
Abstract
The catalytic effects of alkali metal ions (Na+ and K+) on NOx precursor formation during coal pyrolysis were investigated using the N-containing compound pyridine as a model compound. Density functional theory calculations at the B3LYP/6-31G (d, p) level of theory were conducted to elucidate the mechanism of pyridine pyrolysis and the pathways for HCN formation. The calculation results indicate that Na+ and K+ have distinct influences on different pyrolysis reactions; these alkali metal ions facilitate the initial hydrogen transfer from C1 to N and C2, whereas they hinder the other hydrogen migration reactions. Both Na+ and K+ significantly reduce the activation energies for C–C bond breakage and triple-bond formation, whereas they increase the activation energies for the isomerization reactions. The different effects essentially result from the distinct charge distributions induced by the two ions. Due to the distinct influences on the different reactions, the rate-determining steps are modulated, affecting the competitiveness of the different possible pathways of HCN formation. The formation of HCN from pyridine is promoted in the presence of Na+ and K+ because all the overall activation energies are decreased for different pathways. The calculation results agree well with previous experimental studies. Thus, the findings offer a new and promising approach to reveal the formation mechanism of NOx and facilitate the control of NOx for coal utilization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Nitrogen oxide (NOx) emissions are predominantly ascribed to the combustion of fossil fuels, especially coal, and have generated severe adverse effects on the environment and human health (Kampa and Castanas 2008; Wu et al. 2010). In addition to generating acid rain, NOx contributes significantly to the greenhouse effect: its greenhouse effect is 260 times that of carbon dioxide (Tsiliyannis 2016; Chu et al. 2020). Thus, the emission of NOx from coal utilization is a serious environmental concern. During the initial pyrolysis stage of coal combustion, organic N-containing compounds (mainly pyrrole, pyridine, and quaternary compounds) are first converted into small-molecule compounds (e.g., NH3 and HCN), which can react with oxygen to form NOx (Houser et al. 1980; MacNamara and Simmie 2003; Koger and Bockhorn 2005; Zhang et al. 2011; Ye et al. 2019). Both pyrrole and pyridine-based N-containing compounds have been widely used as model compounds to investigate the pyrolysis mechanisms (Liu and Guo 2017; Ma et al. 2019; Mai et al. 2020). Understanding the mechanisms of NH3 and HCN (NOx precursors) formation from organic N-containing compounds is critical to control NOx emissions and has thus attracted widespread attention in recent decades.
Both experimental and computational approaches have been widely applied to explore the pyrolysis mechanisms of various N-containing compounds in coal, including pyridine and its derivatives (Hämäläinen et al. 1994; Molina et al. 2000; Adamczyk et al. 2014; Ikeda and Mackie 1995; Mackie et al. 1990; Hong et al. 2013; Ninomiya et al. 2000; Gu et al. 2013). HCN and cyanoacetylene have been detected as major products of the pyrolysis of pyridine and 2-methylpyridine, based on which Ikeda and Mackie (1995) developed a kinetic model to simulate pyrolysis reactions. Mackie et al. (1990) reported similar findings for pyridine pyrolysis under argon atmosphere. They proposed that the rupture of all C–H bonds in the pyridine ring is feasible, especially the one at the ortho position of the nitrogen atom, which is weaker than the others. Hong et al. (2013) detected intermediates and products from pyridine pyrolysis using tunable synchrotron vacuum ultraviolet photoionization and molecular-beam mass spectrometry. Following the homolytic cleavage of the C–H bond, o-, p-, and m-pyridyls were generated and then decomposed by breaking the pyridine ring. Among the pyridyl compounds, m-pyridyl was determined as the major source for HCN formation. Computational methods have also been applied to investigate the pyrolysis mechanism of pyridine. The semi-empirical PM3 calculations indicated that the C–H bond of pyridine is broken to generate radicals; decomposition fragments are then successively generated by rupturing the weakest bonds of the radicals (Ninomiya et al. 2000). Gu et al. (2013) computationally studied the decomposition mechanism of pyridine using the B3LYP functional and 6-31G(d,p) basis set. They found that the breakage of the C–H bond in pyridine preferentially produced the o-pyridyl radical. In addition, ring-opening via C–N bond cleavage in the o-pyridyl radical was more favorable than ring-opening via C–C bond cleavage, as indicated by the lower activation barrier of the former process. Meanwhile, our density functional theory (DFT) calculations indicated that the concerted hydrogen transfer is more feasible for the breakage of the C–H bond than direct homolysis (Jiang et al. 2017; Liu et al. 2019; Liu et al. 2020a, b).
During the real coal pyrolysis/combustion process, the pyrolysis of pyridine and other N-containing compounds can be affected by the other components in coal, such as metal ions. In addition, metal salts can be used to accelerate the process of coal gasification because the salt ions can create various active species by reacting with the carbonaceous material. Consequently, the effects of alkali metal ions on coal pyrolysis have been extensively studied. Na and K are the major ion-exchangeable alkali metals in low-rank coals, where they exist as carboxylates (–COOmRn, R = Na/K) or as soluble salts (RmCln, R = Na/K) (Liu et al. 2000). Na and K are easily volatilized during pyrolysis, which affects the evolution channels and the product distribution of coal (Durie 2013). Ohtsuka et al. (1997) found that the total conversion of coal–N to NOx precursors (HCN and NH3) and N2 strongly depended on the alkali metal ions at a high temperature of 1000 °C. Franklin et al. (1983) investigated the roles of metal ions by pyrolyzing raw coal in the presence of calcium, sodium, and potassium under helium atmosphere at 1400 K. They found that sodium and potassium ions had stronger effects on coal pyrolysis than calcium ion. This conclusion was supported by Wood and Sancier (1984), who found that alkali metal salts often had significant catalytic effects on pyrolysis reactions.
Currently, the mechanism of HCN formation from the pyrolysis of pure pyridine under uncatalyzed conditions has been comprehensively investigated; in contrast, few studies have reported the effects of alkali metal ions on the mechanism of pyridine pyrolysis. In this study, DFT calculations were conducted to investigate the catalytic effects of two typical alkali metal ions, K+ and Na+, on the formation of HCN via pyridine pyrolysis.
2 Calculation details
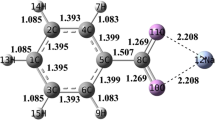
All calculations were performed using Gaussian 16 (Frisch et al. 2016). Many studies have used DFT calculations to shed light on the pyrolysis chemistry of N-containing compounds (e.g., pyrrole, indole, and pyridine) (Liu et al. 2018a, b, 2020). The stable geometries of all reactants, intermediates, transition states, and products were obtained through an initial unconstrained optimization using the B3LYP/6-31G (d, p) method. The geometries of the transition states were located using the TS (Berny) method, which has been confirmed to be accurate for the optimization of organic species (Liu et al. 2018a, b, 2020). In addition, according to Glukhovtsev et al. (1997), B3LYP calculations on iron-containing species gave reliable results comparable with experimental data. Basis set superposition error was also considered by examining the complexation energies of the Na+/K+–pyridine complexes. The results indicate that the method employed in the present work is suitable to explore interactions between pyridine and Na+/K+ (Table S1 in Supplementary Material).
All optimized geometry configurations were further examined by frequency analysis at the same computational level. The vibrations were checked with the aid of the visualization software GaussView 6.0 (Dennington et al. 2016). The reactants, intermediates, and products were verified as the local minima with no imaginary frequencies, while all transition states were confirmed to be first-order saddle points having one imaginary frequency. The corresponding minima and first-order saddle points were verified to be on the same potential energy surface by conducting intrinsic reaction coordinate calculations at the same computational level. The enthalpies at 298.15 K and 1 atm were determine to discuss the energetics (Ling et al. 2009; Beste and Buchanan 2009; Liu et al. 2018a; Liu et al. 2020a, b). The activation energy was defined as the difference in enthalpy or Gibbs free energy (shown in Supplementary Material) between the reactants and the relevant transition state (Glukhovtsev et al. 1997).
3 Results and discussion
In our previous investigation, DFT calculations were conducted to investigate the possible pathways of HCN formation during pyridine pyrolysis under uncatalyzed conditions. Pyridine tends to decompose via the initial hydrogen transfer process, as shown in Figure S1 in Supplementary Material. According to our preliminary work, the pyrolysis of pyridine to form HCN can proceed via five pathways (Fig. 1): path-a, path-b, path-c, path-d, and path-e. Based on the DFT calculation results, the effects of Na+ and K+ on the initial hydrogen transfer and the formation of HCN are discussed in Sects. 3.1 and 3.2, respectively.

Scheme showing the possible pathways of HCN formation during pyridine pyrolysis
3.1 Effects of Na+ or K+ on the initial hydrogen transfer reactions
Table 1 summarizes the activation energies of the initial hydrogen transfer reactions under the uncatalyzed and Na+/K+-catalyzed conditions. The two alkali metal ions have different influences on the five sites of hydrogen transfer. The removal of H from C1 (Reactions a and b) is promoted by Na+ and K+. For Reaction a, Na+ decreases the activation energy to a greater extent than K+, whereas the opposite is true for Reaction b. Notably, the activation energy for the hydrogen transfer from C1 to C2 (Reaction b) decreases by 69.4 and 71.9 kJ/mol in the presence of Na+ and K+, respectively. The differences in the activation energy for Reaction a (C1 to N) are less than 10 kJ/mol for the uncatalyzed and ion-catalyzed conditions. Other hydrogen transfer reactions are hindered in the presence of alkali metal ions, and the inhibition effect is stronger in the presence of Na+ than that of K+. The above results indicate that alkali metal ions affect the decomposition of pyridine and the formation of HCN by influencing the initial hydrogen transfer process, with Na+ having a greater influence than K+. Among the five hydrogen transfer reactions, Reaction b is the most feasible in the presence of alkali metal ions, whereas Reaction a is dominant under uncatalyzed conditions. The detailed effects of alkali metal ions on the pathways of HCN formation are discussed below.
3.2 Effects of Na+ and K+ on the pathways of HCN formation
While the presence of metal ions does not change the possible pathways of HCN formation from pyridine, it does affect the geometries of the transition states and intermediates. The detailed pathways are compared below. In this section, the letters “n” and “k” are used to represent the presence of Na+ and K+, respectively, and the transition states and intermediates are respectively distinguished by the letters “t” and “m”. For example, “a-n-1t” represents the first transition state of path-a in the presence of Na+.
3.2.1 HCN formation via path-a
The energy profiles for HCN formation via path-a with and without catalysis are depicted in Fig. 2. As discussed above, the decomposition of pyridine begins with the transfer of hydrogen from C1 to N, disturbing the stability of the N-heterocycle. The transition states a-1t, a-n-1t, and a-k-1t are involved in this step for the uncatalyzed, Na+-catalyzed, and K+-catalyzed conditions with activation energies of 357.7, 349.6, and 354.4 kJ/mol, respectively. Subsequently, the six-membered intermediate a-1 m isomerizes into the five-membered ring a-2 m via the simultaneous cleavage of the C1–N bond and generation of the C1–C6 bond. The presence of Na+ or K+ increases the activation energy from 462.2 to 468.1 or 470.2 kJ/mol, respectively. Thus, Na+ and K+ inhibit the isomerization reaction of the pyridine ring, and K+ has a stronger effect than Na+.

Energy profiles of uncatalyzed and Na+/K+-catalyzed pyridine pyrolysis to form HCN via path-a (unit: kJ/mol)
Subsequently, the five-membered ring of intermediate a-2 m is cleaved via a concerted ring-opening reaction in which the C=N double bond becomes a triple bond. This step has an activation energy of 511.3 kJ/mol without catalysis. For Na+- and K+-catalyzed pyridine pyrolysis, the activation energies of this ring opening are 460.4 and 467.0 kJ/mol, respectively. This indicates that Na+ and K+ can significantly reduce the activation energy for the concerted ring-opening reaction of pyridine derivatives by ~ 50 kJ/mol. Finally, the C4–C5 bond of intermediate a-3 m is cleaved via hydrogen transfer from C4 to C5, producing a-4 m (vinylacetylene) and HCN. The final step has an activation energy of 561.2 kJ/mol under the uncatalyzed condition. The addition of Na+ or K+ lowers the activation energy by 84.7 or 36.4 kJ/mol, respectively, showing that Na+ and K+ promote the breakage of the C–C bond to form HCN. Moreover, Na+ has a greater effect than K+ in this step. Notably, the final step is the rate-determining step in path-a regardless of the presence of alkali metal ions; the overall activation energies are 561.2, 476.5, and 524.8 kJ/mol for the uncatalyzed, Na+-catalyzed, and K+-catalyzed conditions, respectively.
In summary, alkali metal ions (Na+ and K+) can significantly promote the concerted ring-opening and final hydrogen transfer in path-a. These two reaction steps both involve C–C bond rupture. In comparison, the isomerization reaction and initial hydrogen transfer in path-a are less affected by the two alkali metal ions. Na+ and K+ lower the overall activation energy to promote HCN generation via path-a, with Na+ having a stronger catalytic effect than K+.
3.2.2 HCN formation via path-b
The effects of alkali metal ions on the pyrolysis of pyridine in path-b are shown in Fig. 3. The initial hydrogen transfer from the C1 position to the C2 position is unique in comparison with the initial hydrogen transfer steps in the other pathways. The N-heterocycle is cleaved via C1–C2 bond cleavage, which is accompanied by the hydrogen transfer. As stated above, this reaction is facilitated by the presence of alkali metal ions. The activation energies are 413.9, 344.5, and 342.0 kJ/mol for the uncatalyzed, Na+-catalyzed, and K+-catalyzed conditions. Subsequently, the intermediate b-1 m undergoes isomerization via the simultaneous formation of the C1–C5 bond and breaking of the C5–N bond. The activation energy of this step is 371.5 kJ/mol under the uncatalyzed condition. During catalytic pyridine pyrolysis, the activation energies of the transition states are 395.0 and 389.9 kJ/mol Na+ and K+, respectively, 23.5 and 18.4 kJ/mol higher than the corresponding activation energy without alkali metal ions, respectively. This result shows that Na+ and K+ inhibit the isomerization reaction of the pyridine ring, and that Na+ has a slightly stronger effect than K+. Finally, the newly formed C1–C5 bond is cleaved through a hydrogen transfer reaction to produce b-3 m (vinylacetylene) and HCN. Without catalysis, the activation energy of this step is 561.2 kJ/mol, and it is the rate-determining step of path-b. In the presence of Na+ and K+, the activation energy is decreased by 84.7 and 36.4 kJ/mol, respectively. Even so, this process is still the rate-determining step under the Na+- and K+-catalyzed conditions.

Energy profiles of uncatalyzed and Na+/K+-catalyzed pyridine pyrolysis to form HCN via path-b (unit: kJ/mol)
In summary, the two alkali metal ions promote the formation of HCN by significantly reducing the overall activation energy in path-b. The overall catalytic abilities of the two alkali metal ions are similar, with K+ having a slight advantage over Na+. As expected, the two alkali metal ions promote the breakage of C–C bonds during step 1 and step 3 of path-b.
3.2.3 HCN formation via path-c
Figure 4 shows the energy profiles of path-c under the uncatalyzed and alkali metal ion-catalyzed conditions. The hydrogen transfer from C2 to C1 initiates the decomposition of pyridine in path-c. Under the uncatalyzed condition, transition state c-1t is involved with an activation energy of 389.6 kJ/mol. Under the catalysis of Na+ and K+, transition states c-n-1t and c-k-1t are involved, and the activation energy is increased by 21.4 and 15.1 kJ/mol, respectively. Hence, both Na+ and K+ hinder internal hydrogen transfer (C2 to C1), with Na+ having a slightly greater effect than K+. Following the initial hydrogen transfer, the H at C1 migrates to the N atom (485.1 kJ/mol) under the uncatalyzed condition. The activation energy of this step is increased to 535.2 and 527.6 kJ/mol under the catalysis of Na+ and K+, respectively.

Energy profiles of uncatalyzed and Na+/K+-catalyzed pyridine pyrolysis to form HCN via path-c (unit: kJ/mol)
Different from path-a, the intermediate c-2 m undergoes ring opening to break the C1–N bond without the involvement of a five-membered intermediate. The activation energy decreases slightly from 359.0 kJ/mol (uncatalyzed) to 355.6 (Na+-catalyzed) and 354.2 kJ/mol (K+-catalyzed). Subsequently, the N–H bond rotates into a proper position for the final hydrogen transfer from the N atom to the C4 position. Finally, the concerted cleavage of the C4–C5 bond leads to the production of HCN and b-5 m (vinylacetylene). The presence of Na+ or K+ greatly decreases the activation energy of the final hydrogen transfer from 687.4 kJ/mol (uncatalyzed) to 503.0 kJ/mol (Na+-catalyzed) or 516.6 kJ/mol (K+-catalyzed). Thus, Na+ has a greater promotion effect than K+ in this step.
According to the above results, the rupture of C–C bonds in the ring-opening and final hydrogen transfer reactions are facilitated by the alkali metal ions, similar to in path-a. However, the alkali metal ions have little effect on other reactions that do not involve the rupture of C–C bonds. The two alkali metal ions also alter the rate-determining step of path-c. In the uncatalyzed condition, the final step determines the reaction rate; in contrast, during Na+/K+-catalyzed pyridine pyrolysis, the second step of hydrogen transfer (C1 to N) is the rate-determining step. In summary, In summary, Na+ and K+ significantly promote C–C bond breakage during pyridine pyrolysis to form HCN, and Na+ shows a greater catalytic effect than K+.
3.2.4 HCN formation via path-d
Figure 5 shows the catalytic effects of Na+ and K+ on path-d. As mentioned above, the initial hydrogen transfer from C3 to C2 is hindered by the presence of alkali metal ions. The activation energy increases from 406.6 kJ/mol (uncatalyzed) to 423.0 kJ/mol (Na+-catalyzed) and 419.5 kJ/mol (K+-catalyzed). Subsequently, the C1–N bond breaks accompanied by hydrogen transfer from C5 to the N atom. Unsurprisingly, the activation energy (660.5 kJ/mol) is greatly reduced by alkali ion catalysis to 433.3 kJ/mol for Na+ and 431.6 kJ/mol for K+. The H atom bound to the N atom then migrates to the C4 position to form a C≡N triple bond. Notably, the activation energy of this step is also greatly reduced under the catalysis of the two alkali metal ions. This reaction does not involve the breakage of a C–C bond but a transformation from a double bond to a triple bond. The uncatalyzed activation energy is 587.1 kJ/mol; the activation energy under the Na+-catalyzed condition (454.6 kJ/mol) is decreased by 132.5 kJ/mol, while that under the K+-catalyzed condition (461.8 kJ/mol) is lowered by 125.3 kJ/mol. Similarly, HCN and d-4 m (vinylacetylene) are formed by a final breakage of the C4–C5 bond via hydrogen transfer with an activation energy of 620.9 kJ/mol. The activation energies under Na+ and K+ catalysis (540.4 and 550.6 kJ/mol, respectively) are 80.5 and 70.3 kJ/mol lower than that without catalysis. Thus, Na+ and K+ significantly promote the final C–C bond cleavage to form HCN, with Na+ having a stronger effect than K+.

Energy profiles of uncatalyzed and Na+/K+-catalyzed pyridine pyrolysis to form HCN via path-d (unit: kJ/mol)
In the presence of alkali metal ions, both the scission of the C–C bond and the formation of the triple bond from the double bond are facilitated by catalysis by Na+ and K+. Additionally, whereas the second step (hydrogen transfer) is the rate-determining step of the uncatalyzed process, the second step (hydrogen transfer) is the rate-determining step of path-d under alkali metal catalysis. All in all, both Na+ and K+ alter the rate-determining step of path-d, reduce the activation energy, and promote the formation of HCN, with Na+ having a greater influence than K+.
3.2.5 HCN formation via path-e
Figure 6 illustrates the energy profiles of path-e under the uncatalyzed, Na+-catalyzed, and K+-catalyzed conditions. First, the H atom at the C3 position of pyridine transfers to the C2 position to form e-1m via internal hydrogen transfer. In the absence of alkali metal ions, the transition state is e-1t, which has an activation energy of 389.4 kJ/mol. Both Na+ and K+ inhibit the internal hydrogen transfer reaction. The activation energy under Na+ catalysis is 423.4 kJ/mol, which is 34.0 kJ/mol higher than that without catalysis. K+ has a slightly weaker influence than Na+; K+ catalysis increases the activation energy to 418.0 kJ/mol. Next, the intermediate e-1 m undergoes a concerted ring-opening reaction to break the C1–C2 bond via the transition state e-2t (481.5 kJ/mol). As for the Na+- or K+-catalyzed pyrolysis of pyridine, the activation energy is lowered to 399.6 or 402.5 kJ/mol, respectively. Different from the other four pathways, the breakage of the C5–N bond results in the formation of a C≡N triple bond, producing HCN and e-3 m. Without catalysis, this final step determines the reaction rate of path-e, and the overall activation energy is 625.3 kJ/mol. The presence of Na+ or K+ significantly decreases the activation energy of this reaction by 163.7 or 157.9 kJ/mol, respectively. Even so, the final C–N bond scission is still the rate-determining step, and the overall activation energy is 461.6 or 467.4 kJ/mol less than that under the Na+- or K+-catalyzed condition, respectively.

Energy profiles of uncatalyzed and Na+/K+-catalyzed pyridine pyrolysis to form HCN via path-e (unit: kJ/mol)
The above calculation results indicate that alkali metal ions significantly promote the C–C bond scission and formation of the C≡N triple bond in path-e. Na+ and K+ lower the overall activation energy for pyridine pyrolysis, further promoting the formation of HCN. Overall, Na+ has a slightly greater catalytic effect than K+.
The energy profiles of the above five pathways under the uncatalyzed, Na+-catalyzed, and K+-catalyzed conditions are compared in Fig. S2–S4 in Supplementary Material. Without catalysis, the most favorable pathway for HCN formation is path-a. In contrast, under the catalysis of Na+ or K+, path-e is the most favorable pathway. The presence of the two alkali metal ions alters the competitiveness of the possible HCN formation pathways and greatly decreases the energy barriers of the pyrolysis reactions, suggesting a promotion effect on HCN generation. This finding is consistent with the results of previous experimental studies (Li and Tan 2000; Yan et al. 2005; Liu and Guo 2017; Bai et al. 2019; Liu et al. 2019; Luo et al. 2019), attesting to the rationality of the computational results of the present work. Notably, the energetics of the above pathways are discussed based on enthalpy; however, the same trend was obtained when comparing the Gibbs free energies (Figs. S5–S7 in Supplementary Material).
The influence of temperature was examined by comparing the rate constants of the HCN generation pathways. According to Fig. 7, path-e has the largest rate constant in the temperature range from 273 to 973 K, demonstrating the dominance of this pathway in HCN generation under Na+/K+ catalysis. As the pyrolysis temperature increases, the advantage of path-e gradually weakens because the other four pathways have greater pre-exponential factors. The natural population analysis charges were calculated to further analyze the roles of Na+ and K+ at the electronic level (Figs. S8–S12 in Supplementary Material). As discussed in Sect. 3.1, Na+ has a stronger effect than K+ on all reactions except hydrogen transfer from C1 to C2, which can be ascribed to the distinct variations in charge induced by Na+ and K+. For the initial hydrogen transfer from C1 to C2, the change in charge for the H acceptor (the C2 atom) is larger in the presence of K+ than in the presence of Na+. In contrast, for the other initial hydrogen transfer reactions, the variation in charge for the H acceptor is larger for Na+ than for K+. In addition, Na+/K+ has little effect on the molecular structures but a large influence on charge redistribution. Hence, the energetic changes are strongly correlated to the charge variation induced by the two alkali metal ions.

Comparison of the reaction rate constants of the rate-determining steps of different pathways in the temperature range of 293–973 K under the a Na+-catalyzed condition and b K+-catalyzed condition
4 Conclusions
The present work explored the roles of Na+ and K+ in the pyrolysis of pyridine to produce HCN using DFT calculations. Based on the computational results, the effects of the two alkali metal ions can be summarized as follows.
Na+ and K+ have dual roles in the initial hydrogen transfer reactions. Both ions facilitate the migration from C1 to N or C2 by reducing the activation energy. For Reaction b (hydrogen transfer from C1 to C2), the activation energy is decreased by 69.4 and 71.9 kJ/mol under the catalysis of Na+ and K+, respectively. In contrast, the activation energies of the other initial hydrogen transfer reactions (C2 to C1, C2 to C3, and C3 to C2) are increased by 13.1–54.0 kJ/mol. For all reactions except hydrogen transfer from C1 to C2, Na+ has a greater effect than K+ due to the distinct variations in charge induced by these two alkali metal ions.
Among the elementary reactions involved in the formation of HCN during pyridine pyrolysis, Na+ and K+ significantly decrease the activation energies of C–C bond breakage and triple bond formation. In contrast, the other reactions are only slightly promoted by the two alkali metal ions, and the isomerization reactions are even hindered by them. The different energetic effects essentially result from the distinct charge distributions induced by the two alkali ions.
The presence of Na+ and K+ significantly lowers the overall activation energy and even alters the rate-determining step for both path-c and path-d. Hence, the formation of HCN is promoted by the presence of Na+ or K+. Due to the different effects of Na+/K+ on the individual elementary reactions and pathways, the competitiveness of the possible pathways is altered by these ions. Under the uncatalyzed condition, path-b, which involves two hydrogen transfer reactions along with C–C bond breakage and isomerization, is the most favorable. In the presence of Na+/K+, the most favorable pathway changes to path-e, in which pyridine is decomposed via hydrogen transfer reactions, concerted ring opening, and C–N bond scission.
Therefore, the inclusion of Na+ and K+ in coal should be considered for source-control NOx abatement techniques.
References
Adamczyk WP, Werle S, Ryfa A (2014) Application of the computational method for predicting NOx reduction within large scale coal-fired boiler. Appl Therm Eng 73:343–350
Bai Y, Lv P, Li F, Song X, Su W, Yu G (2019) Investigation into Ca/Na compounds catalyzed coal pyrolysis and char gasification with steam. Energy Convers Manag 184:172–179
Beste A, Buchanan AC III (2009) Computational study of bond dissociation enthalpies for lignin model compounds. Substituent effects in phenethyl phenyl ethers. J Organ Chem 74:2837–2841
Chu F, Su M, Yang G (2020) Heat and mass transfer characteristics of ammonia regeneration in packed column. Appl Therm Eng 176:115405
Dennington R, Keith T, Millam J (2016) GaussView, Version6. Semichem Inc., Shawnee Mission
Durie R (2013) The science of Victorian brown coal: structure, properties and consequences for utilization. Butterworth-Heinemann, Oxford
Franklin HD, Cosway RG, Peters WA, Howard JB (1983) Effects of cations on the rapid pyrolysis of a Wyodak subbituminous coal. Ind Eng Chem Process Des Dev 22:39–42
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16, Revision B.01. Gaussian, Inc., Wallingford
Glukhovtsev MN, Bach RD, Nagel CJ (1997) Performance of the B3LYP/ECP DFT calculations of iron-containing compounds. J Phys Chem A 101:316–323
Gu XH, Mao XF, Zhao Y, Li WB, Zhang XJ (2013) Study on the basic nitrogen compounds from coal-derived oil. J Coal Sci Eng (China) 19:83–89
Hämäläinen JP, Aho MJ, Tummavuori JL (1994) Formation of nitrogen oxides from fuel-N through HCN and NH3: a model-compound study. Fuel 73:1894–1898
Hong X, Zhang TC, Zhang LD, Qi F (2013) Identification of intermediates in pyridine pyrolysis with molecular-beam mass spectrometry and tunable synchrotron VUV photoionization. Chin J Chem Phys 22:204
Houser TJ, Hull M, Always RM, Biftu T (1980) Kinetics of formation of HCN during pyridine pyrolysis. Int J Chem Kinet 12:569–574
Ikeda E, Mackie JC (1995) Thermal decomposition of two coal model compounds—pyridine and 2-picoline. Kinetics and product distributions. J Anal Appl Pyrolysis 34:47–63
Jiang X, Lu Q, Hu B, Liu J, Dong C, Yang Y (2017) A comprehensive study on pyrolysis mechanism of substituted β-O-4 type lignin dimers. Int J Mol Sci 18:2364–2377
Kampa M, Castanas E (2008) Human health effects of air pollution. Environ Pollut 151:362–367
Koger S, Bockhorn H (2005) NOx formation from ammonia, hydrogen cyanide, pyrrole, and caprolactam under incinerator conditions. Proceed Combust Inst 30:1201–1209
Li CZ, Tan LL (2000) Formation of NOx and SOx precursors during the pyrolysis of coal and biomass. Part III. Further discussion on the formation of HCN and NH3 during pyrolysis. Fuel 79:1899–1906
Ling LX, Zhang RG, Wang BJ, Xie KC (2009) Density functional theory study on the pyrolysis mechanism of thiophene in coal. J Mol Struct 905:8–12
Liu J, Guo X (2017) ReaxFF molecular dynamics simulation of pyrolysis and combustion of pyridine. Fuel Process Technol 161:107–115
Liu R, Huang TTS, Tittle J, Xia D (2000) A theoretical investigation of the decomposition mechanism of pyridyl radicals. J Phys Chem A 104:8368–8374
Liu J, Lu Q, Jiang XY, Hu B, Zhang XL, Dong CQ, Yang YP (2018a) Theoretical investigation of the formation mechanism of NH3 and HCN during pyrrole pyrolysis: the effect of H2O. Molecules 23:711
Liu J, Zhang X, Lu Q, Shaw A, Hu B, Jiang X, Dong C (2018b) Mechanism study on the effect of alkali metal ions on the formation of HCN as NOx precursor during coal pyrolysis. J Energy Inst 92:604–612
Liu J, Zhang X, Lu Q, Shaw A, Hu B, Jiang X, Dong C (2019) Mechanism study on the effect of alkali metal ions on the formation of HCN as NOx precursor during coal pyrolysis. J Energy Inst 92(3):604–612
Liu S, Wu Y, Zhou C, Wu J, Zhang Y (2020a) Study on the CO formation mechanism during coal ambient temperature oxidation. Energies 13:2587
Liu J, Zhang X, Hu B, Lu Q, Liu DJ, Dong CQ, Yang YP (2020b) Formation mechanism of HCN and NH3 during indole payrolysis: a theoretical DFT study. J Energy Inst 93:649–657
Luo J, Zou C, He Y, Jing H, Cheng S (2019) The characteristics and mechanism of NO formation during pyridine oxidation in O2/N2 and O2/CO2 atmospheres. Energy 187:115954
Ma Z, Zhang Y, Li C, Yang Y, Zhang W, Zhao C, Wang S (2019) N-doping of biomass by ammonia (NH3) torrefaction pretreatment for the production of renewable N-containing chemicals by fast pyrolysis. Bioresour Technol 292:122034
Mackie JC, Colket MB, Nelson PF (1990) Shock tube pyrolysis of pyridine. J Phys Chem 94:4099–4106
MacNamara JP, Simmie JM (2003) The high temperature oxidation of pyrrole and pyridine; ignition delay times measured behind reflected shock waves. Combust Flame 133:231–239
Mai TVT, Nguyen HT, Huynh LK (2020) Ab initio kinetic mechanism of OH-initiated atmospheric oxidation of pyrrole. Chemosphere 263:127850
Molina A, Eddings EG, Pershing D, Sarofim A (2000) Char nitrogen conversion: implications to emissions from coal-fired utility boilers. Prog Energy Combust Sci 26:507–531
Ninomiya Y, Dong Z, Suzuki Y, Koketsu J (2000) Theoretical study on the thermal decomposition of pyridine. Fuel 79:449–457
Ohtsuka Y, Zhiheng W, Furimsky E (1997) Effect of alkali and alkaline earth metals on nitrogen release during temperature programmed pyrolysis of coal. Fuel 76:1361–1367
Tsiliyannis CA (2016) Emissions and power losses due to biofuel or biomass nitrogen: assessment and prevention mechanisms. Energy Fuels 30:9396–9408
Wood BJ, Sancier KM (1984) The mechanism of the catalytic gasification of coal char: a critical review. Catal Rev Sci Eng 26:233–279
Wu CL, Cao Y, Dong ZB, Cheng CM, Li HX, Pan WP (2010) Mercury speciation and removal across full-scale wet FGD systems at coal-fired power plants. J Coal Sci Eng (China) 16:82–87
Yan X, Che D, Xu T (2005) Effect of rank, temperatures and inherent minerals on nitrogen emissions during coal pyrolysis in a fixed bed reactor. Fuel Process Tehcnol 86:739–756
Ye C, Wang Q, Zheng Y, Li G, Zhang Z, Luo Z (2019) Techno-economic analysis of methanol and electricity poly-generation system based on coal partial gasification. Energy 185:624–632
Zhang K, Li Y, Yuan T, Cai J, Glarborg P, Qi F (2011) An experimental and kinetic modeling study of premixed nitromethane flames at low pressure. Proc Combust Inst 33:407–414
Acknowledgments
This work was supported by the National Key Research Development Program of China (2017YFB0601900), National Nature Science Foundation of China (51806220, 51922040), China Postdoctoral Science Foundation (2019TQ0091), Grants from Fok Ying Tung Education Foundation (161051), and Fundamental Research Funds for the Central Universities (2020MS020, 2020DF01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, J., Zhao, W., Fan, X. et al. Effect of alkali metal ions on the formation mechanism of HCN during pyridine pyrolysis. Int J Coal Sci Technol 8, 349–359 (2021). https://doi.org/10.1007/s40789-021-00427-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40789-021-00427-3