
Abstract
This paper reviews polymer response to shock and discusses how plastics behave differently depending on the strain rate applied. The author uses polyethylene, polytetrafluroethylene, polycarbonate and epoxy as example materials. It is suggested that there are two distinct regimes of polymer response corresponding to low and high shock pressures (weak and strong shock regimes) that must be considered separately. Above a high pressure threshold it is proposed that polymers homogenize to carbon-containing structures which behave similarly. Further, the yield stress of a polymer volume element asymptotes to the theoretical strength of the material as volume shrinks to zero. It is suggested that these two behaviours reflect the same physics and this feature is common to the condition identified earlier for crystalline materials.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As applications of polymers and novel polymer matrix composites grow, they are placed under increasingly more extreme environments such as impact, in space or under high temperature conditions (e.g. [1, 2]). In these environments, composites are engineered with increasing strength and with components at scales from micron fibres to nanoscale carbon inclusions [3, 4]. Further, understanding of the dynamic response of polymers and polymer matrix composites at high pressures and strain rates is vital to fully describe the response of structures [5–12].
Polymers have received relatively less attention at high pressure and strain rate than other classes of material. However within the limited literature available, there have been indications from the first that behaviour was of a different nature to that observed in metals [13]. For example, Champion, in studying polytetrafluroethylene (PTFE), noted a change in slope in the shock velocity, U s, particle velocity (u p) curve, corresponding to an impact stress of about 0.5 GPa, and giving a volume change of 2.2 % [14, 15]. The first comprehensive overview of the shock response of thermoplastics, thermosets, and rubbers was done by Carter and Marsh and more recent work has built on these studies [13].
This paper will review response across a range of distance and time scales. It will thus be useful to describe mechanical deformation and components of structure using a terminology defined here. There are already a number of terms used in differing manners across the physical and engineering sciences; ‘microscale’, ‘mesoscale’ and ‘macroscale’ will be used to define regions within a solid. However, to avoid ambiguity, a framework is defined here to expand on these and relate scale to mechanical response. For each one discussed an order of magnitude dimension is included. The ‘atomic scale’ (ca. 0.1 nm) builds to an assemblage of atoms to define the ‘nanoscale’ (ca. 10 nm). In polymers, chains make links through Van de Waals and London forces that are weaker than the intrachain bonding. They can fold one upon the other to form lamellae which may grow into spherulites (of order 1 µm but often larger) and analogous to grains in crystalline solids. The strength of the solid drops across the boundary as defects may be present there. An assembly of spherulites, defects and second phase particles will combine to form a representative volume element that constitutes a microstructure and defines mechanical response at larger scales. A ‘component’ (ca. 1 mm) is introduced here as an element of a ‘structure’, the largest scale considered here. With this set of definitions the term ‘microscale’ will be used to describe the atomic and nanoscales, the ‘mesoscale’ the transition to the component scale, and the ‘macroscale’ will combine the component and the structural scales. The boundaries between each, between micro- and meso- (at the grain boundary) and between component- and structural- at a joint, represent drops in local cohesion at which failure may occur. At the end of the mesoscale regime strong electrostatic forces cease to dominate response and interactions can be regarded as mechanical (for further discussion see [16] chap. 8).
The polymer responds to a mechanical impulse defined by a particular rise time, duration and amplitude and it sweeps different depths within the microstructure as it propagates. An element of material responds by a deformation mechanism and this has a particular threshold and relaxation time associated with it. A changing stress state must nucleate and allow damage mechanisms to begin. Flow can only proceed as long as release does not occur before the mechanism has completed. The physical region sampled by the pulse and the defects within it will determine the stress required to start plasticity and this strength will approach its theoretical limit as loading time is reduced.
Shock waves are known to display structure within their fronts. At lower stress amplitudes, elastic waves precede an inelastic rise to the final pressure whilst under more extreme loading there is a single shock to peak stress. It has been shown for a range of solids that the transition from defect dominated (weak shock) to homogeneous (strong shock) behaviour has a threshold stress and the term weak shock limit (WSL) has been used to describe it (see discussion in [16–21]). This limit has been shown to correlate with the theoretical strength of the material and free volume has been eliminated beyond this stress. The present work will suggest that as volume sampled is reduced and pressure is increased the strength approaches such a threshold.
Engineers have shown over many years a strong dependence of polymer strength on strain rate when the materials are placed in dynamic environments [22]. A common appreciation of this relates increasing strain-rate to higher elastic moduli since excited polymer chains have reduced relaxation times [23]. This work will aim to review the micromechanics evident in polymer response at high strain rate and to use amorphous materials polycarbonate (PC) and an Epoxy and a partially crystalline material (PTFE) to illustrate features of behaviour under the highest compressions and strain rates whilst referring back to the simplest hydrocarbon polyethylene as a baseline material [24].
Materials
Polymers are a class of primarily amorphous solids whose microstructure has varying degrees of order at the mesoscale generally controlled by the synthesis route [25]. Their response to mechanical loading can be divided into quasistatic, intermediate rate and shock regimes and this paper will focus on the latter. Quasistatically, their strength takes differing values according to microstructural changes accessed as temperature rises [26]. In the glassy regime, polymers exhibit high stiffness (large modulus) that drops across the glass-transition as the microstructure expands and becomes a rubber [27]. At higher temperature response again enters a viscous regime where the polymer starts to flow and at higher values still they eventually decompose. Thus mechanical properties change rapidly with temperature but also with the time that is spent under load since high strain rates and low temperatures restrict the modes of deformation in the chains within the microstructure [28].
Some polymers (e.g. thermosets) are amorphous and show no order; polycarbonate (PC) and epoxy discussed here are in this class. The epoxy tested in [29] was cured from a mixture of an epoxy resin mixture, a flexibilizer, and a hardener. The resin mixture consisted of 14–22 % 1,4-butanedioldiglycidyl ether (C10H18O4) and 78–86 % of the product of reaction between epichlorohydrin (C3H5ClO) and bisphenol to give an approximate empirical formula of C21H24O4 [29]. The thermoplastic PTFE is semicrystalline and consist of spherulites embedded within an amorphous matrix [30, 31]. Each of these classes displays different response to loading at quasistatic rates than under shock as discussed below [32–34]. In a polymer, the molecular building blocks, which forms the chain (the monomers from which it is made) provide only short-range order when a large volume of material is under load but assume more importance as impulses become shorter. The repeat units for PC, epoxy and PTFE are shown in Fig. 1 along with that of the simplest thermoplastic polyethylene (PE) for reference. The chains themselves and their arrangement under differing environments determine statistical limits upon observed behaviours. However, as pulses become shorter (and length scales decrease) the interactions at the microscale control the dynamic behaviour of the polymer and strength is fixed by the confining pressure and determined by the intrachain and Van der Waals forces between chains [25].

Monomer units for a PE, b epoxy, c PTFE and d PC discussed below
Previous work has measured ambient mechanical properties and shock response for the materials discussed here and this is summarised in Table 1.
Shock Compression
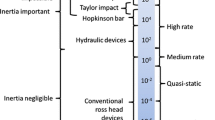
There is a ready supply of mechanical data concerning the response of polymers at low and intermediate strain rates (see [35] for example). However, there is a relative paucity of shock data as the loading becomes more rapid and the impulse amplitude increases [13, 36–38]. In the high shock pressure regime, there is only one extensive study (using explosive lenses by Carter and Marsh) and written up into an openly accessible report by Fritz and Sheffield [13]. This means of loading only subjected targets to high stress levels and Taylor wave loading and so there is little detail for response at lower stress amplitudes where the shock was not overdriven. However, the work nevertheless shows an indication of behaviour across the class of materials. Laser compression with ultrafast spectroscopy as a diagnostic has also probed structural stretching and relaxation processes with polymers subject to femptosecond loads [39–42].
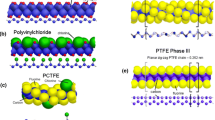
The phase diagram for PTFE is shown in Fig. 2a) [43]. Under laboratory conditions at room temperature, a pressure-induced phase transition has been reported in PTFE at 0.50–0.65 GPa (Phase II–III). Other work has determined the dynamic transition behaviour in the polymer [44–47]. Phase II PTFE consists of a helical conformation with a 13 atom repeat unit and a well-ordered hexagonal packing of the helical chains, while in phase III the helical conformation gives way to a planar zig-zag and the chain packing takes on an orthorhombic or monoclinic lattice structure. Recent work using a high-pressure diamond-anvil cell and near-infrared Raman (NIR) spectroscopy suggests the transition occurs at 0.65 GPa and exhibits around ±0.05 GPa of hysteresis [48–51] The phase transition results in a 13 % local volume change within the crystalline domains and a considerable reduction in compressibility. PTFE also exhibits two atmospheric pressure, crystalline transitions at 19 and 30 °C [52].

It is found for a range of materials (that don’t change phase) that the relationship between the shock velocity U S achieved and the driving velocity u p, is a linear relation related via two constants thus
The constants c 0 and S are generally determined by experiment. However, the first is related to the bulk sound speed (and in metals such as copper can be shown to be so) while the second, S, reflects the rate of change of compressibility of the material with pressure (dK/dP for a material deforming hydrodynamically; see derivation in [53]). In materials that change phase, there is generally a transition state and linear behaviour in regimes either side of the transformation state. In this case S is fit to collected data either side of this point and generally takes different values either side of the transformation state (see [53] for example). Unlike in simple metals, a bulk sound speed, c 0, calculated from ultrasonic measurements of c L and c s does not correspond to the intercept of the plot of Eq. (1) for polymers and generally lies above this value. Secondly in the low pressure (below the 20-30 GPa transition) the curve in many polymers has a non-linear form and may be fitted with a polynomial to represent the data.
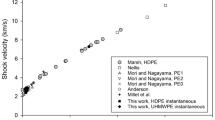
Shock data on PTFE obtained from various commercial and pedigree sources has shown there to be no significant effect of controlled production and purity at least within the experimental scatter (Fig. 2b) [44, 54]. Figure 2 b) shows stress-particle velocity data for the range of materials with a best-fit line superposed onto the data. However, this low-pressure region contains the phase transformation from phase II to phase III which occurs at 0.7 GPa and at a particle velocity of ca. 0.19 mm µs−1 (note the dashed line in the figure).
For many commercially available polymers and specially produced plastics, a linear response is observed above a particle velocity of ca. 0.2 mm µs−1. The intercept on the shock velocity axis c 0 is less than the ambient longitudinal sound speed c L as noted previously. Lines are plotted on the curve showing fits to literature constants for PTFE (given in Table 1) and determined in [13, 36, 37]. There have also been recovery experiments that show details of changes in crystallinity in the low pressure region [45]. Shocking material within phase II (<0.7 GPa) results in a decrease in crystallinity, decrease in Young’s modulus, decrease in yield stress, and little change in microstructure. Conversely, transition from phase II into phase III (>0.7 GPa) results in increased crystallinity, an apparently independence of pressure, increase in Young’s modulus, increase in yield stress and significant microstructural refinement [46].
The low pressure equation-of-state measurements made across a range of polymers reflect the integrated effect of the density of the materials and their strength in the this region which is dominated by Van der Waals’ forces between interacting polymer chains; larger for the fluorinated PTFE than the hydrocarbon polyethylene (PE). Here changes in mechanical response are most closely linked to changes in the percentage of crystallinity and crystalline domain size [46]. Nevertheless Carter and Marsh showed that the low-pressure data can be approximated assuming only a two-dimensional force field is present between chains in a shocked thermoplastic [13].
Nearly all polymers show a further high-temperature (ca. 2000 K) and high-pressure transformation at 20–30 GPa for which the volume change is also large (Fig. 3) [13]. This is seen as a discontinuity in the slope of the U s–u p curve at u p = ca. 3 mm µs−1. This is shown here for the polymers PTFE and PC and vertical dotted lines mark the particle velocities at which the pressure reached 25 GPa; PTFE to the left and PC to the right. As seen above, behaviour in the low pressure regime is dominated by rotation and rearrangements of chains subject to weaker Van der Waals’ and London inter-chain forces. Carter and Marsh suggest that at high pressure, as separation reduces, the observed behaviour approaches that of an homogeneous, diamond like solid and this is supported by the magnitude of the weak shock limit for these materials [13, 55]. By this threshold significant volume change has occurred and the transformation that results is analogous to the carbon-diamond transition where covalent bonds are broken in favour of tetragonal bonds between planes [56]. The reader will notice that the value of S above this point is close to the same in both PTFE and PC and this is found to be the case for a range of polymers for which shock data is available [36]. The slope S = 1.47 in this high pressure region.

U S–u p for PTFE and PC to high pressure; transition at ca. 25 GPa in each polymer. The shock velocity is displaced vertically for PTFE by 1 mm µs−1 to display behaviours more clearly [13]. The vertical dotted lines mark the particle velocities at which the pressure reached 25 GPa; PTFE to the left and PC to the right
In crossing the WSL boundary a shift in behaviour and properties is found and the resulting microstructures may be expected to have markedly different properties to those of the parent polymer. In summary, low pressure shock behaviour is akin to densification of a porous medium whilst the high pressure transition accesses fully dense behaviour with an overdriven strong shock regime in plastics familiar in other classes of material where the response is homogeneous.
Strength
The non-invasive measurement of in-material states of stress and strain within loaded targets is a paradigm that has yet to be fully achieved yet the shear strength within compressed materials represents an integrated history of microstructural evolution within the loaded targets (e.g. [57, 58]). To do this requires sensors to measure these changes during the compression impulse. Mounting these sensors within polymers is simple and reproducible since the material is an insulator and the mounting epoxies are matched to the target. In recent times, great advances have been made using manganin sensors to achieve this goal [59–65]. To appreciate the technique requires a brief digression into stress states behind a shock front when the material has been shocked to a state of macroscopic uniaxial strain. In such a regime there is a biaxial stress system behind the shock consisting of longitudinal and lateral components and sensors have been developed to probe their development [16]. Further details of the flow at low stresses must be taken into account so that a staged refinement of the analysis used to deconvolve the change in resistance back to stress can be used over the range of stresses up to the weak shock limit (where homogeneous and hydrodynamic behaviours ensue) [66–70]. In what follows the evolution of the shear stress during impact in a range of polymers is determined in the lower pressure regime with embedded gauges.
Figure 4 shows the development of the stress field within the thermoset epoxy compressed by impact in the low-pressure regime. Sensors were embedded in the flow, a distance from the impact face to observe the development of the stress field behind the shock [29]. In each case the difference between the two components of stress determines twice the shear stress and these strength histories are represented by solid lines in the figure. The epoxy has an isotropic network structure and so weaker crosslinks dominate response. As shock amplitude increases, shear stress rises behind the front as the pressure in this range increases. The longitudinal and lateral stresses rise rapidly in all cases and whilst the former is flat, the latter slowly relaxes; (although only the longitudinal component is shown here). The figure shows the response at two stress levels with the impulses displayed along with two corresponding shear stress histories. At these lower amplitudes this epoxy strengthens in an almost linear manner over the microsecond or so of the experiment. As the stress level is increased, the rate of rise increases and at higher stresses again the final strength asymptotes to a maximum value. This is interpreted as the compression of a microstructure not yet at full density and the evolving strength corresponds to the interaction of polymer chains at increasing compression.

Response of the thermoset epoxy. Two stress levels 3.5 (solid) and 2 GPa dashed) showing evolution of the shear stress behind the shock [29]
Having described response in a thermoset epoxy, Fig. 5 shows lateral stress histories for PC. Again the longitudinal stress histories show well-formed, flat topped impulses. Those in Fig. 5a) show two experiments in which the sensor was positioned 2 mm from the impact face. They were recorded in two shots at the same impact stress. In both cases the longitudinal stress induced was 1.5 GPa but the experiments represent different loading geometries; in one case, an asymmetric impact with a metal flyer plate and in the second, a symmetric impact where the impactor and target were both polycarbonate. The two stress histories show differing response over the first microsecond but lie one upon the other thereafter; there is a perturbation in the case of the metal flyer over the first microsecond after which the shear stress behind the front relaxes to that seen in the symmetrical case. In that case the lateral stress falls monotonically (the shear stress increases). The behaviour for asymmetric impact when sensors are placed at other depths back from the impact face is shown in Fig. 5b) at 2 mm and c) at 6 mm.

Impact on polycarbonate at two stress levels ca. 1.5 and 3 GPa. a Lateral stress histories for the sensor 2 mm from the impact face for a symmetric (PC flyer/PC target; upper trace) and an asymmetric impact (metal flyer/PC target; lower trace). In both cases the longitudinal stress induced was 1.5 GPa. To allow details of the history to be observed more easily, the symmetrical impact stress has been multiplied by a factor of 1.1 so that the histories lie distinct from each other for clarity. b Metal flyers launched at targets with lateral and longitudinal sensors at 2 mm and c at the 6 mm station. Lateral and longitudinal stress are dashed and shear stress histories are solid curves [81]
The region near the impact face sees an unsteady response for a period of a microsecond or so before the lateral stress equilibrates (Fig. 5b). This does not correspond to any wave transit across the target or within the impactor but to equilibration of the microstructure to the lateral strain that exists at the impact face (in classical terms where the differing Poisson’s ratios of the two materials allow lateral motion). In the bulk of the material (Fig. 5c) the lateral stress drops monotonically showing the shear stress rising behind the front as these polymers develop resistance to flow. Furthermore the shear resistance rises more rapidly at higher stress as the impulse compresses microstructure to achieve high density.
The flow near an impact surface in the weak shock regime is only one-dimensional at the macroscale and then only when flyer and target are of the same material so that flow is symmetric. Brittle materials attain an inelastic state by microfracture driven from the impact face; such a zone also exists in metals but now stress state equilibration by slip is much faster. Indeed there is always such a region where materials of differing impedance interact, in which surface lateral strain must exist and in which assumptions of one-dimensional behaviour breakdown. This region is of greater extent in polymers than is the case for metals and brittle solids as can be seen above. Nevertheless there are features of the behaviour that are due to this asymmetry in the flow in both the other classes of materials. In metals this is seen in variation of microhardness in material in the hundreds of microns near the impact face, whilst in brittle ceramics, fracture waves are initiated when the impact is asymmetric that are not present in the symmetric case [71, 72]. In polymers the surface zone is of a different nature since in this case the response is dominated by densification of the initial open structure; by Van de Waals forces in thermoplastics or compressing cross-linking bonds in thermosets. This densification zone defines the response of plastics to dynamic loads above the yield stress and the shock regime exemplifies this behaviour well as these histories show.
The Limits of Yield
The strength of polymers is controlled by electronic and steric interactions, which in combination act to define response during dynamic loading. In the low-pressure region, molecular spacing and geometrical conformation are key in defining the development of the material’s strength under compression. However, in all the cases investigated here, polymers show increasing shear strength with pressure consistent with static measurements [73]. A change in behaviour is found in almost all plastics when compression reaches the point at which Van der Waals’ repulsions are overcome. In other work this threshold has been attributed to a transformation but it has been suggested above that this is the limit above which a material’s theoretical strength is overcome; the weak shock limit [13, 19]. Material strength data has been collected in a series of tests corresponding to different loaded volumes and as the pulse duration is reduced, the strength increases. This data is collected here to suggest that strength measured approaches the theoretical strength at the pressure where the change in shock behaviour is observed.
Data exists for the quasistatic and intermediate rate testing of the yield stress of polymers at differing strain rates ([74] for example). As strain rate increases, the stress equilibration time for each dynamic test reduces and the impulse delivered samples smaller and smaller regions [75]. This behaviour ensures that determination of strength occurs at three scales; structural response probed at the macroscale, component response probed at the mesoscale and within the constituent components at the microscale in these tests. Even at this scale measurements using traditional means render a series of values with some statistical spread fixed by variations in the packing of the chains [30]. Testing at intermediate rates from quasistatic to Split Hopkinson (or Kolsky) Pressure Bar shows strengthening over a range of strain rates up to ca. 103 s−1 of the same order as seen in metals (see Fig. 6a for PC and PTFE). [32, 35, 74, 76, 77]. Within this data the compressive behaviour of both PTFE and PC show strengthening within both the thermoplastic and the amorphous polymer. Indeed there is an increase of hardening rate around 103 s−1 as observed in other solids [78]. For PC, data exists for not only compressive but tensile strength as a function of strain rate [79, 80]. It will be seen that tensile approach compressive strengths at the highest strain rates.

The strain-rate associated with a test may be transformed into its components in order to recover the loading time and spatial zone accessed within the material. The strain at failure may be determined from the yield stress through the modulus. This is then used along with the tabulated strain rate to calculate a characteristic equilibration time for the process at the scale at which the test is conducted. In addition, tests on small volumes of material or with short pulses can be added to the data to review the strengths deduced in these experiments. Such a process has been conducted on the data from Fig. 6a) to construct the transformed datasets in Fig. 6b). The upper points correspond to data for PTFE, the lower for PC. The logarithm of strength is plotted against the logarithm of the time duration of loading and data for plate impact (ca. 10 ns) and laser shock loading (<1 ns) are included for PC to show behaviour at the smallest pulse lengths [43, 44, 81, 82]. The impulse sweeps a volume and a characteristic swept length scale can be calculated via the wave speed for the material. Since the scale is logarithmic and the wave speed of polymers slow (ca. 1 mm µs−1), the scale can be read in either µm or ns for sweep distance or loading time in this case. The laser shock pulse is probing distances of order a few monomer units within the polymer and records deduce values for the strength that approach the theoretical strength for the material. Two points are shown on the ordinate that correspond to the Frenkel formulation of the theoretical tensile and shear strengths of polycarbonate calculated from the moduli measured under ambient conditions (E 0 and µ 0) [83]. Clearly these data are exceeded at the highest pressures since the modulus itself is rapidly changing during compression and plate impact strengths included here at two nominal low pressures are around this limit for both the polymers considered. Finally a theoretical strength is included at a nominal 0.1 nm calculated using a quoted modulus for carbon nanotubes (E 1) which might be assumed to approximate an idealised strength for the nanoscale phase of compression [55]. Whilst the laser measurement does not reach this value it is close to it indicating that such tests may be used to directly probe the ultimate strengths of condensed phase materials.
The shaded region separates a microscale from a mesoscale response and potentially two different classes of mechanism in each case. Thus intermediate strain rate and shock data probe different bonding and strength properties with a polymer. Further the boundary between these regions separates ordered from disordered regions within the microstructure.
Discussion and Conclusions
The response of polymers to high dynamic pressure emphasizes the need found across materials’ physics to combine results from all the existing platforms and new imaging techniques to understand the complex response of these materials. Advances in new tools and diagnostics have illuminated their response dramatically and this is favoured since they have low density and can be easily penetrated by X-ray and particle sources to image deformation in real time under load [84, 85].
Their response to a dynamic impulse shows behaviour of a different nature to that of metals and ceramics which are close-packed at the bond (0.1 nm) scale. The micromechanics of the response of polymers to weak shock can be described in terms of the packing of the polymer chains at distances of five to ten times these dimensions. While the response of fully dense metals or ceramics in the weak shock regime is dominated in the first moments of load by the creation of localised regions to accommodate macroscopic strain, polymers respond by compressing against the weaker inter-chain Van der Waals forces or the lower density of cross-links [53, 70]. Microscale packing dominates behaviour and with approximately inverse square dependence on chain separation response is sensitive to conformation once compression begins [31, 86, 87].
Strength increasing behind the shock results from the closer proximity of the molecular components with strong attractive bonding and greater steric entanglement that resists further strain as time develops [73]. The strength of the repulsion reflects the electronegativity of the included atoms with sulphur showing less effect than oxygen and with fluorine applying the greatest force and exhibiting the greater strength. At these pressures cross-linkage in thermosets shows negligible difference to the behaviours observed with thermoplastics that have none, which illustrates that these different configurations, so important under ambient conditions, are homogenised as pressure increases. Across all of this densification range, compression of the open structure against the Van der Waals bonding or cross-linking in the cells does work that is converted to heat within the lattice. Thus it is no surprise that the strain input over a particular time is analogous to heating to an equivalent temperature. This behaviour is reflected in the response of PTFE and PC discussed in detail here but also in a wide range of polymers studied under shock. At larger compressions there is a transition to the strong shock regime for the polymer (at ca. 25 GPa in the materials discussed here; Fig. 3) and this represents the weak shock limit for these materials beyond which response is homogeneous [19, 21, 55].
Finally while micromechanics dominates polymer response, mechanisms still remain to be fully understood in depth. Any length scale has three phases of behaviour; localisation, flow and interaction and at least in the densification regime, polymer stress state stability is frequently not achieved in short impulse, low strain, laboratory tests [16]. Thus integrated loading techniques developed for metals (such as Taylor impact) reveal the transit to stress state stability over milliseconds for polymers where microseconds were sufficient in metals [16, 43, 88, 89]. At the present time most polymer constitutive models in use in hydrocodes are top level, semi-empirical fits to behaviour that have severe limitations in their applicability since they borrow concepts based on viscoplasticity and are fit to data in which the stress state is rarely steady (e.g. [47, 52, 88, 90, 91]). Future developments in theory and experiment must advance in tandem if models are to become more accurate and predictive. In all of the idiosyncrasies of polymer response in shock, it is the kinetics of the densification behaviour that must be addressed first in order to full reconcile observed states with the microstructures loaded. The complexity of polymer response has sometimes mediated their use in dynamic environments but greater understanding will ensure polymers become key components of the structures of the future in demanding and extreme environments.
References
Lee J-H, Veysset D, Singer JP, Retsch M, Saini G, Pezeril T, Nelson KA, Thomas EL (2012) High strain rate deformation of layered nanocomposites. Nat Commun. doi:10.1038/ncomms216
Lee JH, Loya PE, Lou J, Thomas EL (2014) Dynamic mechanical behavior of multilayer graphene via supersonic projectile penetration. Science 346(6213):1092–1096
Harris PJF (2011) Carbon nanotube science; synthesis, properties and applications. Cambridge University Press, Cambridge
Ray SS, Okamoto M (2003) Polymer/layered silicate nanocomposites: a review from preparation to processing. Prog Polym Sci 28:1539–1641
Dandekar DP, Hall CA, Chhabildas LC, Reinhart WD (2003) Shock response of a glass-fiber-reinforced polymer composite. Compos Struct 61:51–59
Eatwell M, Millett JCF, Bourne NK, Meziere Y (2006) The behaviour of a glass-fibre epoxy composite during plate impact, In: Furnish MD et al (ed) Shock compression of condensed matter—2005, American Institute of Physics, Melville, pp. 801–804
McDonald SA, Millett JCF, Bourne NK, Bennett K, Milne AM, Withers PJ (2007) The shock response, simulation and microstructural determination of a model composite material. J Mater Sci 42:9671–9678
Millett JCF, Bourne NK, Meziere YJE, Vignjevic R, Lukyanov A (2007) The effect of orientation on the shock response of a carbon fibre-epoxy composite. Compos Sci Technol 67:3253–3260
Vignjevic R, Campbell JC, Bourne NK, Djordjevic N (2008) Modeling shock waves in orthotropic elastic materials. J Appl Phys 104:044904
Chawla KK (2012) Composite materials: science and engineering. Springer, New York
Suwanprateeb J, Tiemprateeb S, Kangwantrakool S, Hemachandra K (1998) The role of filler volume fraction in the strain-rate dependence of calcium carbonate-reinforced polyethylene. J Appl Polym Sci 70:1717–1724
Addessio FL, Aidun JB (1998) Analysis of shock-induced damage in fiber-reinforced composites. In: Davison L, Shahinpoor M (eds) High Pressure Shock Compression of Solids III. Springer, New York, pp 241–275
Carter WJ, Marsh SP (1995) Hugoniot equation of state of polymers. Los Alamos, Los Alamos National Laboratory
Champion AR (1971) Shock compression of teflon from 2.5 to 25 kbar: evidence for a shock-induced transition. J Appl Phys 42:5546–5550
Champion AR (1972) Effect of shock compression on the electrical conductivity of three polymers. J Appl Phys 43:2216–2220
Bourne NK (2013) Materials in mechanical extremes; fundamentals and applications. Cambridge University Press, Cambridge
Bourne NK (2015) On the ultimate strength of condensed matter. Metall Mater Trans A 46(10):4498–4505
Bourne NK (2014) The limits of strength in materials in the condensed phase. In: Buttler W, Furlanetto M, Evans W (eds) 18th APS-SCCM and 24th AIRAPT, Pts 1-19. doi:10.1088/1742-6596/500/11/112010
Bourne NK (2014) The threshold for hydrodynamic behaviour in solids under extreme compression. J Appl Phys 116(9):093505
Bourne NK, Akrology (2012) Materials’ physics in extremes. In: Elert ML et al (eds) Shock compression of condensed matter. American Institute of Physics, Melville, pp 1331–1334
Bourne NK (2011) Materials’ physics in extremes: akrology. Metall Mater Trans A 42A(10):2975–2984
Gray III, G.T., C.M. Cady, and W.R. Blumenthal, Influence of temperature and strain rate on the constitutive behavior of Teflon and nylon, in Plasticity 99: Constitutive and Damage Modeling of Inelastic Deformation and Phase Transformation, A.S. Khan, Editor. 1998, Neat Press: Fulton, Maryland. p. 955-958
Ward IM, Sweeney J (2012) Relaxation transitions: experimental behaviour and molecular interpretation in mechanical properties of solid polymers. Wiley, New York, pp 261–284
Bourne NK, Millett JCF, Goveas SG (2007) The shock response of polyoxymethylene and polyethylene. J Phys D Appl Phys 40:5714–5722
Mills NJ (2005) Plastics: microstructure and engineering applications, 3rd edn. Butterworth-Heinemann, Amsterdam
Hamdan S, Swallowe GM (1996) The strain-rate and temperature dependence of the mechanical properties of polyetherketone and polyetheretherketone. J Mater Sci 31:1415–1423
Ferry JD (1980) Viscoelastic properties of polymers. Wiley, New York
Millett JCF, Bourne NK, Barnes NR (2002) The behavior of an epoxy resin under one-dimensional shock loading. J Appl Phys 92:6590–6594
Ashby MF, Jones DRH (1996) Engineering materials 1: an introduction to properties, applications and design. Elsevier, Amsterdam
Ashby MF, Jones DRH (2005) Engineering materials 2: an introduction to microstructures, processing and design, 3rd edn. Elsevier, Oxford
Walley SM, Field JE, Safford NA (1991) A comparison of the high strain rate behaviour in compression of polymers at 300 K and 100 K. J Phys IV France 1(C3):185–190
Walley SM, Field JE, Scullion IM, Heukensfeldt Jansen FPM, Bell D (1987) Dynamic strength properties and solid particle erosion behaviour of a range of polymers. In: Field JE, Dear JP (eds) Proceedings of the 7th international conference on erosion by liquid and solid impact, Cavendish Laboratory, Cambridge, paper 58
Walley SM, Xing D, Field JE (1995) Mechanical properties of three transparent polymers in compression at a very high rate of strain. In: Williams JG, Pavan A (eds) Impact and dynamic fracture of polymers and composites, Mechanical Engineering Publications Ltd, London, pp 289–303
Walley SM, Field JE (1994) Strain rate sensitivity of polymers in compression from low to high strain rates. DYMAT J 1:211–228
Marsh SP (1980) LASL shock hugoniot data. University of California Press, Berkeley
Johnson JN, Dick JJ, Hixson RS (1998) Transient impact response of three polymers. J Appl Phys 84:2520–2529
van Thiel M (1966) Compendium of shock wave data. Livermore, Lawrence Radiation Laboratory
Dlott DD (1999) Ultrafast spectroscopy of shock waves in molecular materials. Ann Rev Phys Chem 50:251–278
Kim HJ, Hambir SA, Dlott DD (1999) Ultrafast dynamics of shock waves in polymers and proteins: the energy landscape. Phys Rev Lett 83(24):5034–5037
Wakabayashi K, Nakamura KG, Kondo K, Yoshida M (1999) Time-resolved Raman spectroscopy of polytetrafluoroethylene under laser-driven shock compression. Appl Phys Lett 75(7):947–949
Banishev AA, Shaw WL, Dlott DD (2014) Dynamics of polymer response to nanosecond shock compression. Appl Phys Lett 104(15):101914
Bourne NK, Brown EN, Millett JCF, Gray GT III (2008) Shock, release and Taylor impact of the semicrystalline thermoplastic PTFE. J Appl Phys 103:074902
Bourne NK, Gray GT III (2003) Equation of state of PTFE. J Appl Phys 93:8966–8969
Brown EN, Rae PJ, Trujillo CP, Dattelbaum DM, Gray III GT, Bourne NK (2006) Shock and recovery of PTFE above and below the phase II to phase III transition, In: Furnish MD et al (ed) Shock compression of condensed matter—2005, American Institute of Physics, Melville, pp 196–199
Brown EN, Trujilo CP, Gray GT III, Rae PJ, Bourne NK (2007) Soft recovery of PTFE shocked through the crystalline phase II-III transition. J Appl Phys 101:024916
Resnyansky AD, Bourne NK, Millett JCF, Brown EN (2011) Constitutive modeling of shock response of PTFE. J Appl Phys 110:033530
Weir CE (1954) Temperature dependence of compession of linear high polymers at high pressures. J Res Nat Bur Stand 53:245–252
Beecroft RI, Swenson CA (1959) J Appl Phys 39:1793
Flack HD (1972) High-pressure phase of PTFE. J Polym Sci 10:1799–1810
Eby RK, Clark ES, Farmer BL, Piermarini GJ (1990) Crystal structure of poly (tetrafluoroethylene) homo-and copolymers in the high pressure phase. Polymer 31:2227
Rae PJ, Brown EN, Clements BE, Dattelbaum DM (2005) Pressure-induced phase change in PTFE at modest impact velocities. J Appl Phys 98:063521
Davison L, Graham RA (1979) Shock compression of solids. Phys Rep 55:255–379
Robbins DL, Sheffield SA, Alcon RR (2004) Magnetic particle velocity measurements of shocked Teflon. In: Furnish MD, Gupta YM, Forbes JW (eds) Shock compression of condensed matter—2003, American Institute of Physics, Melville, pp 675–678
Bourne NK (2014) The threshold for hydrodynamic behaviour in solids under extreme compression. J Appl Phys 119:093505
Erskine DJ, Nellis WJ (1991) Shock-induced martensitic phase transformation of graphite to diamond. Nature 349:319–417
Bourne NK, Millett JCF, Gray GT III (2009) On the shock compression of polycrystalline metals. J Mater Sci 44:3319–3343
Bourne NK, Gray GT III, Millett JCF (2009) On the shock response of cubic metals. J Appl Phys 106:091301
Macdonald SA, Bourne NK, Millett JCF, Rosenberg Z (2015) Towards a final analysis of lateral gauge response. In: Oleynik I, Peiris S, Ravelo R (eds) Proceedings of shock compression of condensed matter, American Institute of Physics, Melville, in press
Rosenberg Z, Brar NS (1994) Hysteresis of lateral piezoresistive gauges. In: Schmidt SC et al (eds) High Pressure Science and Technology 1993. American Institute of Physics, New York, pp 1707–1710
Rosenberg, Z (2000) Review on lateral stress measurements with piezoresistive gauges. In: Furnish MD, Chhabildas LC, Hixson RS (eds) Shock compression of condensed matter–1999, American Institute of Physics, Melville, pp 1033–1037
Rosenberg Z, Bourne NK, Millett JCF (2007) On the effect of manganin gauge geometries upon their response to lateral stress. Meas Sci Technol 18:1843–1847
Rosenberg Z, Moshel G (2014) J Appl Phys 115:103511
Yaziv D, Rosenberg Z, Partom Y (1980) Release wave calibration of manganin gauges. J Appl Phys 51:6055–6057
Appleby-Thomas GJ, Hazell PJ, Wilgeroth JM, Wood DC (2010) On the interpretation of lateral manganin gauge stress measurements in polymers. J Appl Phys 108:033524
Bourne NK, Gray GT III, Millett JCF (2003) On the dynamic response of four polymers. J Phys IV France 110:815–820
Bourne NK, Millett JCF, Brown EN, Gray GT III (2007) Effect of halogenation on the shock properties of semicrystalline thermoplastics. J Appl Phys 102:063510
Millett JCF, Bourne NK (2000) The deviatoric response of PMMA to one-dimensional shock loading. J Appl Phys 88:7037–7040
Millett JCF, Bourne NK, Brown EN, Gray III GT (2007) Shear strength and its variation according to structure in shock loaded polyethylene. In: Elert M et al (ed) Shock compression of condensed matter—2007, American Institute of Physics, Melville, pp 719–722
Bourne NK, Gray GT III, Cerretta EK (2016) Dynamic failure; theory and experiment. Springer, New York, in press
Murray NH, Millett JCF, Proud WG, Rosenberg Z (2000) Issues surrounding lateral stress measurements in alumina ceramics. In: Furnish MD, Chhabildas LC, RS Hixson (eds) Shock compression of condensed matter—1999, American Institute of Physics, Melville, pp 581–584
Bourne NK, Millett JCF, Rosenberg Z (1997) On the origin of failure waves in glass. J Appl Phys 81:6670–6674
Rackley FA, Turner HS, Wall WF, Haward RN (1974) Preparation of crosslinked polymers with increased modulus by high-pressure polymerization. J Polym Sci 12(7):1355–1370
Walley SM, Field JE, Pope PH, Safford NA (1989) A study of the rapid deformation behaviour of a range of polymers. Phil Trans R Soc Lond A 328:1–33
Mark H (1942) Intermolecular forces and mechanical behavior of high polymers. Ind Eng Chem 34(11):1343–1348
Walley SM, Chapman DJ, Williamson DM, Morley MJ, Fairhead TW, Proud WG (2009) High rate mechanical properties of Dyneema in compression, in Proc. 9th Int. Conf. on the Mechanical and Physical Behaviour of Materials under Dynamic Loading (DYMAT 2009). Les Ulis, France, EDP Sciences, pp 1133–1138
Walley SM, Field JE, Swallowe GM, Mentha SN (1985) The response of various polymers to uniaxial compressive loading at very high rates of strain. J Phys Fr 46(C5):607–616
Bourne NK (2015) On strength at yield in condensed matter. Metall Mater Trans A 46(10):4491–4497
Fu S, Wang Y, Wang Y (2009) Tension testing of polycarbonate at high strain rates. Polym Test 28:724–729
Siviour CR, Walley SM, Proud WG, Field JE (2005) The high strain rate compressive behaviour of polycarbonate and polyvinylidene fluoride. Polymer 46:12546–12555
Millett JCF, Bourne NK (2006) Shock and release of polycarbonate under one-dimensional strain. J Mater Sci 41:1683–1690
Resseguier TD, Deleignies M (1997) Spallation of polycarbonate under laser-driven shocks. Shock Waves 7:319–324
Frenkel J (1926) Z Phys Chem 37:572
Duffy TS (2005) Synchrotron facilities and the study of the Earth’s deep interior. Rep Prog Phys 68:1811–1859
Holtkamp DB, Clark DA, Ferm EN, Gallegos RA, Hammon D, Hemsing WF, Hogan CE, Holmes VH, King NSP, Liljestrand R, Lopez RP, Merrill FE, Morris CL, Morley KB, Murray MM, Pazuchanics PD, Prestridge KP, Quintana JP, Saunders A, Schafer T, Shinas MA, Stacy HL (2004) A survey of high explosive-induced damage and spall in selected metals using proton radiography. In: Furnish MD, Gupta YM, Forbes JW (eds) Shock compression of condensed matter—2003, American Institute of Physics, Melville, pp 477–482
Mills NJ (1976) The mechanism of brittle fracture in notched impact tests on polycarbonate. J Mater Sci 11:363–375
Young RJ, Lovell PA (1991) Introduction to polymers, 2nd edn. Chapman & Hall, London
Resnyansky AD, Bourne NK, Brown EN, Millett JCF, Rae PJ, McDonald SA, Withers PJ (2015) Phase transition modeling of polytetrafluoroethylene during Taylor impact. J Appl Phys 116:223502
Rau C, Parry S, Garcea SC, Bourne NK, McDonald SA, Eastwood DS, Brown EN, Withers PJ (2015) On compression and damage evolution in PTFE and PEEK. In: Chau TCGR, Sewell TD (eds) Shock compression of condensed matter 2015, AIP, Melville, in press
Clements BE (2012) A continuum glassy polymers model applicable to dynamic loading. J Appl Phys 112:083511
Clements BE, Mas EM (2001) Dynamic mechanical behavior of filled polymers. 1: theoretical developments. J Appl Phys 90:5522–5534
Acknowledgments
The author would like to acknowledge the workers and students without whom the work reviewed in this paper would not have been possible. In particular thanks are due to Drs Rusty Gray, Jeremy Millett, Eric Brown, Gareth Appleby Thomas, Paul Hazell and particularly Zvi Rosenberg who has been involved in the development of lateral stress sensors in low impedance materials from our first experiments. Also to the two reviewers who showed considerable forbearance and made useful suggestions for improvements to the text.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bourne, N.K. On the Shock Response of Polymers to Extreme Loading. J. dynamic behavior mater. 2, 33–42 (2016). https://doi.org/10.1007/s40870-016-0055-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40870-016-0055-5