
Highlights
-
A viable bio-derived material engineering strategy is developed based on the use of skin collagen fibers for the crafting of metal-nitride–carbon nanofiber composites.
-
N-doped carbon nanofiber bundles embedded with Fe2N nanoparticles are fabricated using a structural replication process.
-
The composite with a hierarchically ordered, compact, and multicore–shell heterostructure exhibits increased lithium- and potassium-ion storage performances.
Abstract
Despite the significant progress in the fabrication of advanced electrode materials, complex control strategies and tedious processing are often involved for most targeted materials to tailor their compositions, morphologies, and chemistries. Inspired by the unique geometric structures of natural biomacromolecules together with their high affinities for metal species, we propose the use of skin collagen fibers for the template crafting of a novel multicore–shell Fe2N–carbon framework anode configuration, composed of hierarchical N-doped carbon nanofiber bundles firmly embedded with Fe2N nanoparticles (Fe2N@N-CFBs). In the resultant heterostructure, the Fe2N nanoparticles firmly confined inside the carbon shells are spatially isolated but electronically well connected by the long-range carbon nanofiber framework. This not only provides direct and continuous conductive pathways to facilitate electron/ion transport, but also helps cushion the volume expansion of the encapsulated Fe2N to preserve the electrode microstructure. Considering its unique structural characteristics, Fe2N@N-CFBs as an advanced anode material exhibits remarkable electrochemical performances for lithium- and potassium-ion batteries. Moreover, this bio-derived structural strategy can pave the way for novel low-cost and high-efficiency syntheses of metal-nitride/carbon nanofiber heterostructures for potential applications in energy-related fields and beyond.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Considering their high energy densities and long lifetimes, lithium-ion batteries (LIBs) have been extensively investigated and widely used as power sources for portable electronics and electric vehicles [1, 2]. However, the scarcity and uneven distribution of lithium resources in the earth restrict their applications. In this regard, potential alternatives have attracted considerable interest. Among them, potassium-ion batteries (PIBs) attract increasing attention, owing to the abundant reserves of potassium on the earth and its standard hydrogen potential (− 2.93 V vs. Eº) close to that of lithium (− 3.04 V vs. Eº) [3, 4]. Generally, the main challenge for the PIB anode is the large potassium-ion radius (1.38 Å) [5], leading to sluggish kinetics during electrochemical processes. Although extensive studies have been carried out and significant progress has been made in the development of energy storage materials, the development of advanced anode materials for LIBs and PIBs for large-scale energy storage requires further studies. Transition-metal nitrides (TMNs) have been recently reported as promising conversion anode materials for both LIBs and PIBs, because of their unique physicochemical properties [6] and high theoretical specific capacities [7,8,9,10]. Among the TMNs, the earth-abundant and inexpensive iron nitride (Fe2N) has attracted considerable attention. As a conversion-type energy storage material, Fe2N can reversibly react with Li ions by the formation of metal irons (Fe2N + 3Li+ + 3e− ↔ 2Fe + Li3N) [11, 12]. Owing to its high theoretical density of 7.14 g cm−3, it can transfer 2–3 electrons per formula unit, leading to a theoretical specific capacity as high as 900 mAh g−1 and high volumetric energy density of the battery [13,14,15,16]. Moreover, Fe2N exhibits pseudocapacitance and ultrafast charge transfer from surface/subsurface regions because of its stable phase change. Furthermore, Fe2N and other metal nitrides exhibit high conductivities and good ionic diffusions owing to the vacancies within their crystal structures. Therefore, it is expected that Fe2N as an anode material can be an ideal platform to realize high energy storage capacity and power density. Nonetheless, the practical application of Fe2N is still hindered by the common problems of high polarization and large volume changes during lithiation/potassiation processes, inducing a severe electrode pulverization and fast capacity fading as well as short life span during long-term cycling [17, 18].
Nanostructure engineering has paved the way for the development of high-performance Fe2N anode materials. Compared to bulk materials, zero-dimensional Fe2N nanoparticles (NPs) have more active sites and shorter ionic diffusion paths, while the void space between the particles can help cushion the volumetric changes of the Fe2N NPs to some degree [19, 20]. Nevertheless, the simple change in geometric shape and tuning of the particle size are still unsatisfactory for the improvement in cycling lifetime of Fe2N, as particle agglomeration is unavoidable and the large volume expansion during the cycling may eventually lead to electrode pulverization. Hierarchically nanostructured composites consisting of nanoscale Fe2N and conductive carbon materials could overcome the above drawback to a large extent [12, 21]. Carbon materials such as various forms of amorphous carbon, carbon nanotubes, graphene sheets, and carbon fibers can increase the conductivity, prevent the pulverization of active materials, and reduce the undesirable side reactions between the electrode and electrolyte [17, 22,23,24,25]. Nonetheless, this strategy usually involves time-consuming preparation and tedious structure control processes for the composite electrode materials. Therefore, an alternative low-cost and universal approach to the synthesis of uniform Fe2N NPs encapsulated in carbon frameworks with desirable compositions and morphologies is desirable.
The inspiration by biological materials with complex, optimized, and hierarchical microstructures has been one of the most promising subjects in artificial material engineering. Generally, the bio-inspired material synthesis strategy is based on the self-assembly of specific guest species to obtain inorganic analogues of the biological materials [26,27,28,29]. Natural biological materials used as templates for the material synthesis are low cost, abundant, commercially viable, and environmentally benign [30]. On the other hand, biological materials are morphologically complex but composed of uniform organic/inorganic structural subunits, whose imitation on a small scale could pave the way for the fabrication of novel nanostructured materials. Many biological materials such as biomacromolecules self-assemble into gels or fibers, which can be used to direct the growth of inorganic nanomaterials [31, 32]. In most of these cases, biomacromolecules can provide a matrix, which “traps” metal ions in an aqueous phase. The matrix can be used as a precursor for conversion into nanostructured metal oxides, carbides, or sulfides as well as various carbon-composite heterostructures. For example, carrageenan, extracted from red algae, can strongly bind to multiple metal ions (e.g., Ni2+, Co2+, Cu2+, Fe3+) forming carrageenan–metal hydrogels. The organized double-helix structures in the hydrogels could mediate the growth and formation of ultrasmall metal sulfide NPs (e.g., Co9S8, Ni3S4, CuS, FeS) into hierarchically porous carbon aerogels by pyrolysis [32]. Therefore, the bio-inspired synthesis routes are unique and effective. However, the fabrication of novel TMN/carbon composites has rarely been reported because it often involves the utilization of toxic and expensive organic ligands as well as time-consuming synthesis routes. Therefore, further development of effective TMN/carbon composites is necessary to address the critical issues related to energy storage.
Skin collagen fibers (SCFs), one of the most abundant and renewable biomasses in nature, originate mainly from the skins of domestic animals, which have traditionally been used as raw materials in the leather manufacturing industry [33]. In terms of their geometric structures, the SCFs exhibit a hierarchically interwoven fibrous network morphology. As illustrated in Fig. 1a, the protofibrils are formed from rod-like collagen molecules composed of three polypeptide chains with a triple-helical structure. They are packed together longitudinally in a quarter-staggered alignment with a “gap” region (corresponding to a length of 67 nm) and further organized into larger microfibrils (typical nanofibers with diameters of 50–200 nm). Subsequently, these nanofibrous microfibrils self-assemble into collagen fiber bundles and further into larger SCFs [34]. In their chemical structure, abundant reactive functional groups including –COOH, –OH, and –NH2 exist in the side chains of collagen molecules, which provide the SCFs with high affinities toward multivalent metal ions, such as Ni2+, Co2+, Fe3+, Cr3+, Al3+, and Ti4+ [35]. This property suggests that the SCFs can react directly with transition-metal cations in aqueous solutions without the need for further functionalization. Therefore, the unique structural advantages of the SCFs together with their high affinities to inorganic building blocks make them suitable as an ideal template for the development of hierarchically structured composites for conversion-type reaction anode materials of LIBs and PIBs.

a Schematic of the structure of the native SCFs and synthesis strategy for Fe2N@N-CFBs. FESEM images of b, c natural SCFs and d, e Fe2N@N-CFBs. f–i TEM images of Fe2N@N-CFBs at different magnifications. j, k HRTEM images and l–o HAADF-STEM image and corresponding element maps of Fe2N@N-CFBs
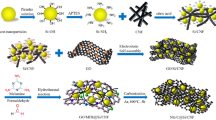
To this end, we report a viable material synthesis strategy for the engineering of a novel one-dimensional (1D) multicore–shell Fe2N–carbon framework heterostructure for superior LIB and PIB anodes, which is composed of hierarchical N-doped carbon nanofiber bundles firmly embedded with Fe2N NPs (denoted as Fe2N@N-CFBs). In our design, natural SCFs extracted from cowhide are used not only as the matrix, which can directly react with Fe3+ ions in an aqueous solution, but also as the biotemplate for structural replication and conversion to Fe2N@N-CFBs through a one-step NH3-driven carbonization/nitridation approach, by which the Fe2N NPs can be formed and uniformly encapsulated into the SCF-derived carbon nanofiber shells with unique multicore–shell heterostructures. This unique architecture design is expected to provide multiple structural advantages to simultaneously address the drawbacks associated with Fe2N during the electrochemical processes. (1) The all-around encapsulation architecture enables a large contact interface between the N-doped carbon nanofiber framework and Fe2N NPs along with continuous electron/ion transport pathways in both radial and axial directions, promoting the electrode reaction kinetics. (2) The 1D long-range N-doped carbon nanofiber framework can provide the elastic buffering function to cushion the volume expansion/contraction of the embedded Fe2N NPs, preserving the integrity of the electrode microstructure. (3) The compact hierarchical structure of the carbon nanofiber bundles is sufficiently robust to ensure the stable ion intercalation/deintercalation of each internal Fe2N@carbon nanofiber contained inside N-CFBs, favoring the cycling stability and high volumetric energy density. The as-developed Fe2N@N-CFB electrode exhibits high reversible capacities of 660 mAh g−1 at 100 mA g−1 for LIBs and 242 mAh g−1 at 50 mA g−1 for PIBs. A remarkable rate performance (202 mAh g−1 at 10.0 A g−1) and ultralarge cycling lifetime (580 and 420 mAh g−1 after 1000 cycles at 1.0 and 2.0 A g−1, respectively) in LIBs have also been achieved.
2 Experimental Section
2.1 Preparation of Materials
Raw materials: White SCFs were prepared using bovine hides according to the previously reported method [36]. The bovine hides were cleaned, unhaired, limed, split, and delimed according to the procedures of leather processing to remove noncollagen components. The skin was then treated with an aqueous solution of acetic acid (16.0 g L−1) three times to remove mineral substances. After the pH of the skin was adjusted to 4.8–5.0 with an acetic acid–sodium acetate buffer solution, the skin was dehydrated by absolute ethyl alcohol, dried in vacuum, ground, and sieved, yielding the SCFs. A chemically pure Fe2(SO4)3 was purchased from the Jinshan Chemical Reagent Factory. Analytical-grade sodium chloride, acetone, and other reagents were purchased from the Chengdu Kelong Chemical Reagent Factory.
Immobilization of Fe3+ on SCFs: 15.0 g of collagen fibers and 6.0 g of NaCl were soaked in 400 mL of deionized water at 25 °C. After continuous stirring for 2 h, the pH value of the suspension was adjusted to 1.7−2.0 using a diluted H2SO4 (1 mol L−1). Subsequently, 22.5 g of Fe2(SO4)3 was added and the suspension was kept constant under stirring for 4 h. The pH value was then slowly increased to ≈ 3.8–4.0 using a saturated sodium bicarbonate solution over more than 2 h. The resultant mixture was reacted at 40 °C for 4 h. Finally, the Fe3+-immobilized SCFs (SCFs@Fe3+) were collected by filtration, followed by washing with deionized water and acetone and drying in air.
Preparation of the Fe2N@N-CFB composite: The as-prepared SCFs@Fe3+ was then treated in a tube furnace with a programmable control of the temperature. The sample was heat-treated stepwise, first at 100 °C in air for 1 h, and subsequently heated to 500 °C (2 °C min−1) in an NH3 atmosphere and held at this temperature for 2 h to generate Fe2N-nanoparticle-impregnated N-doped carbon nanofiber bundles (Fe2N@N-CFBs). For comparison, pure N-doped carbon nanofiber bundles (N-CFBs) were also prepared using HF etching to remove the Fe2N NPs from Fe2N@N-CFBs. Bare Fe2N nanofiber bundles (Fe2N-FBs) were also prepared by a simple two-step calcination method, i.e., SCFs@Fe3+ was precalcined in air for 4 h to remove the organic framework, and then the resultant products were treated in an NH3 atmosphere at 500 °C to yield bare Fe2N-FBs.
2.2 Material Characterization
The crystal structure of the product was analyzed by X-ray diffraction (XRD, Bruker) with Cu Kα radiation (λ = 1.54056 Å). The chemical states and compositions of the samples were characterized by X-ray photoelectron spectroscopy (XPS) using an ESCALAB 250Xi instrument; all binding energy peaks were calibrated by C 1s at 284.5 eV. The peak fittings of all high-resolution core spectra were carried out with the XPSPEAK 4.1 software, using a mixed Gaussian–Lorentzian function. The morphologies and microstructures of the products were observed using field-emission scanning electron microscopy (FESEM, Nova NanoSEM 450), coupled with energy-dispersive X-ray (EDX, Oxford Instrument) spectroscopy, and transmission electron microscopy (TEM, JEOL, JEM-2100F, 200 kV, equipped with an EDX spectrometer). A thermogravimetric analysis (TGA) was carried out using a Netzsch TG209F1 instrument in the range of room temperature to 800 °C at a heating rate of 10 °C min−1 under air atmosphere. Differential scanning calorimetry was performed using a NETZSCH STA 449C instrument in the range of room temperature to 800 °C (10 °C min−1) under Ar atmosphere. Raman spectra were recorded using a JY HR800 Raman spectrophotometer (HORIBA Jobin Yvon, HR800, France) with a 532.17-nm laser radiation. Mössbauer measurements were performed using a conventional spectrometer (Germany, Wissel MS-500) in the transmission geometry and constant acceleration mode. A 57Co(Rh) source with an activity of 25 mCi was used. The velocity calibration was carried out with a room-temperature α-Fe absorber. The spectra were fitted by the Recoil software package using a Lorentzian site analysis.
2.3 Electrochemical Measurements
Electrochemical measurements were carried out using lithium-ion half-cells. The working electrodes were prepared by mixing the active material, Super P, and polyvinylidene fluoride in a mass ratio of 7:2:1 in an N-methyl-2-pyrrolidone solvent, and then the mixed slurry was spread onto a copper foil. The as-prepared electrodes were dried in vacuum at 60 °C for 12 h. The mass loading of active material on each electrode was 1.0 −1.2 mg cm−2. Two-electrode CR2032 coin cells were assembled in an argon-filled glove box to evaluate the electrochemical performances of the samples. In the LIBs, the counter electrode was a disk of lithium foil, while a porous polypropylene film (Celgard 2400) served as the separator. The electrolyte was 1 M of LiPF6 dissolved in a mixture of ethylene carbonate (EC), ethyl methyl carbonate, and dimethyl carbonate (DEC) in a volume ratio of 1:1:1. In the case of the PIBs, CR 2016 coin-type cells were assembled. The electrolyte consisted of a solution of 0.8 M of KPF6 in EC/DEC (volume ratio of 1:1). The counter and reference electrodes were potassium metal, and the separator was a glass fiber. All coin cells were aged at room temperature for 12 h before the test to ensure that the electrode was completely soaked in the electrolyte. Galvanostatic charge/discharge tests were carried out using a Neware battery test system (Neware BTS, Neware, Shenzhen, China) in a voltage window of 0.01−3.0 V. Cyclic voltammetry (CV) measurements were performed at a scan rate of 0.1 mV s−1 in the range of 0.01−3.0 V using a PARSTAT multichannel electrochemical working station (Princeton Applied Research, USA). Electrochemical impedance spectroscopy (EIS) was carried out using a PARATAT electrochemical workstation with an alternating-current (AC) voltage amplitude of 5 mV in the frequency range of 100 kHz to 0.1 Hz.
Calculation of volumetric capacity: The mass density of the electrode was calculated by ρ = M (mg)/V (cm3), where M and V are the mass and volume of the electrode, respectively. The thickness of the electrode was estimated by FESEM. The volumetric capacity (CV) was calculated by CV = Cg × ρ.
3 Results and Discussion
3.1 Fabrication and Structural Characterization
The overall synthesis strategy for the hierarchically ordered multicore–shell Fe2N@N-CFBs is illustrated in Fig. 1a. First, the Fe3+ precursors can be directly adsorbed and immobilized on the SCFs in the aqueous solution, in which the Fe3+ were primarily reacted and coordinated with the –COOH groups in the collagen triple helix through the formation of coordination complexes [37], as indicated by the color change of the SCFs from white to yellow. After the thermal treatment of the resultant SCFs@Fe3+ intermediates in the NH3 atmosphere at 500 °C, the organic SCF framework can be completely decomposed, leaving the N-doped carbon nanofiber bundles, while the coordinated Fe3+ ions were able to react with the active nitrogen released from NH3 through in situ conversion into Fe2N NPs embedded in the SCF-derived carbon nanofiber during the high-temperature nitridation. A unique multicore–shell Fe2N@N-CFB sample was finally obtained. For comparison, two control samples were prepared. One of them consisted of pure N-doped carbon nanofiber bundles (denoted as N-CFBs) obtained by etching out all Fe2N NPs from the Fe2N@N-CFBs, while the other sample was bare Fe2N nanofiber bundles (denoted as Fe2N-FBs) prepared by thermal removal of the SCF organic framework. The corresponding preparation is presented in detail in Experimental Section.
The morphologies and microstructures of the samples were analyzed using FESEM. As shown in Fig. 1b, c, the basic building blocks of the native SCFs are the collagen nanofibers with a distinctive D-period structure (length: ≈ 65 nm) and average diameter of 50–200 nm (Fig. 1c), which self-assemble into a collagen microfibril and even into a larger collagen fiber bundle (5–10 μm, Fig. 1b), confirming the intrinsic hierarchically fibrous structure of the SCFs from the nanoscale to the microscale. Similar to the native SCFs (Fig. 1b, c), it should be noted that the SCFs@Fe3+ intermediates consisting of closely packed nanofibers also exhibit a distinctly fibrous morphology (Fig. S1), suggesting that the Fe3+-complexing process did not destroy the bio-inherent hierarchical structure of the SCFs. The FESEM image of Fe2N@N-CFBs shows that a single fiber bundle with an average diameter of 3–5 µm is composed of densely packed nanofibers (Fig. 1d). The nanofibers with smaller diameters of 50–100 nm are aligned together in a definite and ordered arrangement. Fe2N@N-CFBs still maintains all basic building blocks of the nanofiber morphology of the SCF template. Moreover, the magnified FESEM image in Fig. 1e shows a large number of small Fe2N NPs on the surface of each nanofiber. To further demonstrate the existence of Fe2N NPs, Fe2N@N-CFBs was further subjected to etching by hydrofluoric acid. As shown in Fig. S2, after the treatment by acidic etching, the resultant N-CFBs still consisted of numerous interwoven nanofibers, each of which had a relatively smooth surface, along with a large number of visible pores (marked by yellow circles in Fig. S2b). This implies that the Fe2N NPs can be removed from Fe2N@N-CFBs. They can serve as in situ pore-forming agents to prepare porous carbon nanofiber bundles. It is worth mentioning that the native SCFs without Fe3+-coordination cannot retain their natural fibrous structure if they are carbonized directly under the NH3 atmosphere at 500 °C, as shown in Fig. S3. According to the principles of the traditional mineral tanning chemistry, the tanning interaction of animal hides and skins with metal salts could significantly increase the degree of cross-linking between the collagen macromolecules, thereby improving the thermal shrinkage temperature of collagen (Fig. S4) and leading to their transformation into leather. Therefore, our results also confirm that the precoordination to Fe3+ is indispensable to maintain and stabilize the fibrous structure of the native SCFs to ensure their conversion to ordered CFBs during the high-temperature carbonization process [38].
To further demonstrate the applicability of the SCFs as a template, the Fe3+-immobilized SCFs were directly calcined in air to convert them into Fe2O3 intermediates (Fig. S5) prior to the nitridation treatment. Owing to the template replication action of the SCFs, the resultant Fe2N-FBs composed of numerous Fe2N particles also exhibited a unique fibrous morphology similar to that of the pristine SCFs, as shown in Fig. S6. In contrast to Fe2N@N-CFBs, it should be noted that the bare Fe2N-FBs had a denser and more compact structure and thicker fiber bundles. This indicates that despite the effective replication of the fibrous structure, the absence of the SCF-derived carbon framework led to the conspicuous agglomeration of Fe2N particles in the bare Fe2N-FBs. Figure S7 shows SEM–EDX spectroscopy maps, which verify the uniform distributions of Fe, N, C, and small amount of O element in Fe2N@N-CFBs.
The microstructure of Fe2N@N-CFBs was further analyzed by TEM. Typical TEM images of an individual nanofiber are presented in Fig. 1f, g. The diameter of the individual nanofiber is approximately 100 nm. Numerous well-crystallized Fe2N NPs (black regions) with an average size of ≈ 10–20 nm are uniformly dispersed over and confined within the nanofiber, suggesting the unique multicore–shell nanostructure of Fe2N@N-CFBs. Some Fe2N NPs are dispersed over the outer surface of the nanofiber, as shown in Fig. 1h (marked by white arrows). To analyze the surroundings of these Fe2N NPs, high-resolution TEM (HRTEM) observations were performed. Figure 1i shows a uniform and thin carbon layer with a thickness of ≈ 5 nm covering the Fe2N NP clinging to the nanofiber matrix, which provides a charge transfer interphase enabling a good contact between the carbon nanofiber and Fe2N NPs. Moreover, as shown in Fig. 1j, the Fe2N NPs confined in the thin carbon shells are geometrically isolated from each other by the SCF-derived carbon nanofiber framework, which can also act as an ideal elastic matrix to accommodate the volume expansion of the enclosed Fe2N NPs. In addition, the HRTEM image (Fig. 1k) shows clear lattice fringes with an interplanar spacing of 0.221 nm, well corresponding to the (002) lattice spacing of Fe2N. To further demonstrate the uniform dispersion of the Fe2N NPs, a dark-field image (Fig. 1l) was acquired using high-angle annular dark-field scanning TEM (HAADF-STEM). The EDX spectroscopy elemental mapping corresponding to the HAADF-STEM image (Fig. 1m–o) shows uniform distributions of the Fe, C, and N elements in Fe2N@N-CFBs. Furthermore, Figs. S8 and S9 show TEM and HRTEM images of the two control samples, N-CFBs and Fe2N-FBs, respectively. Notably, a remarkably porous texture and oval pore tunnels can be observed in N-CFBs by the apparent contrast (marked with white arrows, Fig. S8c), originating from the in situ removal of the Fe2N NPs from the carbon nanofiber by the HF etching. This is consistent with the FESEM observations (Fig. S2), further verifying that the Fe2N NPs are embedded in the carbon nanofiber with a multicore–shell nanostructure. The TEM images (Fig. S9) of the bare Fe2N-FBs reveal that a single Fe2N fiber is composed mainly of aggregated Fe2N crystals exhibiting clear lattice fringes separated by 0.21 nm corresponding to the (011) lattice planes of Fe2N.
The crystalline structures of the as-prepared Fe2N@N-CFBs and control samples were investigated by powder XRD. As presented in Fig. 2a, three major characteristic peaks at 40.9°, 42.9°, and 56.7° were observed for Fe2N-FBs and Fe2N@N-CFBs. These peaks can be attributed to the (002), (111), and (112) planes of the ideal Fe2N (JCPDS No. 72-2126), respectively, demonstrating the existence of Fe2N in Fe2N@N-CFBs. Moreover, the diffraction peaks of Fe2N@N-CFBs are smaller than those of the bare Fe2N-FBs, indicating the smaller sizes of the Fe2N NPs in Fe2N@N-CFBs. Additionally, compared with the bare Fe2N-FBs, the diffraction hump between 20° and 30° in the XRD patterns of Fe2N@N-CFBs and pure N-CFBs is attributed to the carbon nanofiber framework. These results indicate the formation of well-crystallized Fe2N NPs as well as N-CFBs. Figure 2b presents the Raman spectra of the pure N-CFBs, bare Fe2N-FBs, and Fe2N@N-CFBs. For the Fe2N@N-CFBs composite, the peaks below 1000 cm−1 match well with those of the bare Fe2N-FBs. For the pure N-CFBs and Fe2N@N-CFBs, the two characteristic broad peaks at approximately 1355 and 1577 cm−1 are related to the sp3-type disordered carbon (D band) and sp2-type ordered graphitic carbon (G band), respectively [39]. According to the calculation, the intensity ratio of the D band to the G band (ID/IG) is 1.12, which reveals that the carbon nanofibers have more defects and more disordered structure after the N-doping. To further identify the exact phase of Fe2N, we employed Mössbauer spectrometry to study the different types of Fe environments in Fe2N@N-CFBs (Fig. 2c). At room temperature, the spin relaxation time of superparamagnetic NPs is on the order of 10−11 to 10−12 s, considerably smaller than the nuclear sensing time (10−8 s), and the sextet collapses into a singlet or doublet [40]. The spectrum of Fe2N@N-CFBs exhibits an asymmetric doublet, indicating its paramagnetic nature. The spectral areas of the two doublets are related to the formation probabilities of the two iron sites. The formation of the Fe-III site is associated with additional nitrogen neighbors of iron, which enables an increase in the isomer shift. The formation of the Fe-II site implies a slight deviation from the ideal Fe2N stoichiometry, i.e., an orthorhombic ζ-Fe2N1−z phase (inset in Fig. 2c). Therefore, the pure paramagnetic spectrum of ζ-Fe2N1−z has to be further fitted with two subspectra for the Fe-III and Fe-II sites, provided that z ≠ 0 [41, 42]. For Fe2N@N-CFBs, the peak area ratio of the Fe-III site to the Fe-II site is 51.4:48.6, corresponding to z of 0.16 and formula of Fe2N0.84. This shows that Mössbauer spectrometry is an accurate approach to determine the phase compositions of Fe2N materials. The weight fraction of Fe2N in Fe2N@N-CFBs was also estimated by a TGA, as shown in Fig. S10. Considering that the Fe:N ratio is 2:0.84, the Fe2N content in Fe2N@N-CFBs is estimated to be 41.5%.

a XRD patterns and b Raman spectra of Fe2N@N-CFBs, N-CFBs, and Fe2N-FBs. c Mössbauer spectrum of Fe2N@N-CFBs at room temperature. The inset shows the crystal structure of the orthorhombic Fe2N. d–f HR XP spectra of the Fe 2p, N 1s, and C 1s peaks of Fe2N@N-CFBs. g Photographs of SCFs, SCFs@Mn+, and MxNy@N-CFBs (M = Co, Ti, Cr)
The surface element composition and chemical state of Fe2N@N-CFBs were further investigated by XPS. The survey scan XP spectrum (Fig. S11) shows the typical Fe, N, C, and O signals, without signals of impurities. The high-resolution XP spectrum of the Fe 2p orbitals (Fig. 2d) exhibits broad peaks around 711 and 725 eV corresponding to the Fe 2p3/2 and Fe 2p1/2 orbitals, respectively. The peaks at 710.3 and 723.6 eV are related to Fe2+, those at 711.9 and 725.5 eV are attributed to Fe3+, and those at 719.2 and 734.1 eV are attributed to satellite peaks (Sat.) [20, 21]. These results indicate that the valence state of Fe in Fe2N@N-CFBs is a mixture of +2 and +3. The high-resolution N 1s XPS peak was further deconvoluted (Fig. 2e), and three types of nitrogen were identified, pyridinic at 398.4 eV, pyrrolic at approximately 400.1 eV, and graphitic at 401.1 eV [43, 44]. Majority (52%) of the nitrogen in Fe2N@N-CFBs belongs to the pyridinic type, which could facilitate the Li-ion intercalation into the carbon [45]. The peak around 397.1 eV is associated with the characteristic peak of metal nitride [46]. The high-resolution XP spectrum of C 1s (Fig. 2f) is further resolved into four peaks at 284.2, 284.6, 285.4, and 288.5 eV corresponding to C=C, C–OH, C–N, and C=O bonds, respectively [47]. The residual oxygen-containing functional groups originating from the carbonization of the organic SCFs are helpful to increase the surface wettability of the electrode.
The chemical structure of the collagen macromolecule enables its reactions with various metal salts, such as Co2+, Fe3+, Cr3+, Al3+, and Ti4+, which are also the fundamental chemical processes for the traditional leather production technology. This inspires us to propose a universal and generalizable synthesis strategy based on the use of the native SCFs as the biotemplate for the engineering of other metal-nitride/carbon nanofiber bundle composites (MxNy@N-CFBs, where M is a metal). As shown in Fig. 2g, when the Fe3+ precursors were replaced by Co2+, Ti4+, and Cr3+ precursors, the corresponding SCFs@Mn+ intermediates can be obtained. As expected, we also successfully prepared various MxNy@N-CFBs products including Co4N@N-CFBs (JCPDS No. 41-0943), TiN@N-CFBs (JCPDS No. 87-0630), and CrN@N-CFBs (JCPDS No. 77-0047), which can be verified by the corresponding XRD patterns (Fig. 3g–i). Their typical FESEM images shown in Fig. 3a–f demonstrate the similar morphological characteristics to those of Fe2N@N-CFBs. Similar 1D hierarchical nanofiber bundle structures can be well obtained using an identical method, while metal particles are uniformly dispersed on each of the carbon nanofibers. Therefore, the proposed SCF-based synthesis approach is versatile and may lead to more convenient and competitive routes for a generalizable fabrication of various metal-nitride-based hierarchically heterostructured electrode materials toward potential energy-related applications.

FESEM images of a, d Co4N@N-CFBs, b, e TiN@N-CFBs, and c, f CrN@N-CFBs prepared using SCFs as templates and g–i corresponding XRD patterns
3.2 Electrochemical Performances in Lithium-Ion Storage
The lithium storage behavior of the as-obtained Fe2N@N-CFBs was initially investigated by CV at a scan rate of 0.1 mV s−1 in the voltage range of 0.01–3.0 V (vs. Li/Li+). As shown in Fig. 4a, the small reduction peak at approximately 1.25 V in the first cathodic scan, which disappears in the subsequent cycles, corresponds to the formation of the solid electrolyte interface (SEI) by the irreversible reaction with the electrolyte [12, 21], while the large reduction peak at approximately 0.7 V can be attributed to the lithiation reactions of Fe2N (Fe2N + 3Li+ + 3e− → 2Fe + Li3N). During the following anodic scan, a broad oxidation peak is observed around 1.0 V, which could be attributed to the oxidation of Fe to Fe2N species (2Fe + Li3N → Fe2N + 3Li+ + 3e−). Notably, an obvious difference between the first and following cycles is observed, i.e., the cathodic peak is smaller after the first cycle, which is attributed to the polarization of the electrode in the initial cycle. Nevertheless, both cathodic and anodic peaks in the CV curves overlap with small changes in the subsequent cycles, indicating the excellent reversibility of the electrode. A similar behavior is observed in the CV curves of the bare Fe2N-FBs and N-CFBs electrodes (Fig. S12). Figure 4b shows typical galvanostatic discharge/charge curves for the initial three cycles of the Fe2N@N-CFBs electrode at a current density of 0.1 A g−1; the specific capacities were calculated based on the total mass of Fe2N@N-CFBs. In the first cycle, Fe2N@N-CFBs delivers high initial discharge and charge capacities of 1062 and 781 mAh g−1, respectively, suggesting an initial Coulombic efficiency (ICE) of 73.5%. The irreversible capacity loss can be attributed mainly to the formation of the SEI layer and irreversible insertion of Li+ into the defects in the carbon fiber [30]. Subsequently, the electrode delivers discharge capacities of 712 and 698 mAh g−1 in the second and third cycles, while the corresponding CE quickly increases to 97.4% and 98.6%, respectively, indicating a relatively stable charge/discharge process with a good electrochemical reversibility. Notably, the ICE of the Fe2N@N-CFBs electrode is considerably higher than those of the Fe2N-FBs (68.7%) and N-CFBs (46.3%) electrodes (Fig. S13). This demonstrates that the unique 1D hierarchical structure of N-CFBs and thin carbon layer can serve as ideal conductive substrate and confinement of the active Fe2N NPs to enable the stable and uniform SEI formation. The increased ICE of the Fe2N@N-CFBs electrode may also be partially associated with the proper surface area and pore size distribution compared with those of the other two electrodes, as shown in Fig. S14. In addition, based on the carbon content (58.5%) as well as the specific capacities of the Fe2N@N-CFBs composite (698 mAh g−1) and pure N-CFBs (452 mAh g−1) at the third cycle (Fig. S13b), the reversible Li+ storage capacity originating from the Fe2N is 434 mAh at 0.1 A g−1 with the deduction in capacity contribution of N-CFBs. The corresponding utilization efficiency of active Fe2N materials in the Fe2N@N-CFBs electrode is then calculated to be 48%, provided that the theoretical specific capacity of Fe2N is 900 mAh g−1. This value is significantly higher than that obtained for the Fe2N-FBs electrode (18%), confirming the crucial role of the N-CFBs substrate acting as the 1D hierarchical dispersant and excellent electric conductor for the active Fe2N NPs.

Lithium-ion storage properties of Fe2N@N-CFBs. a CV curves and b discharge–charge voltage profiles of Fe2N@N-CFBs for the first three cycles. c Cycling performances and d rate capacities of Fe2N@N-CFBs, N-CFBs, and Fe2N-FBs. e Repeated rate and cycling tests and f long-term cycling performance of Fe2N@N-CFBs
Figure 4c shows the cycling performances of the Fe2N@N-CFBs electrode at a current density of 0.2 A g−1. Surprisingly, the Fe2N@N-CFBs electrode maintains a high reversible capacity of 710 mA h g−1 after 200 cycles without decay. In contrast, the bare Fe2N-FBs and pure N-CFBs electrodes exhibit relatively low capacities of 220 and 452 mAh g−1, respectively. Further EIS tests indicate that the Fe2N@N-CFBs electrode not only has the smallest interfacial resistance (Rct, Fig. S15), but also exhibits the highest Li+ diffusion coefficient during 200 cycles (Fig. S16). Furthermore, we observed a gradual decrease in Rct for both Fe2N@N-CFBs and Fe2N-FBs electrodes. In particular, a large decrease from ≈ 90 Ω to only 25 Ω after 200 cycles is observed for the Fe2N@N-CFBs electrode. The continuous decrease in charge transfer resistance and capacity increase during the cycling may be attributed to the gradual activation and electrochemical milling processes, where the inner Fe2N NPs and N-CFBs are gradually wetted by the electrolyte and thus provide sufficient active sites for the surface electrochemical reaction as well as faster electrode/electrolyte interface kinetics because of the efficient contact with the electrolyte. In addition to its remarkable cycling performance, the rate capabilities of Fe2N@N-CFBs were evaluated at various current densities. As shown in Fig. 4d, Fe2N@N-CFBs delivered high specific capacities of 640, 586, 524, 451, 362, 246, and 202 mAh g−1 at 0.1, 0.2, 0.5, 1.0, 2.0, 5.0, and 10.0 A g−1, respectively, considerably higher than those of the bare Fe2N-FBs and pure N-CFBs, revealing the excellent kinetics of Fe2N@N-CFBs. The outstanding rate capability and cycling stability of the Fe2N@N-CFBs electrode are further demonstrated in Fig. 4e. The Fe2N@N-CFBs electrode is also able to accommodate the continuously switched rate and cycling tests at repeated current densities. The quantitative kinetic analysis shows that the excellent rate performance originates mainly from the surface-controlled capacitive charge storage contribution by the 1D hierarchical fibrous structure of Fe2N@N-CFBs (Fig. S17). Figure 4f shows the prolonged cycling stabilities of Fe2N@N-CFBs at high current densities of 1.0 and 2.0 A g−1. Fe2N@N-CFBs delivers discharge capacities of 550 and 420 mAh g−1 after 1000 cycles at 1.0 and 2.0 A g−1, respectively. The EI tests indicate that the Fe2N@N-CFBs electrode exhibits a small impedance evolution during the repeated cycles (Fig. S18). These results show the structural durability of the robust multicore–shell Fe2N@N-CFBs.
To understand the electrochemical lithium storage mechanism of Fe2N@N-CFBs, ex situ TEM and HRTEM along with selected-area electron diffraction (SAED) were employed to analyze the phase evolution. Figure 5 shows TEM, HRTEM, and SAED images of the Fe2N@N-CFBs electrode recorded in the fresh, lithiated, and delithiated states in the voltage range of 0.01−3.0 V (Fig. 5a). The TEM image (Fig. 5b1) shows the multicore–shell structure of Fe2N@N-CFBs, while the HRTEM image (Fig. 5b2) and corresponding SAED pattern (Fig. 5b3) reveal the crystalline structure of the inner Fe2N NPs in the pristine Fe2N@N-CFBs. Upon discharge to 0.01 V, the lithiation of Fe2N@N-CFBs led to the pulverization of most of the Fe2N NPs and even disintegration into smaller particles, while the carbon nanofiber matrix inhibited their aggregation to some extent (Fig. 5c1). In this state, the (110) planes of Fe and (101) facet of Li3N can be observed in the HRTEM image (Fig. 5c2), while the diffraction rings in the SAED pattern (Fig. 5c3) confirm the presence of polycrystalline metallic iron (JCPDS No. 89-4185) and β-Li3N (JCPDS No. 78-2005), which indicates the conversion process for Fe2N NPs. Subsequently, upon the delithiation of the electrode in the range of 0.01–3.0 V, the conversion products change back to the Fe2N phase (Fig. 5d1–3), suggesting that the phase conversion reaction of Fe2N@N-CFBs is highly reversible. Ex situ XRD (Fig. S19) performed after discharging and charging further demonstrates the formation of crystalline Li3N and recovery of Fe2N. Based on the above results, the electrochemical lithium storage mechanism of Fe2N@N-CFBs may be explained by Fe2N + 3Li+ + 3e− ↔ 2Fe + Li3N.

a Initial discharge/charge curves of Fe2N@N-CFBs at 50 mA g−1 in the voltage range of 0.01 −3.0 V. b–d TEM, HRTEM, and SAED images of the Fe2N@N-CFBs electrode recorded in the fresh b1–3, discharged c1–3, and charged d1–3 states
3.3 Electrochemical Properties in Potassium-Ion Storage
The potassium-ion storage properties of Fe2N@N-CFBs were also evaluated. Figure 6a shows the first three CV curves at a scan rate of 0.1 mV s−1 in the voltage range of 0.01–3.0 V (vs. K/K+). Only two typical peaks at 0.5 and 1.0 V are observed in the cathodic and anodic scans in the first cycle, which are associated with the potassiation and depotassiation of Fe2N, respectively. Similar results are observed in the CV tests of the bare Fe2N-FBs electrode (Fig. S20). The charge–discharge curves of Fe2N@N-CFBs at 25 mA g−1 are shown in Fig. 6b. The initial discharge and charge capacities are 710 and 310 mAh g−1, respectively, indicating an ICE of 43.7%. The low CE can be attributed to the formation of the SEI layer and irreversible insertion of potassium in the carbon defect sites of Fe2N@N-CFBs [48, 49]. In contrast, the Fe2N-FBs electrode exhibits considerably lower discharge and charge capacities of 379 and 92 mAh g−1, respectively, with a corresponding considerably lower ICE of 24.3% (Fig. S21). Similar to the LIB voltage profiles, the initial discharge/charge curves of the PIBs are also sloping and no obvious plateaus are observed, which can be attributed to the insufficient activation of the potassiation/depotassiation processes [50, 51]. Additionally, the reversible capacity of Fe2N@N-CFBs for potassium storage is considerably lower than that for lithium storage. The large radius of K+ compared to that of Li+ leads to a larger volume expansion and worse reaction kinetics during the discharge/charge process. The rate capacity of the Fe2N@N-CFBs electrode is shown in Fig. 6c. At the current densities of 25, 50, 100, 200, and 500 mA g−1, the reversible capacities of the composite are approximately 340, 242, 174, 110, and 85 mAh g−1, respectively. When the current density is returned to 25 mA g−1, the reversible capacity of 245 mAh g−1 can be retained and reaches approximately 125 mAh g−1 at 200 mA g−1 during the following cycles, demonstrating the highly reversible potassium-ion storage properties of Fe2N@N-CFBs. The cycling performance of the Fe2N@N-CFBs composite at 200 mA g−1 is shown in Fig. 6d. Although a small decrease in capacity is observed, the Fe2N@N-CFBs electrode maintains a reversible capacity of 102 mAh g−1 after 100 cycles. The CE is still close to 100% after the first few cycles. To the best of our knowledge, this is the first demonstration of iron nitride/carbon composites as anode materials for applications in PIBs. To confirm the structural merits of the multicore–shell Fe2N@N-CFBs, the cycling behavior of the Fe2N-FBs electrode was also evaluated for comparison under the same conditions (Fig. 6d). However, the bare Fe2N-FBs delivered a low reversible capacity of only 64 mAh g−1 in the first cycle and exhibited a gradual capacity fading in the subsequent cycles, leading to a negligible capacity as low as ≈ 22 mAh g−1 at the 100th cycle. The relatively low reversible capacity of Fe2N-FBs is attributed mainly to the absence of elastic carbon framework protection during the continuous electrochemical cycling.

Potassium-ion storage properties of Fe2N@N-CFBs. a CV curves and b discharge–charge profiles at 25 mA g−1 for Fe2N@N-CFBs in the first three cycles. c Rate capabilities of the Fe2N@N-CFBs and Fe2N-FBs electrodes at varied current density. The presented capacities are discharge capacities. d Cycling performances of the Fe2N@N-CFBs and Fe2N-FBs electrodes for 100 cycles at 200 mA g−1
The cycling and rate performances of the 1D multicore–shell Fe2N@N-CFBs in LIBs are comparable to those of most reported TMN-based anode materials (Table S1). To better understand the excellent cycling behaviors, the electrode materials for LIBs after cycling (in the full-delithiation state) were further investigated by FESEM and TEM. As shown in Fig. S22a, b, the hierarchically fibrous structure of Fe2N@N-CFBs is well preserved, with a uniform and thin layer of SEI on the surface of the carbon fiber. The Fe2N NPs are homogeneously dispersed but still encapsulated in the carbon nanofiber framework without shedding or aggregation (Fig. S22c, d), suggesting that the carbon fiber framework in Fe2N@N-CFBs is sufficiently robust to accommodate the volume changes during the repeated charge–discharge process. Compared with the original Fe2N@N-CFBs (Fig. 1g, f), the particle size of Fe2N after the cycling is decreased with more expected nanograins, confirming the activation and electrochemical milling process during the lithiation/delithiation operation, well explaining the capacity increase upon the cycling. In contrast, the Fe2N-FBs electrode, as shown in Fig. S22e, f, not only suffers from a serious structural fracture owing to the internal strain caused by the considerable volume variation of the Fe2N particles, but also exhibits an extremely coarse and uneven surface, which could be associated with the thick and incomplete SEI layer. The electrode thicknesses before and after the cycling were also analyzed by FESEM (Fig. S23a–d). The Fe2N@N-CFBs electrode exhibits a considerably smaller increase in electrode average thickness (from 7.6 to 8.0 μm, ≈ 5.2%) than that of the bare Fe2N-FBs electrode (from 4.7 to 7.1 μm, ≈ 51%) after the 50th cycle discharging at 200 mA g−1 (lithiated state). We further used the volumetric capacity based on the lithiation-state electrode volume (highest thickness/volume during the cycling) to represent the electrochemical performance (see Experimental Section for the details of this calculation). The Fe2N@N-CFBs electrode can deliver a reversible volumetric capacity of 854 mAh cm−3 at the current density of 262 mA cm−3 after the 50th cycle (Fig. S23f), considerably higher than that of the bare Fe2N-FBs electrode (only 223 mAh cm−3).
The remarkable electrochemical performance of Fe2N@N-CFBs can be attributed to the advantages of the 1D multicore–shell structure, as illustrated in Fig. 7. First, the all-around encapsulation architecture enables a large contact interface between the N-doped carbon nanofiber framework and Fe2N NPs along with continuous electron/ion transport pathways in both radial and axial directions, promoting the electrode reaction kinetics. In addition, the growth of the stable SEI layer on the surface of the carbon fiber prevents the continual rupturing and reformation of the SEI film, which commonly occur in all-alloy-based anode materials. Second, the multicore–shell architecture together with the long-range 1D carbon nanofiber framework can provide the elastic buffering function to accommodate the volume expansion/contraction of the impregnated Fe2N NPs. Third, the compact carbon nanofiber bundles are sufficiently robust to ensure the stable lithiation/potassiation of each internal Fe2N@carbon nanofiber contained inside N-CFBs, favoring the achievement of an excellent cycling stability during long-period cycles.

Schematic of the different structural evolutions of the Fe2N@N-CFBs (top panel) and Fe2N-FBs (bottom panel) electrode materials during the electrochemical lithium/potassium storage reaction. The light-yellow layer coating on the surface represents the SEI film. (Color figure online)
4 Conclusion
We developed a viable biomass-based structural engineering strategy based on the use of SCFs for the template crafting of hierarchical multicore–shell Fe2N@N-CFBs as advanced anode materials for both LIBs and PIBs. These intriguing NP-encapsulated 1D conductive nanofiber bundle heterostructures not only retained the intrinsic advantages upon the Fe2N nanosizing, but also offered other benefits, such as the effective restraining of the volume variation of Fe2N and enhanced Fe2N conversion kinetics. Consequently, the Fe2N@N-CFBs composite achieved increased lithium-/potassium-ion storage performances. Furthermore, as the metal ions immobilized on the SCFs were optionally tunable, this facile approach utilizing the chemical reactivity and structural replicability of collagen fibers can be easily extended to the design and crafting of other advanced metal-nitride-based materials toward wider energy storage and conversion applications.
References
J.M. Tarascon, M. Armand, Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001). https://doi.org/10.1038/35104644
B. Dunn, H. Kamath, J.M. Tarascon, Electrical energy storage for the grid: a battery of choices. Science 334, 928–935 (2011). https://doi.org/10.1126/science.1212741
F. Yang, H. Gao, J. Hao, S. Zhang, P. Li et al., Yolk-shell structured FeP@C nanoboxes as advanced anode materials for rechargeable lithium-/potassium-ion batteries. Adv. Funct. Mater. 29(16), 1808291 (2019). https://doi.org/10.1002/adfm.201808291
H. Wu, Q. Yu, C.-Y. Lao, M. Qin, W. Wang et al., Scalable synthesis of VN quantum dots encapsulated in ultralarge pillared N-doped mesoporous carbon microsheets for superior potassium storage. Energy Storage Mater. 18, 43–50 (2018). https://doi.org/10.1016/j.ensm.2018.09.025
Y. Marcus, Thermodynamic functions of transfer of single ions from water to nonaqueous and mixed solvents part 3—standard potentials of selected electrodes. Pure Appl. Chem. 57(8), 1129–1132 (1985). https://doi.org/10.1351/pac198557081129
L. Lai, J. Zhu, B. Li, Y. Zhen, Z. Shen et al., One novel and universal method to prepare transition metal nitrides doped graphene anodes for Li-ion battery. Electrochim. Acta 134, 28–34 (2014). https://doi.org/10.1016/j.electacta.2014.04.073
B. Das, M.V. Reddy, G.V. Subba Rao, B.V.R. Chowdari, Synthesis of porous-CoN nanoparticles and their application as a high capacity anode for lithium-ion batteries. J. Mater. Chem. 22, 17505–17510 (2012). https://doi.org/10.1039/c2jm31969a
X. Li, A.L. Hector, J.R. Owen, S.I.U. Shah, Evaluation of nanocrystalline Sn3N4 derived from ammonolysis of Sn(NEt2)4 as a negative electrode material for Li-ion and Na-ion batteries. J. Mater. Chem. A 4(14), 5081–5087 (2016). https://doi.org/10.1039/C5TA08287K
I. Memona, A.S. Mustansar, A.U. Rehman, A. Nisar, M.M. Waheed et al., Mechanistic insights into high lithium storage performance of mesoporous chromium nitride anchored on nitrogen-doped carbon nanotubes. Chem. Eng. J. 327, 361–370 (2017). https://doi.org/10.1016/j.cej.2017.06.095
Q. Sun, Z.W. Fu, Mn3N2 as a novel negative electrode material for rechargeable lithium batteries. Appl. Surf. Sci. 258(7), 3197–3201 (2012). https://doi.org/10.1016/j.apsusc.2011.11.063
J. Guo, Y. Yang, W. Yu, X. Dong, J. Wang et al., Synthesis of α-Fe2O3, Fe3O4 and Fe2N magnetic hollow nanofibers as anode materials for Li-ion batteries. RSC Adv. 6(112), 111447–111456 (2016). https://doi.org/10.1039/C6RA23601D
P. Yu, L. Wang, F. Sun, D. Zhao, C. Tian et al., Three-dimensional Fe2N@C microspheres grown on reduced graphite oxide for lithium-ion batteries and the Li storage mechanism. Chemistry 21(8), 3249–3256 (2015). https://doi.org/10.1002/chem.201406188
A. Sakuma, Self-consistent calculations for the electronic structures of iron nitrides, Fe3N, Fe4N and Fe16N2. J. Magn. Magn. Mater. 102(1–2), 127–134 (2000). https://doi.org/10.1016/0304-8853(91)90277-H
Y. Zhong, X.H. Xia, F. Shi, J.Y. Zhan, J.P. Tu et al., Transition metal carbides and nitrides in energy storage and conversion. Adv. Sci. 3(5), 1500286–1500313 (2016). https://doi.org/10.1002/advs.201500286
W. Li, G. Wu, C.M. Araújo, R.H. Scheicher, A. Blomqvist et al., Li+ ion conductivity and diffusion mechanism in α-Li3N and β-Li3N. Energy Environ. Sci. 3(10), 1524–1530 (2010). https://doi.org/10.1039/c0ee00052c
M.S. Balogun, W. Qiu, W. Wang, P. Fang, X. Lu et al., Recent advances in metal nitrides as high-performance electrode materials for energy storage devices. J. Mater. Chem. A 3(4), 1364–1387 (2015). https://doi.org/10.1039/C4TA05565A
Z. Li, Y. Fang, J. Zhang, X.W. Lou, Necklace-like structures composed of Fe3N@C yolk-shell particles as an advanced anode for sodium-ion batteries. Adv. Mater. 30(30), 1800525–1800529 (2018). https://doi.org/10.1002/adma.201800525
S.L. Liu, J. Liu, W.J. Wang, L.Y. Yang, K.J. Zhu et al., Synthesis of coral-like Fe2N@C nanoparticles and application in sodium ion batteries as a novel anode electrode material. RSC Adv. 6(89), 8613–86136 (2016). https://doi.org/10.1039/C6RA17251B
X. Gao, B.Y. Wang, Y. Zhang, H. Liu, H.K. Liu et al., Graphene-scroll-sheathed α-MnS coaxial nanocables embedded in N, S Co-doped graphene foam as 3D hierarchically ordered electrodes for enhanced lithium storage. Energy Storage Mater. 16, 46–55 (2019). https://doi.org/10.1016/j.ensm.2018.04.027
P. Ge, H. Hou, S. Li, L. Yang, X. Ji, Tailoring rod-like FeSe2 coated with nitrogen-doped carbon for high-performance sodium storage. Adv. Funct. Mater. 28(30), 1801765–1801776 (2018). https://doi.org/10.1002/adfm.201801765
H. Huang, S. Gao, A.M. Wu, K. Cheng, X.N. Li et al., Fe3N constrained inside C nanocages as an anode for Li-ion batteries through post-synthesis nitridation. Nano Energy 31, 74–83 (2017). https://doi.org/10.1016/j.nanoen.2016.10.059
Y. Zhang, P. Chen, X. Gao, B. Wang, H. Liu et al., Nitrogen-doped graphene ribbon assembled core-sheath MnO@graphene scrolls as hierarchically ordered 3D porous electrodes for fast and durable lithium storage. Adv. Funct. Mater. 26(43), 7754–7765 (2016). https://doi.org/10.1002/adfm.201603716
Y. Chu, L. Guo, B. Xi, Z. Feng, F. Wu et al., Embedding MnO@Mn3O4 nanoparticles in an N-doped-carbon framework derived from Mn-organic clusters for efficient lithium storage. Adv. Mater. 30(6), 1704244–1704255 (2018). https://doi.org/10.1002/adma.201704244
L. Kong, J. Zhu, W. Shuang, X.-H. Bu, Nitrogen-doped wrinkled carbon foils derived from MOF nanosheets for superior sodium storage. Adv. Energy Mater. 8(25), 1801515 (2018). https://doi.org/10.1002/aenm.201801515
W. Shuang, H. Huang, L. Kong, M. Zhong, A. Li et al., Nitrogen-doped carbon shell-confined Ni3S2 composite nanosheets derived from Ni-MOF for high performance sodium-ion battery anodes. Nano Energy 62, 154–163 (2019). https://doi.org/10.1016/j.nanoen.2019.05.030
X. Xu, Z. Meng, X. Zhu, S. Zhang, W.Q. Han, Biomass carbon composited FeS2 as cathode materials for high-rate rechargeable lithium-ion battery. J. Power Sources 380, 12–17 (2018). https://doi.org/10.1016/j.jpowsour.2018.01.057
Y. Wei, H. Chen, H. Jiang, B. Wang, H. Liu et al., Biotemplate-based engineering of high-temperature stable anatase TiO2 nanofiber bundles with impregnated CeO2 nanocrystals for enhanced lithium storage. ACS Sustain. Chem. Eng. 7(8), 7823–7832 (2019). https://doi.org/10.1021/acssuschemeng.9b00012
Y. Liu, A. Qin, S. Chen, L. Liao, K. Zhang et al., Hybrid nanostructures of MoS2/sisal fiber tubular carbon as anode material for lithium ion batteries. Int. J. Electrochem. Sci. 13, 2054–2068 (2018). https://doi.org/10.20964/2018.02.63
H. Jiang, H. Chen, Y. Wei, J. Zeng, H. Liu et al., Biotemplate-mediated structural engineering of rod-like V2O5 cathode materials for lithium-ion batteries. J. Alloys Compd. 787, 625–630 (2019). https://doi.org/10.1016/j.jallcom.2019.02.118
X. Wang, J. Li, Z. Chen, L. Lei, X. Liao et al., Hierarchically structured C@SnO2@C nanofiber bundles with high stability and effective ambipolar diffusion kinetics for high-performance Li-ion batteries. J. Mater. Chem. A 4(48), 918783–991881 (2016). https://doi.org/10.1039/C6TA06622D
Z. Chen, Y. Zhang, X. Wang, W. Sun, S. Dou et al., Fast-pulverization enabled simultaneous enhancement on cycling stability and rate capability of C@NiFe2O4 hierarchical fibrous bundle. J. Power Sources 363, 209–217 (2017). https://doi.org/10.1016/j.jpowsour.2017.07.099
D. Li, D. Yang, X. Yang, Y. Wang, Z. Guo et al., Double-helix structure in carrageenan–metal hydrogels: a general approach to porous metal sulfides/carbon aerogels with excellent sodium-ion storage. Angew. Chem. Int. Ed. 55, 15925–15928 (2016). https://doi.org/10.1002/anie.201610301
H. Chen, H. Liu, Y. Guo, B. Wang, Y. Wei et al., Hierarchically ordered mesoporous TiO2 nanofiber bundles derived from natural collagen fibers for lithium and sodium storage. J. Alloys Compd. 731, 844–852 (2018). https://doi.org/10.1016/j.jallcom.2017.10.116
X.L. Wang, X. Huang, Z.R. Chen, X.P. Liao, C. Liu et al., Ferromagnetic hierarchical carbon nanofiber bundles derived from natural collagen fibers: truly lightweight and high-performance microwave absorption materials. J. Mater. Chem. C 3(39), 10146–10153 (2015). https://doi.org/10.1039/C5TC02689J
D.H. Deng, H. Wu, X.P. Liao, B. Shi, Synthesis of unique mesoporous ZrO2–carbon fiber from collagen fiber. Microporous Mesoporous Mater. 116(1–3), 705–709 (2008). https://doi.org/10.1016/j.micromeso.2008.05.018
X.P. Liao, M.N. Zhang, B. Shi, Collagen-fiber-immobilized tannins and their adsorption of Au(III). Ind. Eng. Chem. Res. 43(9), 2222–2227 (2004). https://doi.org/10.1021/ie0340894
R. Tang, X.P. Liao, X. Liu, B. Shi, Collagen fiber immobilized Fe(III): a novel catalyst for photo-assisted degradation of dyes. Chem. Commun. 47, 4 (2005). https://doi.org/10.1039/b512184a
X. Wang, K. Gao, X. Ye, X. Huang, B. Shi, Close-packing of hierarchically structured C@Sn@C nanofibers for high-performance Li-ion battery with large gravimetric and volumetric energy densities. Chem. Eng. J. 344, 625–632 (2018). https://doi.org/10.1016/j.cej.2018.03.078
L. Xia, S. Wang, G. Liu, L. Ding, D. Li et al., Flexible SnO2/N-doped carbon nanofiber films as integrated electrodes for lithium-ion batteries with superior rate capacity and long cycle life. Small 12(7), 853–859 (2016). https://doi.org/10.1002/smll.201503315
Z.K. Heiba, M.B. Mohamed, H.H. Hamdeh, M.A. Ahmed, Structural analysis and cations distribution of nanocrystalline Ni1−xZnxFe1.7Ga0.3O4. J. Alloys Compd. 618, 755–760 (2015). https://doi.org/10.1016/j.jallcom.2014.08.241
M. Niederdrenk, P. Schaaf, K.P. Lieb, O. Schulte, Characterization of magnetron-sputtered iron-nitride films. J. Alloys Compd. 237(1–2), 81–88 (1996). https://doi.org/10.1016/0925-8388(95)02195-7
Y. Dong, B. Wang, K. Zhao, Y. Yu, X. Wang et al., Air-stable porous Fe2N encapsulated in carbon microboxes with high volumetric lithium storage capacity and a long cycle life. Nano Lett. 17(9), 5740–5746 (2017). https://doi.org/10.1021/acs.nanolett.7b02698
D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo et al., Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 351(6271), 361–365 (2016). https://doi.org/10.1126/science.aad0832
Y. Wang, W. Chen, Y. Nie, L. Peng, W. Ding et al., Construction of a porous nitrogen-doped carbon nanotube with open-ended channels to effectively utilize the active sites for excellent oxygen reduction reaction activity. Chem. Commun. 53(83), 11426–11429 (2017). https://doi.org/10.1039/C7CC07249J
A.L.M. Reddy, A. Srivastava, S.R. Gowda, H. Gullapalli, M. Dubey et al., Synthesis of nitrogen-doped graphene films for lithium battery application. ACS Nano 4(11), 6337–6342 (2010). https://doi.org/10.1021/nn101926g
D.K. Nandi, U.K. Sen, S. Sinha, A. Dhara, S. Mitra et al., Atomic layer deposited tungsten nitride thin films as a new lithium-ion battery anode. Phys. Chem. Chem. Phys. 17(26), 17445–17453 (2015). https://doi.org/10.1039/C5CP02184G
D. Oh, C. Ozgit Akgun, E. Akca, L.E. Thompson, L.F. Tadesse et al., Biotemplating pores with size and shape diversity for Li-oxygen battery cathodes. Sci. Rep. 7(1), 1–11 (2017). https://doi.org/10.1038/srep46808
K. Lei, C. Wang, L. Liu, Y. Luo, C. Mu et al., A porous network of bismuth used as the anode material for high-energy-density potassium-ion batteries. Angew. Chem. Int. Ed. 57(17), 4687–4691 (2018). https://doi.org/10.1002/anie.201801389
H. Yang, R. Xu, Y. Yao, S. Ye, X. Zhou et al., Multicore–shell Bi@N-doped carbon nanospheres for high power density and long cycle life sodium- and potassium-ion anodes. Adv. Funct. Mater. 29(13), 1809195–1809205 (2019). https://doi.org/10.1002/adfm.201809195
Z. Ju, S. Zhang, Z. Xing, Q. Zhuang, Y. Qiang et al., Direct synthesis of few-layer F-doped graphene foam and its lithium/potassium storage properties. ACS Appl. Mater. Interfaces 8(32), 20682–20690 (2016). https://doi.org/10.1021/acsami.6b04763
J. Huang, X. Lin, H. Tan, B. Zhang, Bismuth microparticles as advanced anodes for potassium-ion battery. Adv. Energy Mater. 8(19), 1703496 (2018). https://doi.org/10.1002/aenm.201703496
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (21878192, 51502180), the Fundamental Research Funds for the Central Universities (2016SCU04A18), the 1000 Talents Program of Sichuan Province, the Sichuan Province Science and Technology Support Program (2014GZ0093), and the Australian Research Council (DP160102627).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jiang, H., Huang, L., Wei, Y. et al. Bio-Derived Hierarchical Multicore–Shell Fe2N-Nanoparticle-Impregnated N-Doped Carbon Nanofiber Bundles: A Host Material for Lithium-/Potassium-Ion Storage. Nano-Micro Lett. 11, 56 (2019). https://doi.org/10.1007/s40820-019-0290-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40820-019-0290-0