
Abstract
Wnt signaling plays important roles in stem cell self-renewal and differentiation in adults as well as in embryonic development. Mutations that activate canonical Wnt/β-catenin signaling also initiate and maintain several cancer states, including colorectal cancer and leukemia, and hence Wnt inhibitors are currently being explored as therapeutic options. In this review, we summarize previous studies and update recent findings on canonical Wnt signaling and its components, as well as their roles in somatic stem cell homeostasis and maintenance of cancer-initiating cells.
Similar content being viewed by others

Introduction
Wnts are secreted glycoproteins that play multiple roles in development and adult stem cell homeostasis, and mutations that activate the canonical Wnt/ß-catenin pathway are commonly associated with a variety of carcinomas [1, 2]. Wnt is a portmanteau of the names of the founding members of the gene family, wingless (wg1, a classical mutant in Drosophila) and int1 (activation of the int1 gene by viral integration leads to mammary cell transformation) [3–5]. Wnt signaling regulates proliferation, specification, and patterning during embryonic development and regulates the balance between self-renewal and differentiation in embryonic stem cells as well as multiple somatic stem cells including hematopoietic, epidermal, neuronal, mesenchymal, and intestinal stem cells. The Wnt family in mammals comprises 19 genes encoding secreted glycoproteins that bind to Frizzled receptors and lipoprotein receptor-related protein (LRP) co-receptors. The role of the pathway in normal development, somatic stem cells, and human disease has been extensively reviewed [6–15] and information is also available in an authoritative website (http://web.stanford.edu/group/nusselab/cgi-bin/wnt/). In this review, we briefly summarize the previous studies on Wnt signaling and present an update on the most recent findings of the role of Wnt signaling in regulating stem cells and various disease states.
Canonical Wnt Signaling
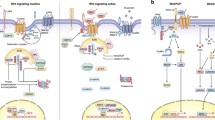
The canonical Wnt pathway is a series of double negatives, but, overall, binding of Wnts at the cell surface stabilizes cytosolic ß-catenin, which translocates to the nucleus to activate Wnt target gene expression. In the absence of Wnts, ß-catenin is targeted for proteasomal degradation by a complex of the scaffold Axin, casein kinase-1 (CK1), glycogen synthase kinase-3 (GSK-3), and adenomatous polyposis coli (APC) (Fig. 1). Sequential phosphorylation of ß-catenin by CK1 and GSK-3 leads to ß-TRCP dependent ubiquitylation and degradation of ß-catenin [16–18]. Additional complexity of the degradation complex is illustrated at the highly informative Wnt homepage referred to above (http://web.stanford.edu/group/nusselab/cgi-bin/wnt/). Binding of Wnts to a Frizzled family receptor and its co-receptor LRP5 or LRP6 causes recruitment of Axin to the membrane, phosphorylation of LRP, and subsequent inhibition of GSK-3-dependent phosphorylation of ß-catenin. Unphosphorylated β-catenin accumulates and translocates to the nucleus where it displaces the transcriptional repressor groucho/transducin-like enhancer of split (TLE) proteins from TCF/LEF (T-cell factor/lymphoid enhancer-binding factor) family transcription factors and thereby activates Wnt target genes [19, 20]. Several mechanisms for GSK-3 inhibition have been proposed, including rapid dissociation of APC from the Axin complex (APC enhances GSK-3 activity; dissociation impairs GSK-3 activity) [21, 22], direct inhibition of GSK-3 by the phosphorylated LRP5/6 C terminus [23], and sequestration of GSK3 into multivesicular endosomes [24]. Importantly, phosphorylation of the N terminus of GSK-3, as seen in response to receptor tyrosine kinase signaling, does not play a role in canonical Wnt signaling [25–27].

a The canonical Wnt/ß-catenin pathway and additional branches downstream of Axin/GSK-3. In the absence of Wnt (ligand), a destruction complex consisting of Axin, APC, GSK-3, and CK1 catalyzes sequential phosphorylation of ß-catenin, leading to ubiquitylation by ß-TRCP ubiquitin ligase and subsequent proteasomal degradation. Independently of ß-catenin, GSK-3 destabilizes other proteins that are regulated by Wnt-STOP (Wnt-stabilization of proteins) signaling and also suppresses mTORC1 and the Hippo pathway effectors YAP and TAZ. b Upon Wnt binding to Frizzled and LRP co-receptors, Axin is recruited to LRP and APC dissociates from the complex. GSK-3 phosphorylation of ß-catenin is inhibited; unphosphorylated ß-catenin accumulates, translocates to the nucleus, and binds to TCF, leading to Wnt target gene expression. Wnt-mediated inhibition of GSK-3 also activates mTORC1 (proliferation, growth, translation), YAP/TAZ (Hippo signaling), and Wnt-STOP (protein stabilization) independently of but in parallel with ß-catenin
Canonical Wnt/ß-catenin signaling typically induces target genes that encode feedback inhibitors of the pathway. These include secreted inhibitors such as Dickkopf 1 (Dkk1) [28] and intracellular inhibitors such as Axin2 [29] and Naked [30]. Dkk1 binds directly to LRP5/6 [31] and either inhibits formation of the ternary complex between LRP5/6, Frizzled, and Wnt or promotes internalization of LRP5/6 [32]. Wnt inhibitory factor1 (Wif1) on the other hand binds to and sequesters Wnts, thereby inhibiting both canonical and non-canonical signaling pathways [33]. Secreted frizzled-related proteins (Sfrp) resemble the ligand binding domain of Frizzleds and hence bind to and similarly sequester Wnts [34]. Axin2, similar to Axin1, inhibits the pathway by promoting ß-catenin turnover. Naked (naked cuticle) antagonizes canonical Wnt signaling in Drosophila and Xenopus [30], but its mechanism of inhibition is not well characterized [35].
Divergent Wnt Pathways
Most of the core components of the canonical Wnt pathway appear to be borrowed from other pathways, including metabolic regulators (e.g. CK1 and GSK-3), cell adhesion (ß-catenin), cell and tissue polarity (Frizzled and Disheveled), and mitotic spindle function (APC, GSK-3). Furthermore, Wnt signaling pathways can branch at multiple points, from Wnt interaction with non-canonical receptors to Disheveled, which regulates alternative pathways, to the Axin/APC complex, which regulates multiple signaling effectors apart from ß-catenin. Although the term “canonical Wnt signaling” often implies a ß-catenin-mediated response, this is perhaps more accurately termed Wnt/ß-catenin signaling; Wnts can also utilize “canonical” Wnt/Frizzled signaling through the Axin complex but then diverge from ß-catenin. These divergent pathways include the regulation of mitotic spindle polarity in C. elegans [36], activation of mammalian target of rapamycin complex 1 (mTORC1) signaling [37], regulation of the Hippo effectors YAP and TAZ, and the Wnt-STOP pathway, which regulates protein stability through GSK-3 phosphorylation [38•] (Fig. 1).
Canonical Wnt signaling inhibits GSK-3 activity [39–41] and, through GSK-3 inhibition, activates mTORC1 [37]. mTORC1 is a metabolic sensor also known for its regulation by nutrients and growth factor/receptor tyrosine kinase signaling pathways [42]. mTORC1 is inhibited by an upstream complex comprising the tuberous sclerosis complex (TSC) proteins TSC1 and TSC2. GSK-3 phosphorylates TSC2 and enhances its function. Hence, inhibition of GSK-3 by canonical Wnt signaling impairs TSC function to activate mTORC1, and this has an important effect on proliferative responses to canonical Wnt pathway activation, as described below [43, 44, 45•].
Canonical Wnt signaling also regulates the Hippo pathway, a highly conserved regulator of organ size. Similar to Wnt/ß-catenin signaling, Hippo signaling involves inhibition of protein kinases and stabilization of transcription activators YAP and TAZ. Although the proposed mechanisms of Wnt regulation differ in detail, as reported by different groups, in general, YAP/TAZ is destabilized by the Axin/APC complex, in part by phosphorylation by GSK-3. Thus, similar to ß-catenin, activation of canonical Wnt signaling or inhibition of GSK-3 stabilizes YAP and TAZ. YAP is required for Wnt/APC-dependent intestinal tumorigenesis [46•, 47•]. Park et al. showed that Wnt5a, acting through a non-canonical ROR/G-protein coupled mechanism, increases YAP/TAZ transcription, which then indirectly antagonizes canonical Wnt signaling by inducing secreted pathway inhibitors [48•].
Non-canonical Wnt signaling is a broad term that has been applied to a variety of pathways that employ Wnts and/or Frizzleds. The best characterized pathway genetically and phenotypically is the planar cell polarity pathway, which uses Frizzleds, Disheveled, and in some organisms, a subgroup of Wnt genes to control the polarity of cells within a planar tissue. This review will focus mainly on canonical Wnt signaling; for thorough and thoughtful reviews on non-canonical signaling see [49, 50].
Wnt Signaling in Stem Cells
Stem cells have two distinct properties: (1) self-renewal: the ability of stem cells to divide and generate new stem cells and (2) multilineage potential: the ability to differentiate into distinct cell types. In many cases, a distinct progenitor population (also called transit amplifying cells) exists that demonstrates a high rate of proliferation, early lineage commitment, and loss of the capacity for long-term self-renewal. These progenitor cells then give rise to a terminally differentiated cell. Regulation of this balance between self-renewal and differentiation is central to the maintenance and regeneration of tissues such as the hematopoietic system, epidermal, mammary, and gastrointestinal epithelial cells, mesenchymal derivatives, and specific populations of adult neurons. Canonical Wnt signaling has been strongly implicated in each of these contexts, and overactivation of Wnt signaling in at least some of these cell populations is an early step in carcinogenesis.
Embryonic stem cells (ESCs) are derived from the pluripotent cells of the inner cell mass of mammalian embryos. ESCs are selected in cell culture for indefinite self-renewal and maintenance of pluripotency, the ability to differentiate into all cells of the embryo and adult organism. ESCs have provided a powerful in vitro system for studying pluripotency, lineage specification, and tissue regeneration, as well as for the generation of genetically engineered animal models. Pluripotency is maintained in part by expression of pluripotency factors, Nanog [51], Sox2 [52], and Oct4 [53], which repress differentiation. Activation of Wnt signaling sustains the expression of these transcription factors maintaining the pluripotent state of ESCs [54] and supports stem cell expansion [55]. Knockout of the Wnt antagonists Gsk3a and Gsk3b (similar but distinct genes that encode GSK-3α and GSK-3β, respectively) constitutively activates Wnt signaling and similarly enhances expansion of pluripotent cells that have limited potential for differentiation unless GSK-3 activity is restored [27]. These findings led to the use of GSK-3 inhibitors (combined with a MEK inhibitor) to culture mouse ESCs in the absence of leukemia inhibitory factor (LIF) or support cells (termed 2i culture) [56]. APC mutations similarly increase β-catenin protein abundance and block differentiation of ESCs [57]. This effect of GSK-3 inhibition and ß-catenin stabilization may be mediated in part by a novel mechanism involving ß-catenin-dependent degradation of TCF3 (encoded by Tcf7l1) [58, 59].
Differentiation of pluripotent stem cells, such as human ESCs, to different cell types can be achieved by modulating Wnt/β-catenin signaling at different stages in culture. For example, activation of Wnt signaling leads to definitive endoderm formation and subsequent inhibition of Wnt signaling is required for foregut endoderm formation [60]. Reactivation of Wnt signaling at later stages, combined with inhibition of TGF-β and Notch signaling, promotes generation of hepatocytes [61].
Intestinal stem cells (ISCs) are present in the crypts of the small intestine and give rise to transit amplifying cells that ultimately differentiate into enterocyte, goblet cells, Paneth cells, and cells of enteroendocrine lineage [62]. Intestinal epithelial cells turnover at a high rate, and individual crypts generate as many as 200 new cells/day [11]. In the small intestine, two populations of cells are capable of generating new cells to populate the crypt: the crypt base columnar (CBC) cells, an actively cycling Lgr5+ population, and a reserve stem cell population at the +4 position (marked by Bmi1 and Hopx), which remains quiescent but is activated upon tissue damage [63, 64•]. Wnt3a secreted by neighboring Paneth cells is an essential niche signal that maintains the Lgr5+ cells [65]. Wnt3a is localized in the basolateral plasma membrane of the crypts, is transferred from Paneth cells to Lgr5+ cells, and is then retained by the Frizzled receptor. Wnts spread in a stem cell division-dependent manner in the crypt which creates a gradient, regulated by E3 ligases Rnf43/Znrf3 and Wnt signaling agonist, Lgr4–5/R-Spondin [66]. Similarly, loss of Wnt/β-catenin signaling impairs intestinal homeostasis by causing forced differentiation of stem cells [67]. Overall, these studies demonstrate the essential role of canonical Wnt signaling in maintenance of ISC homeostasis. For a more detailed discussion of Wnt signaling and intestinal epithelial homeostasis, see a recent review by Mah et al. [68].
Hematopoietic stem cells (HSCs) form all circulating blood cells and are capable of life-long self-renewal. Evidence for canonical Wnt signaling in HSC self-renewal has been well summarized in previous reviews [11, 69, 70] and includes evidence that (1) Wnts are expressed in HSCs and in hematopoietic stromal cells, (2) Wnt reporters are active in HSCs, (3) soluble Wnts, including Wnt3a, enhance ex vivo proliferation of HSCs [11, 71–74], (4) Wnt3a deficiency reduces the number and reconstitution capacity of fetal liver HSCs [75] and self-renewal of bone marrow HSCs [72], (5) inhibiting Wnt signaling impairs HSC proliferation ex vivo [71, 76–78], and (6) conditional knockout of ß-catenin at an early stage of HSC development (using vav-cre) impairs competitive repopulation of adult HSCs [9, 79].
However, the role of canonical Wnt signaling in HSC maintenance has been controversial. Knockout of ß-catenin and γ-catenin in adult HSCs (with mx1-cre) has no effect on HSC homeostasis in vivo [76, 80, 81], and overexpression of a stabilized form of ß-catenin diminishes repopulating activity and leads to hematopoietic failure in vivo [82, 83], observations that are difficult to reconcile with a requirement for canonical Wnt signaling in adult HSC self-renewal, at least for baseline HSC homeostasis.
However, these negative findings have led to intriguing counterarguments. An elegant study by the Staal group shows that the effect of Wnt signaling on HSC function is dosage dependent. Mild levels of Wnt activation (twofold) enhance the repopulating capacity of HSCs whereas a further increase in Wnt activity impairs HSC self-renewal. Intermediate levels of Wnt activation (fourfold) enhance myeloid differentiation potential whereas high levels of Wnt activation (22-fold) induce T-cell differentiation potential [84]. Thus, either complete absence or excessively high levels of Wnt activity impair HSC function [75, 84]. Furthermore, the observations that HSCs can respond to ectopic Wnt activators indicate that the signaling machinery is present in HSCs and can function “on demand,” perhaps under stress conditions such as transplant or radiation-induced injury repair [9].
In addition, we propose here a model that could reconcile the seemingly contradictory knockout data: Vav-cre deletes ß-catenin in embryonic HSCs whereas mx1-cre deletes ß-catenin in adult HSCs. As Wnt signaling can establish chromatin architecture at Wnt responsive regulatory elements that can be maintained epigenetically and read out at later stages [85], Wnt signaling could be essential for establishing responsive chromatin architecture during the ontogeny of HSCs that is maintained in adults and is essential for HSC homeostasis. As long as ß-catenin dependent chromatin marks deposited during development are maintained, ß-catenin may no longer be required in adult HSCs.
The role of canonical Wnt signaling in HSCs may also be complicated by the function of ß-catenin independent pathways, especially mTORC1. Inhibition of GSK-3, which mimics Wnt signaling, not only enhances HSC renewal (ß-catenin dependent) but also leads to an eventual loss of self-renewing cells due to activation of mTORC1 signaling [43, 44] (mTORC1 activation through loss of Pten also drives lineage commitment and HSC exhaustion [86, 87]). These antagonistic effects of Wnt activation could confound a simple interpretation of the role of canonical Wnt signaling in HSCs.
This dichotomous signaling activity can be exploited to improve ex vivo maintenance of HSCs. Thus, HSCs with long-term marrow repopulating ability can be maintained in a defined, cytokine-free, serum-free medium using inhibitors of GSK3 and mTOR [43, 44]. GSK3 inhibitors activate Wnt/β-catenin signaling, which maintains self-renewal of HSCs. As GSK-3 inhibition also activates mTORC1, addition of an mTOR inhibitor (rapamycin) prevents HSC exhaustion while preserving self-renewal. Valproic acid along with lithium (GSK3 inhibitor) also increases the repopulating potential of HSCs by increasing stem cell-related genes and repressing differentiation-related genes [88]. In zebrafish and murine HSCs, PGE2 enhances Wnt signaling by regulating the destruction of β-catenin in a cAMP/PKA dependent manner [89]. Pretreatment of human umbilical cord blood with PGE2 also enhances clinical engraftment in HSC transplants [90, 91].
Wnt Signaling in Cancer
Aberrant activation of Wnt signaling is associated with multiple cancers, including colon, breast, hepatocellular, endometrial, ovarian, and anaplastic thyroid cancers [1, 92]. The first mammalian Wnt gene to be identified, int-1/Wnt1, was discovered in an insertional mutagenesis screen for genes that confer a transformed growth phenotype on mammalian breast epithelial cells in culture. The subsequent demonstration that loss of function mutations in APC associated with familial and sporadic colorectal cancer causes stabilization of ß-catenin and activation of Wnt target genes established the Wnt pathway as key mechanism in carcinogenesis [93].
Familial adenomatous polyposis (FAP) is caused by truncating mutations in the APC gene. Heterozygotes develop multiple colonic polyps due to loss of heterozygosity that for unclear reasons favors growth of colonocytes in humans. Virtually, all patients with FAP will develop colorectal cancer unless the colon is removed. Somatic truncation mutations are also found in more than 80 % of sporadic colorectal cancers. Oncogenic APC mutations cluster in the mutation cluster region (MCR) and result in premature truncations that delete the Axin interaction domain and the domain that confers turnover of ß-catenin. These truncating mutations cause stabilization of ß-catenin and constitutive activation of Wnt target genes. Conversely, overexpression of a region that only contains the ß-catenin regulatory and Axin interaction domains of APC is sufficient to reduce ß-catenin protein levels, Wnt target gene expression, and multiple APC loss of function phenotypes in vivo [94–96]. Of the ~20 % of sporadic colorectal cancers (CRCs) that have intact APC genes, many contain mutations in the N-terminal phosphorylation sites in ß-catenin that mediate proteosomal degradation, a compelling genetic argument that APC suppresses intestinal neoplasia through inhibition of the canonical Wnt/ß-catenin pathway.
A working hypothesis is that aberrant activation of Wnt signaling in ISCs leads to expansion of ISC-like cells. Deletion of Apc in Lgr5+ cells leads to tumorigenesis while deletion in Lgr5− cells does not; although under certain conditions associated with inflammation, Lgr5− cells may be able to dedifferentiate into Lgr5+ stem cells and then contribute to neoplastic transformation (as cited in [92]).
APC deletion is not sufficient for carcinogenesis, however. APC mutations are an early step in neoplasia, leading to increased proliferation and microadenomas, but additional mutations, for example in p53, PTEN, K-Ras, PIK3CA, and SMAD4 [92], are required for carcinogenesis. Furthermore, nuclear ß-catenin is reportedly not detectable in early adenomas associated with APC mutations, despite increased cytosolic ß-catenin [97–99]. While it is possible that the immune detection methods were not sensitive enough to detect nuclear ß-catenin, it is also possible that an alternative pathway downstream of APC mediates the early proliferative response. For example, mTORC1 is activated by loss of APC, and this could contribute to the early proliferative response [45•]. Indeed, mTORC1 inhibitors reduce both size and number of adenomas in mice bearing truncating mutations in Apc [100–105]; mTORC1 inhibitors also reverse many of the developmental defects seen in apc mutant zebrafish [45•].
Two mechanisms for mTORC1 activation by APC mutation have been proposed: mTORC1 is activated by the reduced GSK-3 activity associated with loss of APC, as observed in Apc mutant mice, zebrafish [21, 45•], and adenomas from FAP patients (PSK, unpublished observations). mTOR is also a Wnt target gene and expression of the kinase is increased by loss of APC [100, 102]. Activation of mTORC1 in the setting of APC mutations suggests an intriguing parallel between FAP and other familial syndromes that are similarly associated with mTOR activation, including Peutz-Jeghers syndrome, Cowden’s syndrome, and tuberous sclerosis.
The Wnt target genes c-Myc and Ccnd1 (encodes cyclin D1) are important effectors of Wnt dependent proliferation in CRC cells in vitro and in adenomatous lesions in vivo, as deletion of c-Myc blocks tumor formation in the setting of Apc mutations in mice and loss of ccnd1/cyclin D1 (as well as cyclin D2) reduces the frequency of adenomas [106–109]. As discussed above, the Hippo pathway gene YAP is also required for intestinal tumorigenesis after Apc disruption.
Wnt Signaling in Leukemia
Acute myeloid leukemia (AML) is a disease affecting white blood cells causing an abnormal accumulation of myeloid blasts at the expense of normal blood cells, anemia, thrombocytopenia, and ultimately death in about 40 % of patients. Activating mutations in canonical Wnt pathway components are rare in human leukemias and were not described in the cancer genome atlas (TCGA) analysis of somatic mutations in AML [110]. However, considerable evidence has accrued supporting a role for Wnt signaling in AML cells from patients and in experimental models of acute and chronic myelogenous leukemias and myelodyplastic syndromes. Thus, activation of the pathway may contribute to leukemia pathogenesis as a downstream consequence of known AML-associated mutations, such as Flt3-ITD or chromatin modifiers, or indirectly through mutations that occur in stromal cells. Here, we will discuss evidence supporting a role for Wnt signaling in AML and CML. An in-depth review of the topic can be found in [69, 70].
Combined overexpression of Hoxa9 and Meis1 in hematopoietic stem and progenitor cells (HSPCs) in mice leads to AML and an associated increase in the level of unphosphorylated (“activated”) ß-catenin in leukemic granulocyte-macrophage progenitors (L-GMPs). Induction of AML was blocked by ß-catenin knockout indicating that ß-catenin is required for this leukemic transformation. Furthermore, overexpression of Hoxa9 and Meis1 did not induce AML in more committed GMPs unless they were also co-expressed with a stabilized form of ß-catenin [111, 112]. This work demonstrates the requirement for ß-catenin in an experimentally induced murine AML; however, ß-catenin also functions in cell adhesion as a binding partner of cadherins, and further work will be needed to demonstrate that the Wnt/ß-catenin pathway per se is involved in the pathogenesis of AML, for example by disrupting other components of the pathway, and a requirement for Wnt/ß-catenin signaling in human AML has not yet been demonstrated.
Flt3-ITD, one of the most common mutations associated with AML, activates Wnt/ß-catenin signaling in a leukemia cell line and in primary AML cells [113]. The mechanism remains unclear but may involve increased expression of Frizzled-4 and/or ß-catenin protein induced by FLT3-ITD. Although Flt3-ITD activates AKT, AKT phosphorylation of GSK-3 does not contribute to canonical Wnt signaling, so an alternative pathway likely plays a role in this setting.
Kousteni and co-workers elegantly showed that conditional activation of ß-catenin signaling within the osteoblast lineage in mice causes clonal expansion of hematopoietic cells leading to AML (through increased expression of the Notch ligand Jagged-1 in osteoblasts), along with chromosomal breaks/translocations analogous to chromosomal translocations frequently observed in human AML. They also found increased Wnt/ß-catenin signaling in osteoblasts and increased Notch signaling in hematopoietic cells in patients with MDS or AML [114•]. These data support a role for Wnt signaling in niche cells that may have been overlooked in TCGA sequencing analysis.
Myelodysplasia (MDS) is a clonal hematopoietic disorder that often progresses to AML. MDS is characterized by hypercellular bone marrow (in most cases), dysplastic morphology, and reduced peripheral blood counts. The APC gene is present in a region of human chromosome 5q that is frequently deleted in MDS and in AML. Haploinsufficiency for Apc in mice increased engraftment in primary transplants but impaired repopulation in secondary transplants and led to an MDS/myeloproliferative phenotype [115, 116]. These findings are similar to the effect of GSK-3 inhibition or depletion in hematopoietic cells. Gsk3 knockdown [43, 44] or conditional knockout [117•] of Gsk3b causes expansion of HSPCs and an increase in mature granulocytes; complete KO of Gsk3a and Gsk3b causes a markedly hyperproliferative phenotype with increased frequency of blasts in the marrow and an abundance of mature granulocytes in peripheral blood and tissues [117•]. The mechanism downstream of GSK-3 in these settings is in part through Wnt/ß-catenin signaling as well as a parallel activation of mTORC1 [43, 44], although contribution from other effectors of GSK-3 seems likely as well.
Wnt/ß-catenin signaling has also been proposed to play a role in CML [79, 118], ALL [119], and CLL [69, 70]. For example, GMPs from patients with CML in blast crisis or patients with imatinib resistant CML have elevated levels of unphosphorylated ß-catenin [118]. Furthermore, missplicing of GSK3Β leading to an in-frame deletion of exons 8 and 9 was observed in 4 of 7 patients with CML in blast crisis. This misspliced form was also associated with increased ß-catenin protein levels and enhanced serial engraftment, both of which were restored to normal levels by overexpression of wild-type GSK3B [120]. Furthermore, deletion of β-catenin synergizes with imatinib to delay recurrence of CML [121]. These findings again suggest a correlation between increased ß-catenin protein and aggressive growth of CML and are consistent with a role for canonical Wnt/ß-catenin signaling in CML, but additional work will be required to prove a requirement for the Wnt pathway per se, as opposed to other functions of ß-catenin.
Conclusions
Wnt signaling plays important roles in regulating stem cell homeostasis in health and disease. New insights into the pathway have revealed additional complexity to the regulation and output of the pathway at multiple steps. Our understanding of the logic of the canonical Wnt pathway, worked out from genetic and biochemical analyses in diverse model organisms, has developed from a linear pathway to a branching network of multiple downstream effectors in addition to, and independent of ß-catenin, including mTOR and Hippo signaling. These new findings indicate that canonical Wnt signaling comprises multiple pathways, even without considering planar cell polarity and other non-canonical Wnt pathways.
Dysregulation of the canonical Wnt pathway can be catastrophic, as overactivation causes colorectal carcinoma, multiple other epithelial cancers, and likely plays a significant role in hematopoietic neoplasms. However, extrinsic modulation of Wnt signaling also offers an opportunity for therapeutic interventions, some of which are already in clinical trials. Inhibition of Wnt signaling is being explored to treat malignancies, and this approach is advancing rapidly as new “druggable” targets are discovered, including upstream inhibitors of Wnt ligand generation and function as well as downstream inhibitors of ß-catenin stability. Small molecule control of Wnt signaling is also at various stages of development for the promotion of stem cell self-renewal, expansion, and engraftment. Even small increases in HSCs in umbilical cord blood, for example, could revolutionize the clinical use of HSC transplantation by making this therapeutic modality available to many more adults in need of stem cell transplants. Small molecule approaches that maintain stem cell capacity for self-renewal will also be important factors for ex vivo manipulation of somatic stem cells, for example to allow genome editing without concomitant differentiation and loss of long-term self-renewal during ex vivo manipulations. Hence, further studies on how to control the level of Wnts and Wnt signaling components in primary patient samples will remain a critical area of future study.
References
Papers of particular interest, published recently, have been highlighted as:• Of importance
Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14(15):1837–51.
Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205.
Sharma RP, Chopra VL. Effect of the wingless (wg1) mutation on wing and haltere development in Drosophila melanogaster. Dev Biol. 1976;48(2):461–5.
Nusse R et al. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature. 1984;307(5947):131–6.
Rijsewijk F et al. The drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell. 1987;50(4):649–57.
Malhotra S, Kincade PW. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell. 2009;4(1):27–36.
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127(3):469–80.
Wend P et al. Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol. 2010;21(8):855–63.
Lento W et al. Loss of beta-catenin triggers oxidative stress and impairs hematopoietic regeneration. Genes Dev. 2014;28(9):995–1004.
Lien WH, Fuchs E. Wnt some lose some: transcriptional governance of stem cells by Wnt/beta-catenin signaling. Genes Dev. 2014;28(14):1517–32.
Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–50.
Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26.
Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13(7):513–32.
Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014;346(6205):1248012.
Lento W et al. Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb Perspect Biol. 2013;5(2).
Hart M et al. The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr Biol. 1999;9(4):207–10.
Kitagawa M et al. An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of beta-catenin. EMBO J. 1999;18(9):2401–10.
Winston JT et al. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999;13(3):270–83.
Behrens J et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382(6592):638–42.
Molenaar M et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86(3):391–9.
Valvezan AJ et al. Adenomatous polyposis coli (APC) regulates multiple signaling pathways by enhancing glycogen synthase kinase-3 (GSK-3) activity. J Biol Chem. 2012;287(6):3823–32.
Tran H, Polakis P. Reversible modification of adenomatous polyposis coli (APC) with K63-linked polyubiquitin regulates the assembly and activity of the beta-catenin destruction complex. J Biol Chem. 2012;287(34):28552–63.
Piao S et al. Direct inhibition of GSK3beta by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta-catenin signaling. PLoS One. 2008;3(12):e4046.
Taelman VF et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143(7):1136–48.
Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem. 2000;275(42):32475–81.
McManus EJ et al. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24(8):1571–83.
Doble BW et al. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12(6):957–71.
Bafico A et al. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/arrow. Nat Cell Biol. 2001;3(7):683–6.
Jho EH et al. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22(4):1172–83.
Zeng W et al. Naked cuticle encodes an inducible antagonist of Wnt signalling. Nature. 2000;403(6771):789–95.
Glinka A et al. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;391(6665):357–62.
Mao B et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 2002;417(6889):664–7.
Hsieh JC et al. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999;398(6726):431–6.
Lin K et al. The cysteine-rich frizzled domain of Frzb-1 is required and sufficient for modulation of Wnt signaling. Proc Natl Acad Sci U S A. 1997;94(21):11196–200.
Wharton Jr KA et al. Vertebrate proteins related to Drosophila Naked cuticle bind Dishevelled and antagonize Wnt signaling. Dev Biol. 2001;234(1):93–106.
Rocheleau CE et al. Wnt signaling and an APC-related gene specify endoderm in early C. elegans embryos. Cell. 1997;90(4):707–16.
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–90.
Acebron SP et al. Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol Cell. 2014;54(4):663–74. Wnt/STOP signaling regulates protein stability and cell size in a β-catenin independent manner.
Cook D et al. Wingless inactivates glycogen synthase kinase-3 via an intracellular signalling pathway which involves a protein kinase C. EMBO J. 1996;15(17):4526–36.
Ruel L et al. Functional significance of a family of protein kinases encoded at the shaggy locus in Drosophila. EMBO J. 1993;12(4):1657–69.
Ruel L et al. Regulation of the protein kinase activity of shaggy(Zeste-white3) by components of the wingless pathway in Drosophila cells and embryos. J Biol Chem. 1999;274(31):21790–6.
Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(Pt 20):3589–94.
Huang J et al. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat Med. 2012;18(12):1778–85.
Huang J et al. Pivotal role for glycogen synthase kinase-3 in hematopoietic stem cell homeostasis in mice. J Clin Invest. 2009;119(12):3519–29.
Valvezan AJ et al. Oncogenic mutations in adenomatous polyposis coli (Apc) activate mechanistic target of rapamycin complex 1 (mTORC1) in mice and zebrafish. Dis Model Mech. 2014;7(1):63–71. mTORC1 is activated by Apc mutations independently of ß-catenin in colorectal cancer.
Azzolin L et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell. 2014;158(1):157–70.
Cai J et al. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010;24(21):2383–8.
Park HW et al. Alternative Wnt signaling activates YAP/TAZ. Cell. 2015;162(4):780–94. References 46-48 show that YAP/TAZ functions downstream of APC in intestinal crypt overgrowth and regeneration.
Wallingford JB, Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011;25(3):201–13.
Minami Y et al. Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: their implications in developmental morphogenesis and human diseases. Dev Dyn. 2010;239(1):1–15.
Mitsui K et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113(5):631–42.
Ivanova N et al. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442(7102):533–8.
Nichols J et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95(3):379–91.
Sato N et al. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10(1):55–63.
Corrigan PM et al. Patterns of Wnt/Fzd/LRP gene expression during embryonic hematopoiesis. Stem Cells Dev. 2009;18(5):759–72.
Ying QL et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453(7194):519–23.
Kielman MF et al. Apc modulates embryonic stem-cell differentiation by controlling the dosage of beta-catenin signaling. Nat Genet. 2002;32(4):594–605.
Shy BR et al. Regulation of Tcf7l1 DNA binding and protein stability as principal mechanisms of Wnt/beta-catenin signaling. Cell Rep. 2013;4(1):1–9.
Wray J et al. Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat Cell Biol. 2011;13(7):838–45.
Hoepfner J et al. Biphasic modulation of Wnt signaling supports efficient foregut endoderm formation from human pluripotent stem cells. Cell Biol Int. 2016;40(5):534–48.
Touboul, T., et al., Stage-specific regulation of the WNT/beta-catenin pathway enhances differentiation of hESCs into hepatocytes. J Hepatol. 2016.
Marshman E, Booth C, Potten CS. The intestinal epithelial stem cell. BioEssays. 2002;24(1):91–8.
Clevers H. Stem cells. What is an adult stem cell? Science. 2015;350(6266):1319–20.
Li N et al. Single-cell analysis of proxy reporter allele-marked epithelial cells establishes intestinal stem cell hierarchy. Stem Cell Reports. 2014;3(5):876–91. This study provides evidence for presence of two distict populations of intestinal stem cells.
Sato T et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–8.
Farin HF et al. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature. 2016;530(7590):340–3.
Fevr T et al. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol. 2007;27(21):7551–9.
Amanda Mah, T., Kelley Yan, S., and C. J. Kuo. Wnt pathway regulation of intestinal stem cells. The Journal of Physiology, 2016.
McCubrey JA et al. Multifaceted roles of GSK-3 and Wnt/beta-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia. 2014;28(1):15–33.
Staal FJ, Clevers HC. WNT signalling and haematopoiesis: a WNT-WNT situation. Nat Rev Immunol. 2005;5(1):21–30.
Reya T et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409–14.
Willert K et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423(6938):448–52.
Austin TW et al. A role for the Wnt gene family in hematopoiesis: expansion of multilineage progenitor cells. Blood. 1997;89(10):3624–35.
Van Den Berg DJ et al. Role of members of the Wnt gene family in human hematopoiesis. Blood. 1998;92(9):3189–202.
Luis TC et al. Wnt3a deficiency irreversibly impairs hematopoietic stem cell self-renewal and leads to defects in progenitor cell differentiation. Blood. 2009;113(3):546–54.
Jeannet G et al. Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood. 2008;111(1):142–9.
Schaniel C et al. Wnt-inhibitory factor 1 dysregulation of the bone marrow niche exhausts hematopoietic stem cells. Blood. 2011;118(9):2420–9.
Renstrom J et al. Secreted frizzled-related protein 1 extrinsically regulates cycling activity and maintenance of hematopoietic stem cells. Cell Stem Cell. 2009;5(2):157–67.
Zhao C et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12(6):528–41.
Cobas M et al. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199(2):221–9.
Koch U et al. Simultaneous loss of beta- and gamma-catenin does not perturb hematopoiesis or lymphopoiesis. Blood. 2008;111(1):160–4.
Scheller M et al. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol. 2006;7(10):1037–47.
Kirstetter P et al. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7(10):1048–56.
Luis TC et al. Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell. 2011;9(4):345–56.
Blythe SA et al. Beta-catenin primes organizer gene expression by recruiting a histone H3 arginine 8 methyltransferase, Prmt2. Dev Cell. 2010;19(2):220–31.
Yilmaz OH et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441(7092):475–82.
Zhang J et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441(7092):518–22.
Walasek MA et al. The combination of valproic acid and lithium delays hematopoietic stem/progenitor cell differentiation. Blood. 2012;119(13):3050–9.
Goessling W et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136(6):1136–47.
Cutler C et al. Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood. 2013;122(17):3074–81.
Goessling W et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011;8(4):445–58.
Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26(3):570–9.
Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–70.
Munemitsu S et al. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci U S A. 1995;92(7):3046–50.
Shih IM et al. The beta-catenin binding domain of adenomatous polyposis coli is sufficient for tumor suppression. Cancer Res. 2000;60(6):1671–6.
Hurlstone AF et al. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003;425(6958):633–7.
Anderson CB, Neufeld KL, White RL. Subcellular distribution of Wnt pathway proteins in normal and neoplastic colon. Proc Natl Acad Sci U S A. 2002;99(13):8683–8.
Blaker H et al. Analysis of somatic APC mutations in rare extracolonic tumors of patients with familial adenomatous polyposis coli. Genes Chromosomes Cancer. 2004;41(2):93–8.
Kobayashi M et al. Nuclear translocation of beta-catenin in colorectal cancer. Br J Cancer. 2000;82(10):1689–93.
Fujishita T et al. Inhibition of the mTORC1 pathway suppresses intestinal polyp formation and reduces mortality in ApcDelta716 mice. Proc Natl Acad Sci U S A. 2008;105(36):13544–9.
Koehl GE et al. Rapamycin inhibits oncogenic intestinal ion channels and neoplasia in APC(Min/+) mice. Oncogene. 2010;29(10):1553–60.
Metcalfe C et al. Dvl2 promotes intestinal length and neoplasia in the ApcMin mouse model for colorectal cancer. Cancer Res. 2010;70(16):6629–38.
Hardiman KM et al. Rapamycin inhibition of polyposis and progression to dysplasia in a mouse model. PLoS One. 2014;9(4):e96023.
Wang XW, Zhang YJ. Targeting mTOR network in colorectal cancer therapy. World J Gastroenterol. 2014;20(15):4178–88.
Faller WJ et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature. 2015;517(7535):497–500.
Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev. 2005;19(8):877–90.
Cole AM et al. Cyclin D2-cyclin-dependent kinase 4/6 is required for efficient proliferation and tumorigenesis following Apc loss. Cancer Res. 2010;70(20):8149–58.
He TC et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–12.
Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422–6.
Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med, 2013. 368(22): p. 2059–74.
Wang Y et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327(5973):1650–3.
Thorsteinsdottir U et al. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol Cell Biol. 2001;21(1):224–34.
Tickenbrock L et al. Flt3 tandem duplication mutations cooperate with Wnt signaling in leukemic signal transduction. Blood. 2005;105(9):3699–706.
Kode A et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506(7487):240–4. Activating mutations in β-catenin within the niche (osteoblasts) induce AML.
Lane SW et al. The Apc(min) mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS. Blood. 2010;115(17):3489–97.
Wang J et al. Haploinsufficiency of Apc leads to ineffective hematopoiesis. Blood. 2010;115(17):3481–8.
Guezguez B et al. GSK3 deficiencies in hematopoietic stem cells initiate pre-neoplastic state that is predictive of clinical outcomes of human acute leukemia. Cancer Cell. 2016;29(1):61–74. Combined deletion of Gsk3a and Gsk3b causes myeloproliferative/ myelodysplastic state and possible AML in mice.
Jamieson CH et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–67.
Guo W et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature. 2008;453(7194):529–33.
Abrahamsson AE et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci U S A. 2009;106(10):3925–9.
Heidel FH et al. Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10(4):412–24.
Acknowledgments
The authors acknowledge support from NIH grants 1R01HL110806, 1R01MH100923, and 1R24DK103001. We also receive support relevant to this work from the Institute for Regenerative Medicine, the Institute for Translational Research and Applied Therapeutics, and the Orphan Disease Center at the Perelman School of Medicine at the University of Pennsylvania. We thank Dr. Rachit Badolia, Temple University, for his help with Fig. 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dheeraj Bhavanasi and Peter S. Klein declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Role of Classical Signaling Pathways in Stem Cell Maintenance
Rights and permissions
About this article

Cite this article
Bhavanasi, D., Klein, P.S. Wnt Signaling in Normal and Malignant Stem Cells. Curr Stem Cell Rep 2, 379–387 (2016). https://doi.org/10.1007/s40778-016-0068-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-016-0068-y