A, Tamman H, Ainelo A, Konijnenberg A, Hadzi S, Sobott F, Garcia-Pino A, Horak R, Loris R (2019) A dual role in regulation and toxicity for the disordered N-terminus of the toxin GraT. Nat Commun 10:972
Tamman H, Ainelo A, Ainsaar K, Horak R (2014) A moderate toxin, GraT, modulates growth rate and stress tolerance of Pseudomonas putida. J Bacteriol 196:157–169
Tamman H, Ainelo A, Tagel M, Horak R (2015) Stability of the GraA antitoxin depends on growth phase, ATP Level, and global regulator MexT. J Bacteriol 198:787–796
Tripathi A, Dewan PC, Siddique SA, Varadarajan R (2014) MazF-induced growth inhibition and persister generation in Escherichia coli. J Biol Chem 289:4191–4205
Turnbull KJ, Gerdes K (2017) HicA toxin of Escherichia coli derepresses hicAB transcription to selectively produce HicB antitoxin. Mol Microbiol 731(104):781–792
Wang X, Kim Y, Hong SH, Ma Q, Brown BL, Pu M, Tarone AM, Benedik MJ, Peti W, Page R, Wood TK (2011) Antitoxin MqsA helps mediate the bacterial general stress response. Nat Chem Biol 7:359–366
Wang X, Lord DM, Cheng HY, Osbourne DO, Hong SH, Sanchez-Torres V, Quiroga C, Zheng K, Herrmann T, Peti W, Benedik MJ, Page R, Wood TK (2012) A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat Chem Biol 8:855–861
Wang Y, Zhang SP, Zhang MY, Kempher ML, Guo DD, Han JT, Tao X, Wu Y, Zhang LQ, He YX (2019) The antitoxin MqsA homologue in Pseudomonas fluorescens 2P24 has a rewired regulatory circuit through evolution. Environ Microbiol 21:1740–1756
Wilcox B, Osterman I, Serebryakova M, Lukyanov D, Komarova E, Gollan B, Morozova N, Wolf YI, Makarova KS, Helaine S, Sergiev P, Dubiley S, Borukhov S, Severinov K (2018) Escherichia coli ItaT is a type II toxin that inhibits translation by acetylating isoleucyl-tRNAIle. Nucleic Acids Res 46:7873–7885
Wintjens R, Rooman M (1996) Structural classification of HTH DNA-binding domains and protein-DNA interaction modes. J Mol Biol 262:294–313
Wu WF, Zhou YN, Gottesman S (1999) Redundant in vivo proteolytic activities of Escherichia coli lon and the ClpYQ (HslUV) protease. J Bacteriol 181:3681–3687
Xie Y, Wei Y, Shen Y, Li X, Zhou H, Tai C, Deng Z, Ou HY (2018) TADB 2.0: an updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res 46:D749–D753
Xu K, Dedic E, Brodersen DE (2016) Structural analysis of the active site architecture of the VapC toxin from Shigella flexneri. Proteins 84:892–899
Zhang Y, Zhang J, Hoeflich KP, Ikura M, Qing G, Inouye M (2003) MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol Cell 12:913–923
Zhu L, Inoue K, Yoshizumi S, Kobayashi H, Zhang Y, Ouyang M, Kato F, Sugai M, Inouye M (2009) Staphylococcus aureus MazF specifically cleaves a pentad sequence, UACAU, which is unusually abundant in the mRNA for pathogenic adhesive factor SraP. J Bacteriol 191:3248–3255
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (31770535 and 31300616), the Natural Science Foundation of Gansu Province (17JR5RA208), and Fundamental Research Funds for the Central Universities from Lanzhou University (lzujbky-2017-151).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Si-Ping Zhang, Qian Wang, Shuo-Wei Quan, Xiao-Quan Yu, Yong Wang, Ding-Ding Guo, Liang Peng, Hu-Yuan Feng, and Yong-Xing He declare that they have no conflicts of interest.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, SP., Wang, Q., Quan, SW. et al. Type II toxin–antitoxin system in bacteria: activation, function, and mode of action. Biophys Rep 6, 68–79 (2020). https://doi.org/10.1007/s41048-020-00109-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41048-020-00109-8