
Abstract
Hematopoiesis is characterized by a lifelong balance between hematopoietic stem cell (HSC) self-renewal and differentiation into mature blood populations. Proper instruction of cell fate decisions requires tight homeostatic regulation of transcriptional programs through a combination of epigenetic modifications, management of cis-regulatory elements, and transcription factor activity. Recent work has focused on integrating biochemical, genetic, and evolutionary data sets to gain further insight into these regulatory components. Long noncoding RNA (lncRNA), post-translational modifications of transcription factors, and circadian rhythm add additional layers of complexity. These analyses have provided a wealth of information, much of which has been made available through public databases. Elucidating the regulatory processes that govern hematopoietic transcriptional programs is expected to provide useful insights into hematopoiesis that may be applied broadly across tissue types while enabling the discovery and implementation of therapeutics to treat human disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adult hematopoiesis is sustained throughout an organism’s lifetime by a reservoir of quiescent hematopoietic stem cells (HSCs) that have the capacity to self-renew. HSC produce a series of progenitor subsets that ultimately give rise to all mature blood populations. Proper function of the hematopoietic system inherently demands a balance between these three compartments. Balanced production of blood is achieved by maintaining a tight control over transcriptional programs in each cell type. These transcriptional programs are governed through sophisticated coordination of multiple layers of complexity; including epigenetics, DNA regulatory elements (cis-regulation), and transcription factors (trans-regulation). While these regulatory nodes are known to be important, how they are initiated and maintained and the consequences of their deregulation remain to be fully understood.
Developing an understanding of the mechanisms that underlie HSC biology and lineage differentiation programs will not only aid in elucidating the nature of hematopoiesis but also offer a therapeutic opportunity for ameliorating a myriad of hematopoietic diseases. Recent advances have enabled the production of hematopoietic progenitors in vitro [1, 2], including the discovery and implementation of inducible pluripotent stem cells (iPSCs) [3•]. Further refinement of iPSC technology in combination with the growing utilization of genome editing (CRISPR/Cas9, TALENS, etc.) presents the possibility to generate genetically corrected HSCs for autologous delivery back to the patient to cure disease. Thus, understanding the transcriptional programs that dictate HSC self-renewal or differentiation is essential to developing iPSC-derived cellular therapeutics.
In this review, we discuss recent discoveries that further define how HSCs and differentiation programs are transcriptionally regulated. The focus is on epigenetics and cis- and trans-regulation with special highlights on recent findings related to long noncoding RNAs (lncRNAs) and circadian rhythm.
Epigenetics and Epigenetic Modifiers in Hematopoiesis
Epigenomics is the study of genome-wide heritable traits that influence the transcription of genes which are regulated by mechanisms apart from changes in the underlying genetic code. The discovery and wide implementation of high-throughput massively parallel DNA sequencing has revolutionized the field of biological research, especially that of epigenomics. Epigenetic traits include cytosine methylation (hereafter referred to as DNA methylation), post-translational histone modifications, inclusion of histone variants, and histone positioning/occupancy. Several large collaborative initiatives have been rallied to integrate a large number of human epigenomic data sets from healthy and diseased individuals with the goal of generating reference epigenomes for widespread use by the scientific community. While the Roadmap Project integrates data sets from all tissue types [4], the BLUEPRINT Consortium was founded with the specific goal of generating human reference genomes for the research of blood-based diseases. To date, BLUEPRINT has published five data sets with integrated genome-wide epigenetic analyses from a range of primary hematopoietic cell types and in a variety of contexts. Their collaboration has recently yielded high-impact results that have significant translational potential [5–7].
DNA Methylation in Hematopoiesis
DNA methylation provides an important and reversible gene regulatory mechanism for protein-coding and noncoding genes (e.g., lncRNA) during hematopoiesis [8••]. Methyl residues are added to cytosines by a family of proteins called DNA methyltransferases, and each member performs unique functions. The loss of the de novo DNA methyltransferase, Dnmt3a, blocks HSC differentiation [9] and predisposes leukemic transformation [10]. Alternatively, the loss of the maintenance methyltransferase, Dnmt1, impairs HSC self-renewal by promoting myeloid/erythroid differentiation [11] while blunting leukemogenesis [12]. While studies have analyzed global DNA methylation in multipotent and lineage-restricted progenitors [13], the methylome in HSCs has not been previously discovered. Through the use of tagmentation-based whole-genome bisulfite sequencing (TWGBS), Cabezas-Wallscheid et al. identified over 15,000 differentially methylated regions (DMRs) during the early stages of HSC differentiation [8••]. Integrating gene expression and DNA methylation data provides insight into elucidating the regulatory mechanisms that lead to the differential expression of key genes at the early stages of HSC differentiation. Although there is an overall loss of methylation from the HSC to the MPP1 stage, methylation increased as cells progressed to the more lineage-primed multipotent progenitor populations. These methylation trends are unidirectional (i.e., if methylation of a locus increases from one cell type to the next, it continues this trend as differentiation progresses). A majority of DMRs (52.8 %) overlap with known regulatory elements [14, 15] (e.g., DNase hypersensitive sites, promoters, enhancers, and/or known transcription factor binding sites), and comparing gene expression and DNA methylation levels at individual loci uncovered a strong anticorrelation between the two. These results indicate that DNA methylation represents a regulatory mechanism of cis-regulatory elements in order to modulate the gene expression of a large number of genes in early hematopoietic progenitors. It is possible that during early HSC differentiation, genes involved in proliferation and broad lineage decision programs are initially launched in the MPP1 population through demethylation of the cis-regulatory elements of key genes (e.g., Sfpi1 and Gata2); however, once a cell decides to become primed for a specific lineage, genes involved in the opposing lineage are shut off permanently through the methylation of their cis-regulatory elements. It is clear that integrating gene expression and DNA methylation data provides insight into elucidating the regulatory mechanisms that lead to the differential expression of key genes at the early stages of HSC differentiation. Since DNA methylation has been shown to be dynamically regulated during the later stages of differentiation in multiple lineages [16–18], this integrated approach will also serve as an important step toward understanding key lineage decision events downstream of HSCs as well.
While most studies to date have focused on 5-methylcytosine modifications, 5-hydroxymethylcytosine (5hmc) residues also appear to play a role in regulating transcription, as it is enriched at enhancers and known transcription factor binding sites [19]. Conversion of 5-methylcytosine to 5hmc is mediated by the TET proteins, which is required for normal myeloid [20] and erythroid [21] differentiation. While the exact mechanism of gene regulation mediated by 5hmc is not known, it may serve as an intermediate that facilitates demethylation of cytosines at select loci [22]. 5hmc is further implicated in transcriptional regulation, since changes in 5hmc levels exhibit a stronger correlation with gene expression than 5-methylcytosine at some loci. 5hmc exhibits a positive correlation with gene expression, further setting it apart from 5-methylcytosine modifications [23]. With the ability to specifically identify 5hmc [24] from 5-methylcytosine [25] residues, further classification of the targets and mechanisms of 5hmc-mediated regulation in comparison to 5-methylcytosine modifications will certainly reveal new regulatory pathways in hematopoiesis.
Long Noncoding RNAs in Hematopoiesis
While genes are classically viewed as coding for messenger RNAs that are translated into proteins, a large portion of genes code for RNAs that do not result in the production of protein products, termed noncoding RNAs. One subset of noncoding RNAs that are growing in recognition within the field are long noncoding RNAs (lncRNAs), some of which have recently been found to serve as individual biomarkers in human hematologic malignancy [26]. LncRNAs have also been implicated as regulators of normal hematopoietic development. Transcriptional comparison between HSCs and multipotent progenitors (MPP1-4) identified 79 lncRNAs that are differentially expressed in the early stages of hematopoietic differentiation [8••]. Of these, 12 were significantly enriched in HSCs, although none have yet been functionally analyzed in hematopoiesis. Additionally, 14 lncRNAs were enriched in the more self-renewing HSC/MPP1 cluster, one of which was H19. This lncRNA is part of the imprinted gene network (IGN) that has been implicated in the regulation of embryonic growth [27], and deletion of other members of this pathway impairs HSC self-renewal [28, 29]. Further investigation of the entire IGN family showed that it is significantly enriched in HSCs and their expression levels decrease during differentiation, strongly implicating the IGN family in the regulation of HSC self-renewal and quiescence. It is clear that lncRNAs are involved in normal and malignant hematopoiesis, and the elucidation of their function will be critical to the understanding of how blood development is regulated.
LncRNAs regulate multiple cellular processes, including influencing alternative splicing [30], acting as microRNA sponges, and encoding endogenous siRNAs [31]. Additionally, recent evidence suggests that lncRNAs are capable of influencing gene expression by binding DNA at individual loci to recruit epigenetic/trans-activating proteins or by facilitating long-range DNA interactions. Zhang et al. developed a method to detect RNA-DNA interactions, such as those made between lncRNAs and DNA [32]. This procedure, termed RNA-guided chromosome conformation capture (R3C), involves fixation and isolation of genomic DNA complexes, reverse transcription of the RNA using biotinylated nucleotides, restriction enzyme digest to fragment DNA, and ligation of the newly formed cDNA to the bound genomic DNA. Isolation of the biotinylated complexes can then be analyzed using PCR-based detection or sequencing to determine the genomic locus that is bound by the lncRNA. This method was first utilized to identify a mechanism through which the lncRNA Kcnq1ot1 silences the Kcnq1 locus. Kcnq1ot1 was found to bind the Kcnq1 locus to form an intrachromosomal loop that recruits Ezh2 to silence the locus. The R3C method was also utilized to identify RUNXOR, a 216-kb intragenic lncRNA that interacts with the RUNX1 promoter [33]. Transcribed from a unique promoter located upstream of the RUNX1-P1 promoter, RUNXOR transcripts extend the entirety of the RUNX1 coding sequence, remain unspliced, and are upregulated in AML patient bone marrow cells. RUNXOR interacts with RUNX1 cis-regulatory regions to facilitate long-range intrachromosomal loops, and directly binds to RUNX1 and EZH2, recruiting them to regulate RUNX1. Furthermore, RUNXOR was found to associate with the genomic loci of common RUNX1 translocation partners, EVI1, and ETO, implicating this lncRNA in the malignant translocation process. Alternatively, some lncRNAs work to counteract cancer through epigenetic mechanisms. The promoter of the TCF21 tumor suppressor has been silenced through hypermethylation in a number of solid tumors. Arab et al. found that TARID, a lncRNA transcribed from the TCF21 locus, promotes its expression by mediating recruitment of TET family DNA demethylation enzymes [34]. These studies highlight the diverse and complex nature of gene regulation facilitated by lncRNAs and merit further in-depth investigation of lncRNAs during hematopoiesis.
Although low evolutionary conservation makes analysis difficult for most lncRNAs, some are conserved among mammals [35]. Paralkar et al. recently described over a thousand murine and roughly 600 human lncRNAs expressed throughout mega-erythroid differentiation, but only 15 % of these are conserved [36]. This lack of conservation does not indicate that they are nonfunctional, however, as the majority of lncRNAs analyzed in a functional screen were found to be necessary for erythroid differentiation. These findings expand upon the work of Alvarez-Dominguez et al., who compiled an online database of over 600 lncRNAs that are differentially expressed during murine fetal liver erythropoiesis [37]. Disruption of 12 of these arrested erythroblast maturation, of which half exhibited conservation in humans. While these studies further implicate lncRNAs as controllers of differentiation, they also uncover that conservation and function do not always go hand in hand. This insight will be important to keep in consideration while the murine model system is used to elucidate the in vivo functions of more of these conserved lncRNAs, an approach that will be critical to furthering our understanding of transcriptional regulation in human hematopoiesis.
The Role of DNA Regulatory Elements and Epigenetic Modification in Cis-Regulation of Transcription
Now that the human genome has been successfully sequenced, the focus has turned to the study of functional DNA elements. By using biochemical, evolutionary, and genetic approaches, the Encyclopedia of DNA Elements (ENCODE) has generated a vast number of data sets of RNA transcripts, transcription factor binding sites, and chromatin states in individual cell types that can be used to study human disease [38]. Their results exemplify the need to utilize multiple complementary approaches in order to elucidate how the genome functions in human biology and disease.
DNA elements that serve as regulatory nodes that are involved in modulating the expression of surrounding genes can be subdivided based upon their roles and classified as either promoters or enhancers. While promoters are located in close proximity to a gene’s transcriptional start site and are involved in the recruitment of the core transcriptional machinery to drive gene expression, enhancers are somewhat more loosely defined and are not necessarily near the genes they regulate. Moreover, enhancers can be located several kilobases away and even reside in the coding regions of neighboring genes. Specific epigenetic marks have been identified as markers of promoters (H3K4me3) and enhancers (H3K4me1/2) and can even report their status as active, poised, or silent. These histone marks represent rally points at which transcriptional machinery can be assembled. In fact, there are unique classes of protein domains called chromodomains and bromodomains that specifically bind to methylated or acetylated lysine marks, respectively. Thus, histone marks and their underlying DNA elements have been recently found to be dynamically regulated during hematopoietic differentiation.
While the current model supports a differentiation-dependent decrease in chromatin accessibility that results in a gradual closing of regulatory potential, recent findings suggest that this is not entirely true. Lara-Astiaso et al. were able to characterize the differential usage of DNA regulatory elements during hematopoietic development by utilizing a novel indexing-first chromatin immunoprecipitation (iChIP) method followed by next-generation sequencing to globally track four histone marks (H3K4me1/2/3 and H3K27Ac) through 16 stages in blood differentiation [39••]. Their iChIP technique overcomes typical input limitations by indexing the sheared chromatin of sorted cell populations prior to the ChIP step for individual histone marks. This allows for the pooling of samples from different cell types into the same ChIP reaction, increasing cross-sample reproducibility and yielding high-resolution global coverage with as few as 500 cells. Combined promoter and transcriptional analysis grouped cell types into four clusters (progenitor, lymphoid, myeloid, and erythroid); however, chromatin changes at enhancers revealed that most regulatory elements exhibit lineage-specific activity as each lineage’s progenitors shared stronger enhancer similarity with its own differentiated progeny rather than with progenitors from other lineages. Further analysis of enhancer dynamics identified that although a majority of enhancers found in the mature populations are initially marked in HSCs, a large number of enhancers are also established de novo during differentiation. These de novo lineage-specific enhancers are acquired in a stepwise manner, as they are gained at the first stage of lineage commitment with an increase in chromatin accessibility but only become active (H3K27Ac) upon terminal differentiation. This corroborates several recent studies in the field that describe stepwise gain of do novo cis-regulatory elements [40, 41]. Alternatively, enhancer loss was found to be a more gradual process beginning at the stage of lineage-committed progenitors and ending with a substantial loss occurring at the last stage of terminal differentiation. These results corroborate a study of human hematopoiesis by Abraham et al., which describes the stepwise gain of chromatin bivalency at lineage-specific enhancers during differentiation that is later resolved to an active state in the specific lineage in which the gene is expressed prior to its silencing in the alternative lineage [42]. This suggests that the transcriptional regulatory potential of a cell (i.e., the total number of genes that a cell is able to express) reaches its peak in progenitors that have just become committed to a specific lineage rather than in the HSC. Final differentiation decisions may then depend upon expression of lineage-determining transcription factors to launch terminal differentiation gene programs while silencing the alternative lineages. Thus, it appears that the primary overall function of enhancers is to provide access to sublineage differentiation decisions. Since enhancers can be marked in the HSC, enhancer loss-of-function studies will be useful in determining the role that enhancers play in enabling the multilineage potential of the HSC. These roles could be achieved either through directly modulating lineage choices or indirectly by influencing the gaining of de novo enhancers which then in turn direct lineage decisions.
Phenotypic differences throughout evolution can be viewed as the direct result of disparities in cumulative gene expression levels, which can be viewed as a readout of cis-regulatory element activity. Interestingly, DNA regulatory elements have been found to be differentially conserved throughout evolution. Cheng et al. expanded upon this by utilizing biochemical and comparative genomic approaches in tandem to investigate the consequences of the evolutionary conservation or divergence of regulatory regions [43]. Transcription factor occupancy sites in multiple human and murine cell types were determined by ChIP-seq to investigate how regulatory elements have changed during evolution. Individual transcription factors predominantly reside at either proximal or distal regulatory elements, a preference that is highly conserved. Also, the epigenetic states surrounding each transcription factor binding site are conserved, suggesting that the function of each DNA element is also conserved. Furthermore, transcription factor occupancy is better conserved in proximal compared to distal regions, suggesting that promoters are under stricter selection than enhancers. Thus, comparing regulatory elements across evolutionary clades provides useful insights that can explain how some hematopoietic features are conserved (i.e., similar mature blood populations in humans and mice) while others differ greatly (i.e., lymphoid skew in mice and myeloid skew in humans). Lara-Astiaso et al. showed that progenitor and erythroid-specific elements (e.g., cKit, Meis1 and Gypc, Gata1, respectively) display a high degree of mammalian conservation, while those specific to myeloid and lymphoid lineages exhibit lower conservation (e.g., Cebpa, Cd11b and Pax5, Prf1, respectively) [39••]. As promoters are necessary to initiate gene expression, they are under strict evolutionary pressure that results in overall conservation of differentiation pathways. Alternatively, the divergent nature of enhancers may explain how transcription is fine tuned in a spatiotemporal manner to result in the phenotypic differences that exist between human and mouse hematopoietic systems. This is supported by the observation that during human hematopoietic differentiation, promoter activity remains stable (H3K4me3) while enhancer silencing varies greatly (H3K27me3) [42].
Putting Cis-Regulatory Modules into Context
The implications of the activation or silencing of a single DNA regulatory element are put into context when the transcription factors that interact with each element can be annotated. While ChIP can determine the set of genes that are directly bound by a single transcription factor of interest, it is not the ideal method for the identification of all the proteins bound at a single locus. Several advancements have been made to address this deficiency and present valuable tools for determining gene regulatory networks throughout hematopoietic differentiation.
Since cis-regulatory elements modulate gene expression by assembling large protein complexes that contain numerous transcription factors, identification of all of the individual protein components is important to understanding how they modulate gene expression. To address this problem, Mohammed et al. developed a protocol for the tandem analysis of protein-protein and protein-DNA interactions in an endogenous context from the same sample, termed rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) [44•]. Following formaldehyde crosslinking, chromatin is isolated and sheared, and then subjected to immunoprecipitation. A portion of sample is submitted to DNA sequencing to identify genome-wide binding loci while the remainder is digested with proteases and run through mass spectrometry to identify all of the potential interacting proteins that are isolated by the co-IP. By intersecting these two data sets, all proteins involved in the transcription factor complex are reported and put into perspective by assigning specific genes as targets of the complexes. This is especially useful for discovering proteins involved in gene regulation that do not have DNA binding domains. Additionally, RIME can identify the differential usage of binding partners at each bound locus, thus providing further insights into regulatory networks (including potential signaling modules). Alternatively, engineered DNA-binding molecule-mediated chromatin immunoprecipitation mass spectrometry (enChIP-MS) was developed to study the entire repertoire of transcriptional regulatory mechanisms at a single locus of interest. [45•] EnChIP-MS utilizes the CRISPR/Cas9 system to isolate individual genomic loci for the identification of regulatory proteins and chromatin states. By co-expressing a guide RNA targeted to a locus of interest along with a tagged, catalytically inactive Cas9 protein, immunoprecipitation of the tagged Cas9 in crosslinked cells can isolate the bound locus. Proceeding with mass spectrometry can identify the list of regulators, including transcription factors, histone variants, and all histone modifications. This protocol can be adapted such that the product of the immunoprecipitation can be analyzed for DNA and RNA content as well. This will enable the identification of long-range DNA interactions, bound lncRNAs, or newly transcribed RNA from the purified locus.
Although much work has been done to define transcription factor binding motifs, there currently is no software package available to allow calculations of potential binding motifs for an individual transcription factor of interest. Two recent studies confronted this current limitation. Weirauch et al. determined the DNA binding motifs of over 1000 eukaryotic transcription factors while identifying 54 classes of DNA binding domains that were compiled into the publicly available cis-BP database [46]. They showed that the primary amino acid sequence could accurately predict motif preference while proving functionality by showing that the determined motifs could predict which proteins’ binding would be affected by a human disease risk allele (e.g., ETS1 in lupus or GATA1 in alpha thalassemia). Pujato et al. developed the TF2DNA algorithm, which predicts DNA binding sites based upon their determined protein structure [47]. By building a 3D protein model from a primary amino acid sequence, the program constructs all possible TF-DNA structures and then eliminates those that are biochemically unfavorable to determine the possible binding sequences and their relative binding affinities. The TF2DNA algorithm was successful in predicting the DNA binding motif of the poorly characterized T cell leukemia homeobox 3 (TLX3) transcription factor, which was later validated biochemically.
Transcription Factors in Hematopoiesis
Transcriptional Activation in Hematopoiesis
Cabezas-Wallscheid et al. recently utilized integrated protein, transcriptional, and epigenetic analyses to dissect regulatory networks that distinguish HSCs from MPP1 cells [8••]. Proteomic comparison between the two populations revealed that HSCs were enriched for members of the Hmga transcription factor family, which has been shown to regulate self-renewal through the balance of metabolism and proliferation as downstream mediators of the Lin28-Let7 pathway [48]. Alternatively, MPP1 cells were enriched for proteins involved in DNA replication and cell-cycle progression as well as the maintenance DNA methyltransferase, Dnmt1, which is essential during the early stages of differentiation [49]. Transcriptional analysis of the HSC and MPP1 populations revealed a high correlation between RNA levels and the proteins they encode, as detected by mass spectrometry. A few exceptions which only showed slight differences in messenger RNA (mRNA) levels despite exhibiting large changes at the protein level include glucose metabolic enzymes and members of the Hgma family, which are highly regulated at the post-transcriptional level. Additionally, mRNAs of several histone variants that are involved in chromosomal organization (e.g., H2afz, H2afy2) are downregulated in MPP1. Although no changes were detected in protein levels of these histones, this is most likely due to their long protein half-lives. Differences in splicing between HSCs and MPP1 were also analyzed, revealing a set of 46 differentially spliced transcription factors between the two populations. This group included Foxj3, which showed an increased retention of the first exon in HSCs, which may play a role in DNA-binding preference to promote self-renewal. It is clear that the maintenance of the HSC state and the decision to exit toward differentiation involve a complex interplay between DNA methylation, chromosomal organization, cellular metabolism, microRNA, and RNA splicing. The strong correlation between transcript and protein suggests that post-transcriptional regulation is used for only a few pathways during the HSC to MPP1 transition, and the majority of gene expression changes occur at the transcriptional level. Additionally, since mRNA and protein levels do not always correspond with one another, it is important to analyze both while being careful not to draw conclusions about one without information on the other. While these analyses are insightful in distinguishing the highly expressed genes that are important in maintaining HSC stemness from those that promote differentiation, transcripts expressed at low levels may be missed due to the nature of bulk sequencing.
Single-Cell Analysis of Transcription in Hematopoiesis
Analysis at the single-cell level may assist in further dissecting the changes that are made during the decision to exit the true HSC state. Additional work must be done to enhance the sensitivity of protein detection methods before they can be used to detect the protein (or post-translationally modified protein) content of a single cell; however, recent technological advances have made this possible for RNA detection. Single-cell analysis has revealed that transcription does not occur in a smooth and continuous pattern but actually exhibits waves of transcription between long periods of inactivity, termed “bursting,” initially described in E. coli [50]. Wills et al. recently performed qRT-PCR analysis on a large set of primary human naive B cells to dissect the transcriptional heterogeneity of WNT pathway genes within this immune cell population [51]. Through single-cell analysis, they distinguished bursting of individual genes that is affected by SNPs between individuals. These differences would otherwise be masked by bulk cell analysis, highlighting the importance of biological investigation at the single-cell level.
Massively parallel single-cell RNA sequencing (MARS-seq) has gained tremendous popularity within the field, as it offers the ability to explore global transcriptional heterogeneity within distinct cell populations that have classically been viewed as homogeneous. Recent comparison between single cell and bulk RNA-seq within the same sample revealed that the single-cell data could accurately recapitulate the bulk transcriptome complexity and gene expression distributions [52]. This assessment confirms that single-cell RNA-seq is a viable method for making quantitative measurements of single-cell transcriptomes. Jaitin et al. recently utilized MARS-seq to identify subsets of splenic dendritic cells whose global transcriptional signature puts them outside of the known cell-type hierarchy [53••]. By analyzing the transcriptomes of individual Cd11c+ splenocytes, cells could be clustered into distinct populations based on the levels of expression of surface markers and other genes. Traditional sequencing analysis of individual bulk sorted populations based upon these identified surface markers recapitulated the gene expression signatures of each cluster from the single-cell analysis, confirming the accuracy of the method. Although transcriptionally distinct subpopulations do not prove the existence of additional functional subtypes, single-cell analysis provides a platform for the discovery of such heterogeneity for future study and functional validation.
The advent of single-cell RNA-seq has been accompanied by new analytical methods that assist in organization and making sense of the resulting highly complex data sets. Trapnell et al. have developed Monocle, an algorithm that is designed to analyze several single-cell RNA-seq data sets collected over a time course (e.g., differentiation, proliferation, drug treatment, etc.) in order to generate kinetic gene expression profiles throughout a biological process [54]. Derived from an algorithm used to temporally align microarray data sets, it has been extended to allow for single-cell variation and multiple branching cell fate events. First, the algorithm represents each cell’s expression profile in a high dimensional space, with pairwise comparisons of each gene. Then, independent component analysis is utilized to reduce the data into a low dimensional space for easier visualization and interpretation. Next, a relationship tree is assembled that forms the longest “differentiation path” possible through transcriptionally similar cells. Finally, this tree is used to propose a cell’s path through differentiation. Implementing Monocle permits the discovery of trends in expression of individual genes that can identify key events and processes over time as cells travel through individual cell states. Another tool recently implemented to analyze single-cell RNA-seq data is single-cell clustering upon bifurcation analysis (SCUBA), developed by Marco et al. SCUBA is designed to specifically align multiple single-cell RNA-seq data sets at time points along stages of tissue differentiation [55]. Cells are initially clustered, and then cells from the next time point are analyzed as a single cluster or two separate clusters, with statistical testing to decide if that cell underwent a bifurcation event or not. This analysis continues until a developmental tree is formed, and then a series of mathematical calculations are used to further refine the tree. These highly sophisticated analytical methods present valuable tools that glean as much information as possible out of expensive single-cell RNA-seq data sets as well as construct developmentally relevant gene expression networks.
Transcriptional Repression in Hematopoiesis
While the process of differentiation involves the activation of individual lineage programs that lead to cellular fate determinations, an equally important process involves repressing the genetic programs that maintain the hematopoietic stem/progenitor cells (HSPCs) in an undifferentiated state and promote self-renewal. One such repressor is the histone H3K4 demethylase, Lsd1, which binds to and helps repress genes targeted by a number of hematopoietic transcription factors, including Gfi1, Gfi1b [56], Scl1/Tal1 [57], Bcl11a [58], Sall4 [59], and Runx1 [60]. Two recent studies describe the vital role of Lsd1 in the epigenetic regulation of HSC and myeloid differentiation programs in vivo [61, 62]. Genetic deletion or knockdown of Lsd1 caused HSC differentiation defects as well as impaired production of granulocytes and erythrocytes. Transcriptional analysis revealed an elevation of the stem/progenitor gene signature, and ChIP-seq confirmed that Lsd1 binds and demethylates histone H3K4 at a set of genes that is highly enriched for stem/progenitor regulation. These data implicate Lsd1 as an epigenetic regulator that is critical for the appropriate silencing of stem/progenitor genes during the differentiation process.
It is well known that Lsd1 repression is achieved through binding and collaboration with the corepressor CoREST/REST corepressor (Rcor1) [63]; however, Upadhyay et al. recently elucidated the role of Rcor2 and Rcor3 in regulating Lsd1 activity during erythropoiesis [64]. Interestingly, while Rcor2 acts redundantly to Rcor1, Rcor3 lacks a SANT2 domain shared by its paralogues, thus acting as an inhibitor of Lsd1 demethylation activity. They showed that Rcor1 and Rcor3 expression levels are inversely correlated during mega-erythroid differentiation to modulate Lsd1 target gene expression. Further evidence that Rcor1 is required for erythropoiesis was recently presented by Yao et al. Germline deletion of Rcor1 was embryonic lethal due to a block in erythropoiesis at the proerythroblast stage [65]. Transplantation and in vitro colony forming assays identified Rcor1 to be a cell intrinsic requirement for erythropoietic lineage commitment, as Rcor1 null committed erythroid progenitors exhibited lineage switching, and formed myeloid colonies in vitro. ChIP analysis of control fetal liver proerythroblasts revealed that Rcor1 normally binds cooperatively with Gfi1b at nonerythroid genes to suppress the stem/progenitor signature and the myeloid lineage fate. Thus, not only is the proper regulation of Lsd1 critical during differentiation, but modulation of its cofactors can influence differentiation and terminal lineage commitment. It is clear that transcription factors do not operate alone, but instead assemble as complexes to cooperatively regulate target genes. As such, disruption of one partner in a complex may significantly hinder or alter the function of the complex as a whole. Determination of all of the proteins contained in a complex and which genes they regulate (e.g., via RIME) is necessary to fully understand the function of a single transcription factor.
Circadian Control of Transcription in Hematopoiesis
Circadian rhythm is the physical, mental, and behavioral changes of an organism due to natural fluctuations in environmental light levels, ruled by a master clock in the brain; however, many peripheral tissues maintain their own 24-h clocks as well, including the hematopoietic system [66]. Molecularly, the circadian clock is regulated by the core transcription factors CLOCK and BMAL1 that bind E-box elements to activate downstream genes, some of which in turn negatively feedback to inhibit CLOCK and BMAL1 activity. This negative feedback loop creates oscillations in protein levels that help maintain proper timing of the circadian clock [67, 68].
Correct immunological function depends upon clock maintenance in order to coordinate the differentiation and localization of hematopoietic cells. Recently, Nguyen et al. has identified that monocytes maintain an internal clock, where Bmal1 acts as a traffic cop to properly direct them to migrate from the bone marrow to the peripheral blood or spleen at different times of the day [69]. They also found that Ly6Chi inflammatory monocytes exhibited varying recruitment capacities to sites of inflammation at different times of the day, influencing the balance between a proper immune response and overactivation. Loss of the Bmal1 cell autonomously disrupted chemokine signaling and thus correct monocyte recruitment, making mice susceptible to infection and intensifying metabolic disease. Granulocytes are also influenced by circadian rhythm, as the localization of neutrophils has been shown to exhibit diurnal fluctuations to influence bone marrow niche function [70]. As the most abundant leukocyte in the body, the short-lived lifespan of the neutrophil demands that their production and removal from circulation be tightly controlled. Casanova-Acebes et al. recently identified CD62L as a marker for neutrophil aging, which was utilized to define diurnal time periods when young neutrophils were released from the bone marrow and when aged neutrophils return to the bone marrow for elimination. Interestingly, the elimination of aged neutrophils modulated bone marrow niche cell numbers and their Cxcl12 production to control HSPC release into the peripheral blood. Sophisticated in vivo imaging techniques revealed that aged neutrophils home to bone marrow macrophages and are engulfed, triggering activation of cholesterol-responsive LXR transcription factor signaling in the macrophages. Genes downstream of LXR normally exhibit circadian rhythm corresponding with the elimination of neutrophils, and antibody-mediated depletion of neutrophils blunts this expression pattern. Furthermore, LXR signaling was linked to decreased Cxcl12 production in the niche to promote HSPC mobilization. Thus, both the homing of aged neutrophils back to the bone marrow and their subsequent removal by macrophages are critical to help coordinate HSPC trafficking into the blood, which is known to be important for immunosurveillance and the colonization of peripheral tissues [71]. Interestingly, through the use of parabiosis experiments, this HSPC mobilization proved to be independent of GCSF signaling, which has previously been shown to promote mobilization by triggering the reduction of Cxcl12 in the bone marrow [72]. As GCSF is known as the major granulocytic cytokine [73], it is possible that the effect of GCSF signaling exhibits thresholding, where varying levels of cytokine are able to execute different functions. Low levels of GCSF normally maintained at steady state may maintain sufficient granulopoiesis, but during “emergency situations,” where GCSF levels are higher (including in clinically administered dosing), progenitors are stimulated to increase granulocytic output and are mobilized to address the infection in the periphery.
Although the core clock circuit has been well established, the roles of other proteins in circadian rhythm maintenance are constantly being elucidated. Post-translational modifications also play vital roles in regulating circadian genes, as histone-modifying enzymes and protein kinase Cα (PKCa) are required for proper clock rhythm [74, 75]. The transient nature of post-translational modifications presents a mechanism to rapidly augment protein activity while remaining easily and quickly reversible, allowing cells to respond to stimuli rapidly without requiring de novo protein synthesis. Post-translational modifications (e.g., phosphorylation and acetylation) are sensitive to energy levels and thus the underlying cellular metabolic processes. Clock genes control the ratio of glycolytic vs. oxidative gene transcription, demonstrating circadian control of cellular metabolism [76]. These data strongly link post-translational protein modifications as mediators of circadian rhythm.
Recent work by Nam et al. identified LSD1 as a phosphorylation target of PKCa, enabling LSD1 to bind to CLOCK/BMAL1 to activate the transcription of E-box genes [77]. Knockin mice that were generated to endogenously express a mutant Lsd1 protein that cannot be phosphorylated by Pkca (serine to alanine mutation, termed Lsd1 SA/SA) showed deregulated circadian gene expression and defects in rhythm. Phosphorylation of Lsd1 was shown to be essential not only to its binding to Clock/Bmal1 but also for Clock/Bmal1 retention at E-box elements and H3K9Ac-mediated activation of the target genes. Interestingly, Lsd1 catalytic activity is not required for its co-activation of clock genes, a characteristic shared by Jarid1, another lysine demethylase that cooperatively activates clock genes without utilizing its catalytic activity [78]. Lsd1 and Jarid1 seem to be exceptions to the rule, as other CLOCK/BMAL1 binding epigenetic modifiers activate target gene expression through their catalytic activity (e.g., MLL1 [79], MLL3 [80]). These nonenzymatic roles of Lsd1 and Jarid1 may somehow be regulating CLOCK/BMAL1 activity through a process that is dependent upon their post-translational modification. A close family member to Jarid1 that normally lacks a catalytic domain, Jarid2, was found to promote the activity of the PRC2 complex to which it binds upon becoming methylated itself [81]. It is possible that Jarid1 undergoes post-translational modification to facilitate activation of the CLOCK/BMAL1 complex in a similar manner as discovered with Jarid2. Ultimately, these new findings highlight the importance of post-translational modifications on transcription factors by directing their binding locations, binding partners, and overall impact upon target gene expression, especially in the context of maintaining circadian rhythm. As all of these genes are expressed in the hematopoietic system, it will be interesting to investigate the roles that they and their modified forms play in controlling hematopoietic differentiation. However, cellular context should be kept in consideration, since despite MLL1 catalytic activity is required for CLOCK function in fibroblasts [79], it has recently been found to be dispensable for maintaining HSC fitness [82].
Conclusions
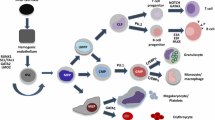
With a myriad of tools available for the study of hematopoiesis, HSCs can serve as a model system for stem cell biology such that knowledge, biochemical approaches, and analytical methods gained through the study of HSCs can be applied to stem cells of other tissues (Table 1). Widespread use and optimized whole-genome sequencing protocols enable multiple biochemical approaches (i.e., histone, DNA methylation, transcription factor ChIP, and RNA-seq) to be integrated with genetic and evolutionary analyses to rigorously dissect the complex nature of transcriptional regulation (Fig. 1). Histone modifications function to regulate the accessibility of DNA regulatory elements by opening or closing chromatin. These DNA regulatory elements, through their underlying nucleic acid composition, can only bind a subset of all transcription factors present in the genome. Not only do these cis-regulatory elements change throughout evolution, but their ability to recruit trans-activating proteins is modulated by their methylation status. The functional impact of a single enhancer region is restricted to the set of transcription factors expressed in a given cell that are able to bind the DNA element. Further complexity is generated by the ability of lncRNAs to bind cis-regulatory enhancers to recruit proteins or create long-range DNA interactions. Finally, the activity of transcription factors may depend on sequential or combinatorial binding of multiple proteins or post-translational modifications. As cell fate decisions are made on a cell by cell basis, single-cell analytical techniques are critical for the dissection of transcriptional programs underlying hematopoietic stem and progenitor potential. Translation of the knowledge gained by these studies can be utilized for the development and implementation of therapeutics to combat human disease.

Epigenetic, cis- and trans-regulatory mechanisms underlying transcriptional control. Proper control of transcription is achieved through the coordination of epigenetic modifications, enhancer elements, and transcription factor activity. The availability of DNA regulatory elements dictates transcription factor binding and recruitment of epigenetic regulators to further regulate chromatin accessibility. Additionally, transcription factor activity can be modulated post-translationally in coordination with circadian rhythmicity. Finally, lncRNAs facilitate long-range chromatin interactions to further modulate transcriptional activity
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Pereira CF et al. Induction of a hemogenic program in mouse fibroblasts. Cell Stem Cell. 2013;13:205–18. doi:10.1016/j.stem.2013.05.024.
Riddell J et al. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014;157:549–64. doi:10.1016/j.cell.2014.04.006.
Elcheva I et al. Direct induction of haematoendothelial programs in human pluripotent stem cells by transcriptional regulators. Nat Commun. 2014;5:4372. doi:10.1038/ncomms5372. Screens in human iPSCs identified two combinations of transcription factors that induce the hematopoietic program and produce pan-myeloid and mega/erythroid lineages.
Kundaje A et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30. doi:10.1038/nature14248.
Cheng SC et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (New York, NY). 2014;345:1250684. doi:10.1126/science.1250684.
Saeed S et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (New York, NY). 2014;345:1251086. doi:10.1126/science.1251086.
Chen L et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science (New York, NY). 2014;345:1251033. doi:10.1126/science.1251033.
Cabezas-Wallscheid N et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014;15:507–22. doi:10.1016/j.stem.2014.07.005. This study identified signaling networks during the early stages of HSC differentiation by parallel integration of multiparameter analyses on rare cell populations.
Challen GA et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31. doi:10.1038/ng.1009.
Mayle A et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2014. doi:10.1182/blood-2014-08-594648.
Broske AM et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009;41:1207–15. doi:10.1038/ng.463.
Trowbridge JJ et al. Haploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012;26:344–9. doi:10.1101/gad.184341.111.
Ji H et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–42. doi:10.1038/nature09367.
Shen Y et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–20. doi:10.1038/nature11243.
Stamatoyannopoulos JA et al. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol. 2012;13:418. doi:10.1186/gb-2012-13-8-418.
Ronnerblad M et al. Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation. Blood. 2014;123:e79–89. doi:10.1182/blood-2013-02-482893.
Rodriguez RM et al. Regulation of the transcriptional program by DNA methylation during human alphabeta T-cell development. Nucleic Acids Res. 2015;43:760–74. doi:10.1093/nar/gku1340.
Shearstone JR et al. Global DNA demethylation during mouse erythropoiesis in vivo. Science (New York, NY). 2011;334:799–802. doi:10.1126/science.1207306.
Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi:10.1186/gb-2011-12-6-r54.
Ko M et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. doi:10.1038/nature09586.
Ge L et al. TET2 plays an essential role in erythropoiesis by regulating lineage-specific genes via DNA oxidative demethylation in a zebrafish model. Mol Cell Biol. 2014;34:989–1002. doi:10.1128/mcb.01061-13.
Hackett JA et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science (New York, NY). 2013;339:448–52. doi:10.1126/science.1229277.
Rampal R et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9:1841–55. doi:10.1016/j.celrep.2014.11.004.
Song CX et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi:10.1038/nbt.1732.
Adey A, Shendure J. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 2012;22:1139–43. doi:10.1101/gr.136242.111.
Bai W et al. Long non-coding RNA NR_045623 and NR_028291 involved in benzene hematotoxicity in occupationally benzene-exposed workers. Exp Mol Pathol. 2014;96:354–60. doi:10.1016/j.yexmp.2014.02.016.
Varrault A et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell. 2006;11:711–22. doi:10.1016/j.devcel.2006.09.003.
Berg JS et al. Imprinted genes that regulate early mammalian growth are coexpressed in somatic stem cells. PLoS One. 2011;6, e26410. doi:10.1371/journal.pone.0026410.
Rossi L et al. Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell. 2012;11:302–17. doi:10.1016/j.stem.2012.08.006.
Bardou F et al. Long noncoding RNA modulates alternative splicing regulators in Arabidopsis. Dev Cell. 2014;30:166–76. doi:10.1016/j.devcel.2014.06.017.
Chan WL, Huang HD, Chang JG. lncRNAMap: a map of putative regulatory functions in the long non-coding transcriptome. Comput Biol Chem. 2014;50:41–9. doi:10.1016/j.compbiolchem.2014.01.003.
Zhang H et al. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J Cell Biol. 2014;204:61–75. doi:10.1083/jcb.201304152.
Wang H et al. An intragenic long noncoding RNA interacts epigenetically with the RUNX1 promoter and enhancer chromatin DNA in hematopoietic malignancies. Int J Cancer J Int Cancer. 2014;135:2783–94. doi:10.1002/ijc.28922.
Arab K et al. Long noncoding RNA TARID directs demethylation and activation of the tumor suppressor TCF21 via GADD45A. Mol Cell. 2014;55:604–14. doi:10.1016/j.molcel.2014.06.031.
Guttman M et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. doi:10.1038/nature07672.
Paralkar VR et al. Lineage and species-specific long noncoding RNAs during erythro-megakaryocytic development. Blood. 2014;123:1927–37. doi:10.1182/blood-2013-12-544494.
Alvarez-Dominguez JR et al. Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood. 2014;123:570–81. doi:10.1182/blood-2013-10-530683.
Kellis M et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci U S A. 2014;111:6131–8. doi:10.1073/pnas.1318948111.
Lara-Astiaso D et al. Immunogenetics. Chromatin state dynamics during blood formation. Science (New York, NY). 2014;345:943–9. doi:10.1126/science.1256271. This study uses a novel method for ChIP-seq on as few as 500 cells (iChIP) to describe enhancer usage during hematopoietic differentiation.
Schlesinger F, Smith AD, Gingeras TR, Hannon GJ, Hodges E. De novo DNA demethylation and noncoding transcription define active intergenic regulatory elements. Genome Res. 2013;23:1601–14. doi:10.1101/gr.157271.113.
Luyten A, Zang C, Liu XS, Shivdasani RA. Active enhancers are delineated de novo during hematopoiesis, with limited lineage fidelity among specified primary blood cells. Genes Dev. 2014;28:1827–39. doi:10.1101/gad.240101.114.
Abraham BJ, Cui K, Tang Q, Zhao K. Dynamic regulation of epigenomic landscapes during hematopoiesis. BMC Genomics. 2013;14:193. doi:10.1186/1471-2164-14-193.
Cheng Y et al. Principles of regulatory information conservation between mouse and human. Nature. 2014;515:371–5. doi:10.1038/nature13985.
Mohammed H et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013;3:342–9. doi:10.1016/j.celrep.2013.01.010. A novel method (RIME) was developed to coordinate the detection of protein-protein interactions and genomic binding coordinates for elucidation of steady-state and inducible transcriptional networks.
Fujita T, Fujii H. Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem Biophys Res Commun. 2013;439:132–6. doi:10.1016/j.bbrc.2013.08.013. This study describes a novel method in which ChIP is followed by mass spectrometry to identify all transcription factors and chromatin states at a specific genomic locus.
Weirauch MT et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–43. doi:10.1016/j.cell.2014.08.009.
Pujato M, Kieken F, Skiles AA, Tapinos N, Fiser A. Prediction of DNA binding motifs from 3D models of transcription factors; identifying TLX3 regulated genes. Nucleic Acids Res. 2014;42:13500–12. doi:10.1093/nar/gku1228.
Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf expression. Cell. 2008;135:227–39. doi:10.1016/j.cell.2008.09.017.
Hu Z et al. Decitabine maintains hematopoietic precursor self-renewal by preventing repression of stem cell genes by a differentiation-inducing stimulus. Mol Cancer Ther. 2010;9:1536–43. doi:10.1158/1535-7163.mct-10-0191.
Golding I, Paulsson J, Zawilski SM, Cox EC. Real-time kinetics of gene activity in individual bacteria. Cell. 2005;123:1025–36. doi:10.1016/j.cell.2005.09.031.
Wills QF et al. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat Biotechnol. 2013;31:748–52. doi:10.1038/nbt.2642.
Wu AR et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11:41–6. doi:10.1038/nmeth.2694.
Jaitin DA et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science (New York, NY). 2014;343:776–9. doi:10.1126/science.1247651. This study utilized a novel technique (MARS-Seq) to analyze the RNA content of individual cells in order to delineate multiple hematopoietic cell types out of the complex heterogeneity of the spleen.
Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6. doi:10.1038/nbt.2859.
Marco E et al. Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proc Natl Acad Sci U S A. 2014;111:E5643–50. doi:10.1073/pnas.1408993111.
Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell. 2007;27:562–72. doi:10.1016/j.molcel.2007.06.039.
Hu X et al. LSD1-mediated epigenetic modification is required for TAL1 function and hematopoiesis. Proc Natl Acad Sci U S A. 2009;106:10141–6. doi:10.1073/pnas.0900437106.
Xu J et al. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci U S A. 2013;110:6518–23. doi:10.1073/pnas.1303976110.
Liu L et al. Histone lysine-specific demethylase 1 (LSD1) protein is involved in Sal-like protein 4 (SALL4)-mediated transcriptional repression in hematopoietic stem cells. J Biol Chem. 2013;288:34719–28. doi:10.1074/jbc.M113.506568.
van Riel B et al. A novel complex, RUNX1-MYEF2, represses hematopoietic genes in erythroid cells. Mol Cell Biol. 2012;32:3814–22. doi:10.1128/mcb.05938-11.
Sprussel A et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia. 2012;26:2039–51. doi:10.1038/leu.2012.157.
Kerenyi MA et al. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. eLife. 2013;2, e00633. doi:10.7554/eLife.00633.
Yang M et al. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell. 2006;23:377–87. doi:10.1016/j.molcel.2006.07.012.
Upadhyay G, Chowdhury AH, Vaidyanathan B, Kim D, Saleque S. Antagonistic actions of Rcor proteins regulate LSD1 activity and cellular differentiation. Proc Natl Acad Sci U S A. 2014;111:8071–6. doi:10.1073/pnas.1404292111.
Yao H et al. Corepressor Rcor1 is essential for murine erythropoiesis. Blood. 2014;123:3175–84. doi:10.1182/blood-2013-11-538678.
Bollinger T, Bollinger A, Oster H, Solbach W. Sleep, immunity, and circadian clocks: a mechanistic model. Gerontology. 2010;56:574–80. doi:10.1159/000281827.
Gekakis N et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science (New York, NY). 1998;280:1564–9.
Shearman LP et al. Interacting molecular loops in the mammalian circadian clock. Science (New York, NY). 2000;288:1013–9.
Nguyen KD et al. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science (New York, NY). 2013;341:1483–8. doi:10.1126/science.1240636.
ohCasanova-Acebes M et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. 2013;153:1025–35. doi:10.1016/j.cell.2013.04.040.
Massberg S et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008. doi:10.1016/j.cell.2007.09.047.
Petit I et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–94. doi:10.1038/ni813.
Lieschke GJ et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737–46.
Naruse Y et al. Circadian and light-induced transcription of clock gene Per1 depends on histone acetylation and deacetylation. Mol Cell Biol. 2004;24:6278–87. doi:10.1128/mcb.24.14.6278-6287.2004.
Jakubcakova V et al. Light entrainment of the mammalian circadian clock by a PRKCA-dependent posttranslational mechanism. Neuron. 2007;54:831–43. doi:10.1016/j.neuron.2007.04.031.
Peek CB et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science (New York, NY). 2013;342:1243417. doi:10.1126/science.1243417.
Nam HJ et al. Phosphorylation of LSD1 by PKCalpha is crucial for circadian rhythmicity and phase resetting. Mol Cell. 2014;53:791–805. doi:10.1016/j.molcel.2014.01.028.
DiTacchio L et al. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science (New York, NY). 2011;333:1881–5. doi:10.1126/science.1206022.
Katada S, Sassone-Corsi P. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat Struct Mol Biol. 2010;17:1414–21. doi:10.1038/nsmb.1961.
Valekunja UK et al. Histone methyltransferase MLL3 contributes to genome-scale circadian transcription. Proc Natl Acad Sci U S A. 2013;110:1554–9. doi:10.1073/pnas.1214168110.
Sanulli S et al. Jarid2 methylation via the PRC2 complex regulates H3K27me3 deposition during cell differentiation. Mol Cell. 2015. doi:10.1016/j.molcel.2014.12.020.
Mishra BP et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 2014;7:1239–47. doi:10.1016/j.celrep.2014.04.015.
Acknowledgments
The authors thank Diane Krause, Maria (Ken) Figueroa, and Gang Huang for critical reading, edits, and insightful comments. This work was partially supported by NIH R01 CA159845 (to H.L.G.).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
David E. Muench and H. Leighton Grimes have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any previously unpublished studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Stem Cell Switches and Regulators
Rights and permissions
About this article

Cite this article
Muench, D.E., Grimes, H.L. Transcriptional Control of Stem and Progenitor Potential. Curr Stem Cell Rep 1, 139–150 (2015). https://doi.org/10.1007/s40778-015-0019-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-015-0019-z