
Abstract
Introduction
The safety of tofacitinib in psoriatic arthritis (PsA) and rheumatoid arthritis (RA) has been demonstrated in clinical studies of ≤ 4 and 9.5 years, respectively. Post-marketing surveillance (PMS) data for tofacitinib from spontaneous and voluntary adverse event (AE) reports have been published for RA, but not PsA. To inform the real-world safety profile of tofacitinib in PsA, we evaluated AE reports submitted to the Pfizer safety database (including RA data for context).
Methods
Endpoints included AEs, serious AEs (SAEs), AEs of special interest (AESIs; serious infections, herpes zoster, cardiovascular events, malignancies, venous thromboembolism), and fatal cases. Exposure was estimated using IQVIA global commercial sales data. Number, frequency, and reporting rates (RRs; number of events/100 patient-years’ [PY] exposure) were summarized by indication and formulation (immediate release [IR] 5 or 10 mg twice daily], modified release [MR] 11 mg once daily, or all tofacitinib). The data-collection period differed by indication (PsA: 14 December 2017 [US approval, IR/MR] to 6 November 2021; RA: 6 November 2012 [US approval, IR] to 6 November 2021; MR approval, 24 February 2016).
Results
A total of 73,525 case reports were reviewed (PsA = 5394/RA = 68,131), with 20,706/439,370 PY (PsA/RA) of exposure. More AEs were reported for IR versus MR (IR/MR: PsA = 8349/7602; RA = 137,476/82,153). RRs for AEs (IR/MR: PsA = 59.6/113.4; RA = 44.0/64.8) and SAEs (PsA = 8.1/13.6; RA = 8.0/9.5) were higher with MR versus IR. AE RRs (RA) in the first 4 years after IR approval were 95.9 (IR; 49,439 PY) and 147.0 (MR; 2000 PY). Frequency of SAEs, AESIs, and fatal cases was mostly similar across formulations and indications. The most frequently-reported AE Preferred Terms (PsA/RA) included drug ineffective (20.0%/17.8%), pain (9.7%/10.6%), condition aggravated (9.9%/10.5%), headache (8.8%/7.9%) and, for PsA, off-label use (10.5%/3.4%).
Conclusions
Tofacitinib PMS safety data from submitted AE reports were consistent between PsA and RA, and aligned with its known safety profile. Exposure data (lower MR versus IR; estimation from commercial sales data), reporting bias, reporter identity, and regional differences in formulation use limit interpretation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Reports of the real-world safety profile of tofacitinib in psoriatic arthritis (PsA) are limited, although the safety of tofacitinib in patients with PsA and rheumatoid arthritis (RA) has been demonstrated in clinical studies of up to 4 and 9.5 years, respectively. |
To date, post-marketing surveillance (PMS) safety data for tofacitinib from spontaneous and voluntary adverse event (AE) reports have been published for RA, but not for PsA. |
This analysis informs the real-world safety profile of tofacitinib in PsA using AE reports submitted to the Pfizer safety database, with RA data included for context. |
What was learned from the study? |
The data collected in this PMS study were aligned with the established safety profile of tofacitinib, and were consistent between PsA and RA. |
While these results should be interpreted in the context of the limitations of PMS studies and spontaneous AE reporting, these data provide insight to the clinician regarding expected real-world safety outcomes in patients with PsA treated with tofacitinib. |
Introduction
Psoriatic arthritis (PsA) is a chronic inflammatory disease (CID), manifesting as skin and nail lesions, peripheral arthritis, inflammation of entheseal insertion points, swollen digits, and spondylitis [1], which is estimated to affect 133 patients per 100,000 population worldwide [2]. Patients with PsA have an increased risk of comorbid cardiovascular disease, obesity, type 2 diabetes, hypertension, metabolic syndrome, malignancy, and infection, and poor health-related quality of life compared to the general population [1, 3]. Rheumatoid arthritis (RA) is a CID associated with joint pain, damage, and long-term disability [4], which has higher prevalence than PsA (estimated at 460 per 100,000 population worldwide [5]). Patients with RA have an elevated risk of cardiovascular morbidity and mortality, malignancy, and infection compared with the general population [6, 7]. PsA has been associated with a generally lower comorbidity burden than RA, including lower rates of thyroid disease, malignancy, infection, and venous thromboembolism (VTE) [8,9,10,11]. Risk of cardiovascular disease is generally described as lower in PsA than in RA, although cardiometabolic risks such as obesity and type 2 diabetes are more frequently observed in PsA [12,13,14].
International treatment guidelines for PsA and RA recommend initial therapy with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), such as methotrexate [15,16,17,18,19]. In patients without sufficient clinical response to csDMARD treatment, advanced therapies are recommended, such as biologic DMARDs (bDMARDs, e.g., tumor necrosis factor inhibitors [TNFi]) or, in certain scenarios, targeted synthetic DMARDs (tsDMARDs, e.g., Janus kinase [JAK] inhibitors). Studies of patients receiving bDMARDs for RA or PsA have reported reductions in risk of cardiovascular disease, possibly owing to control of systemic inflammation [6, 13], similar rates of malignancies [20], and increased risk of infection [6, 21, 22], compared with patient populations not receiving bDMARDs.
Tofacitinib is an oral JAK inhibitor for the treatment of PsA and RA. Tofacitinib is approved for PsA and RA as a 5-mg immediate release (IR) tablet taken twice daily (BID), with other formulations available in some countries: 10 mg BID IR (for RA in Russia, Switzerland [until 2020], and Botswana) and 11 mg once daily (QD) modified release (MR). In PsA, the efficacy and safety of tofacitinib 5 and 10 mg BID have been demonstrated in phase 3 trials in combination with csDMARDs in patients with an inadequate response to TNFi [23] or csDMARDs (with or without prior TNFi treatment) [24, 25], and in a long-term extension (LTE) study with up to 48 months of observation [26]. In RA, the efficacy and safety of tofacitinib 5 and 10 mg BID administered as monotherapy or in combination with csDMARDs, mainly methotrexate, in patients with moderately to severely active RA, have been demonstrated in phase 3 [27,28,29,30,31,32,33], phase 3b/4 [34, 35], and LTE studies with up to 114 months of observation [36,37,38]. The safety profile of tofacitinib in clinical studies has been found to be comparable across PsA and RA [39]. The efficacy and safety profile of the MR formulation have also been characterized in two phase 3 and 3b/4 studies in RA [40, 41].
In the course of the tofacitinib clinical development program, increases in serum lipid levels and malignancies were observed [42,43,44], which prompted a head-to-head, FDA-mandated post-authorization safety study of tofacitinib and TNFi. ORAL Surveillance was a randomized, open-label study conducted from March 2014 to July 2020, which enrolled patients with RA ≥ 50 years of age with ≥ 1 additional cardiovascular risk factor [35]. In this event-driven study, non-inferiority criteria were not met in the comparison of tofacitinib (combined 5-mg BID and 10-mg BID doses) versus TNFi for the co-primary endpoints of adjudicated major adverse cardiovascular events (MACE) and adjudicated malignancies excluding nonmelanoma skin cancer (NMSC) [35]. Following communication of the ad hoc ORAL Surveillance safety analyses in 2019 and the final readout in 2021, there has been continuing interest in monitoring the safety of tofacitinib in real-world use in RA, including in patients with cardiovascular risk factors [45,46,47]. While comparable data for other indications including PsA are lacking, the results of ORAL Surveillance have led to revisions of regulatory labeling across all approved indications for JAK inhibitors, and have been considered in the development of treatment guidelines for RA, PsA, and axial spondyloarthritis [16, 19, 48].
Post-marketing surveillance (PMS) monitors drug safety in real-world use following market release, and complements data from clinical trials. Types of PMS include spontaneous/voluntary reporting of adverse events (AEs), post-marketing observational studies, and active surveillance. PMS data using spontaneous/voluntary AE reports have been previously published for tofacitinib in RA (including data up to November 2015) [49] and in ulcerative colitis (UC) [50], although no similar report exists for PsA, and reports of real-world safety of tofacitinib in PsA are limited [51]. The aim of this analysis was to inform the global real-world safety profile of tofacitinib in PsA, while providing context with data for RA, using spontaneous AE reports submitted to the Pfizer safety database.
Methods
Study Design
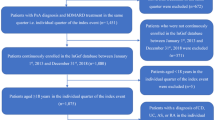
This was a retrospective analysis of worldwide PMS data collected from the Pfizer safety database from 14 December 2017 to 6 November 2021 (for PsA) and from 6 November 2012 to 6 November 2021 (for RA). The 5-mg BID (IR) and 11-mg QD (MR) doses were approved for PsA in the US on 14 December 2017 (in combination with nonbiologic DMARDs) and in the EU on 25 June 2018 and 20 August 2021, respectively (in combination with methotrexate). The 5-mg BID dose was first approved for RA in the US on 6 November 2012, followed by the 11-mg QD dose on 24 February 2016. In the EU, the 5-mg BID and 11-mg QD doses were approved for RA on 22 March 2017 and 16 December 2019, respectively. Spontaneous and voluntary reports of AEs occurring during or after exposure to tofacitinib were collected from patients, healthcare professionals (HCPs), regulatory authorities, post-marketing trials, non-interventional studies, solicited reports from patient support programs and market research programs, and reports extracted from the literature.
The spontaneously reported PMS AE data reported in this analysis were not collected as part of a clinical study and were non-interventional; therefore, no ethics approval was required. All data were reported in aggregate form in summary reports; no individual case-level data were evaluated or reported.
Cumulative Exposure
Cumulative exposure to tofacitinib for PsA or RA was calculated from a combination of audited unit sales from IQVIA’s Multinational Integrated Data Analysis System (commercial sales) database and prescription data from IQVIA’s Prescriber Insights database (https://www.iqvia.com/). Data were available from 61 countries and one region (Central America, which was available as aggregated data). The average daily dose (AVDOS) of tofacitinib was used to convert unit sales into patient-days (cumulative days of therapy) and further divided by 365.25 (days in a year) to obtain patient-years (PY) of exposure. PY for tofacitinib IR were calculated using the AVDOS of 2 units and the combined sales of tofacitinib 5 mg BID and 10 mg BID. PY for tofacitinib MR were calculated using the AVDOS of 1 unit for 11 mg QD tofacitinib sales, then adding the individual PY to generate a cumulative exposure number for tofacitinib during the relevant timeframe. Cumulative exposure data from 6 November 2012 to the third quarter of 2021 were available from IQVIA and were reported by quarter; cumulative exposure was extrapolated to the end of the reporting period (e.g., 6 November 2021) using the average cumulative exposure from the respective previous three quarters. PY of exposure by indication, sex, and age were derived through prescription share obtained from IQVIA’s Prescriber Insights database, and applying the factor to the overall PY calculation obtained from the commercial sales database. International Classification of Diseases 10th revision (ICD-10) codes were used to define the indications, as follows: ‘L405 Arthropathic psoriasis’ for PsA and ‘M06 Other Rheumatoid Arthritis’ and ‘M05 Seropositive Rheumatoid Arthritis’ for RA.
Data Analysis
AE reports in patients with PsA or RA received by the Pfizer safety database were coded using Medical Dictionary for Regulatory Activities (MedDRA) version 25.0, and summarized by type and frequency according to System Organ Class (SOC) and Preferred Term (PT). Data were summarized by indication (PsA/RA) and tofacitinib formulation (IR, MR, or all tofacitinib [sum of IR + MR]). Case reports for which the tofacitinib formulation was not reported were excluded from the ‘all tofacitinib’ group, as no cumulative exposure data were available, and they are therefore reported separately.
Patient sex, age, indication, geographical origin, and reporter identity (HCP [e.g., physician, pharmacist, other HCP] or non-HCP [consumer, lawyer, other non-HCP]) were collected and summarized. Each case could report one or more AEs. Cases were categorized by outcome (fatal, recovered, recovering, unknown, etc.). AEs (types, most frequent AEs occurring in ≥ 2% of patients, serious AEs [SAEs], and AEs of special interest [AESIs]) were summarized by number, frequency, and reporting rate (RR; number of events per 100 PY of estimated exposure). SAEs were defined as AEs resulting in death, hospitalization, or prolongation of hospitalization, persistent or significant disability/incapacity, or congenital anomaly/birth defect; or considered to be life-threatening or an important medical event. AESIs included serious infections (including Coronavirus disease 2019 [COVID-19]), herpes zoster (HZ), cardiovascular events, NMSC, VTE, and malignancies excluding NMSC (search criteria, defined using MedDRA version 24.1, are detailed in the Supplementary Methods).
Sensitivity analyses evaluated AEs received during the following time intervals, to evaluate temporal trends in AE reporting: 2015–2017 (RA only), 2017–2019, and 2019–2021. To avoid double reporting of the same AE across multiple time intervals, case reports and their associated AEs were categorized by time interval according to the date when the case report was first received. Therefore, additional AEs reported subsequently under existing case reports may have actually occurred in later time intervals. AE reports were also evaluated during the first 4 years post-approval for RA (6 November 2012 to 6 November 2016), to align with the duration of available post-approval PsA data, and in subgroups by sex (male versus female) and age (≥ 65 versus < 65 years).
Results
Patient Characteristics
In total, 73,525 case reports were reviewed, comprising 5394 for PsA and 68,131 for RA. Of these, 368 (6.8%) and 4239 (6.2%), respectively, did not report a tofacitinib formulation and were excluded. In PsA, the number of case reports received for the IR versus MR formulations was similar, whereas in RA, the number of case reports received was higher for IR than for MR (Table 1). PY of exposure were higher for the IR than for the MR formulation for both indications. For both indications and formulations, AE reports were more commonly submitted for females, patients < 65 years of age, and patients from North America (Table 1). Similar trends in demographics were observed for the reports with no tofacitinib formulation specified (Table S1). Most reports originated from North America (almost all for MR), and the proportion of reports originating from Europe and the rest of the world was higher with IR than with MR (Table 1).
Approximately half of all reports were submitted by HCPs, with the remainder submitted by non-HCPs such as consumers (Table 2). A small percentage of reports included multiple indications (19.4% with PsA and 4.6% with RA) (Table 2). For PsA, the most reported co-indications were RA (46.5% of those with multiple indications), psoriasis (17.5%), and ankylosing spondylitis (3.7%). The most reported co-indications for RA were PsA (15.8% of those with multiple indications), osteoarthritis (10.1%), and arthritis (8.8%).
AEs
For both PsA and RA, a higher number of AEs were reported for tofacitinib IR (PsA, n = 8349; RA, n = 137,476) versus MR (PsA, n = 7602; RA, n = 82,153) (Table 3). For both indications, RRs for total AEs and SAEs were higher with the MR versus IR formulation, although frequency of SAEs (percentage of AEs reported as serious) was similar. No clear trends across formulations were observed in frequency or RR of AESIs and fatal cases (Table 3). Results for reports with no tofacitinib formulation specified are shown in Table S2. Over the full duration of data collection, a higher RR for total AEs was observed in PsA than in RA (Table 3). In the sensitivity analysis evaluating only the first 4 years post-approval, AE RRs were higher for RA (Table S3) than in the 4-year data for PsA (Table 3) for both formulations, and higher for MR versus IR in both indications. There was limited exposure to the MR formulation in the first 4 years for RA (November 2012 to November 2016) as it was approved in February 2016 (2000 PY, versus 49,439 PY for IR).
When case reports were evaluated by 2-year time intervals, the number of case reports, AEs, and PY of exposure increased over time for both indications (Table 4). For PsA, RRs of AEs, SAEs, AESIs, and fatal cases were similar across the two time intervals examined, as were the frequencies of SAEs, AESIs, and fatal cases. For RA, RRs of AEs, SAEs, most AESIs, and fatal cases were highest in the first time interval (November 2015 to November 2017) and lower thereafter; frequencies of SAEs, AESIs, and fatal cases were comparable across time intervals (Table 4).
In patients with PsA, the most frequently reported PTs overall were drug ineffective, off-label use, condition aggravated, pain, and headache (Fig. 1). In patients with RA, the most frequently reported PTs overall were drug ineffective, pain, condition aggravated, and headache (Fig. 2). For both PsA and RA, the most frequently reported PTs were similar across IR and MR formulations (Figs. 1, 2). Results for reports with no formulation specified are shown in Fig. S1.

Most frequent AEs occurring in ≥ 2% of patients with PsA (by PT). Percentages were calculated from the total case reports per formulation. AE adverse event, COVID-19 Coronavirus disease 2019, IR immediate release, MR modified release, PsA psoriatic arthritis, PT Preferred Term

Most frequent AEs occurring in ≥ 2% of patients with RA (by PT). Percentages were calculated from the total case reports per formulation. AE adverse event, COVID-19 Coronavirus disease 2019, HZ herpes zoster, IR immediate release, MR modified release, PT Preferred Term, RA rheumatoid arthritis
Across PsA and RA, RRs for AEs were higher for female patients than for male patients, although RRs for SAEs, AESIs, and fatal cases were generally consistent (Table 5). Similarly, in both PsA and RA, RRs for AEs were higher in patients < 65 years of age than in patients ≥ 65 years of age, with RRs for SAEs, AESIs, and fatal cases remaining comparable across the age categories.
AESIs
Across indications and formulations, the most reported AESI was serious infection, followed by HZ (Table 3). Within the AESI category of serious infections, the most reported PTs were pneumonia, lower respiratory tract infection, and COVID-19 or COVID-19 pneumonia (Table 6). The frequency and RR for COVID-19 infections in PsA were 0.92% and 0.71, respectively, with 10.2% (15/147) of these infections being reported as serious. Frequency and RR for COVID-19 infections in RA were 0.58% and 0.29, respectively, with 39.1% (494/1263) of these infections reported as serious. The most reported PTs in the HZ AESI category were HZ, ophthalmic HZ, and HZ disseminated (Table 6). Of the total HZ AEs reported, 10.2% (IR) and 8.6% (MR) for PsA and 24.1% (IR) and 9.8% (MR) for RA were considered serious. The most reported PT meeting cardiovascular event AESI criteria was cerebrovascular accident, followed by myocardial infarction and pulmonary embolism (Table 6). Excluding the nonspecific PT of neoplasm malignant, breast cancer or breast cancer female was the most reported PT meeting AESI criteria for malignancies excluding NMSC, followed by lung neoplasm malignant and colon cancer (Table 6). The most reported NMSC PT was skin cancer, followed by basal cell carcinoma and squamous cell carcinoma (Table 6). The majority of VTEs reported were pulmonary embolism, followed by deep vein thrombosis and pulmonary thrombosis (Table 6).
Discussion
In this analysis, post-marketing safety of tofacitinib in PsA and RA ascertained from AE reports submitted to the Pfizer safety database was aligned with the known safety profile of tofacitinib. This analysis, with 4 years of data, is the first PMS report for tofacitinib in PsA, and extends the RA PMS data across a longer timeframe (9 years) than previously published data. Trends in reporting of overall AEs, including types and seriousness, were comparable across the PsA and RA indications. PMS data for AESIs and the most commonly reported AEs provide complementary information to the safety results of previous clinical and observational trials.
Prior post-marketing data in RA [49] and UC [50] generally showed safety reporting trends similar to those of the present study. For example, the most frequently reported AEs (by PT) were mostly consistent with those reported in the earlier RA PMS study and the UC PMS study. In all three studies, drug ineffective, condition aggravated, headache, diarrhea, and fatigue were among the top ten most frequently reported AEs. In contrast to these findings for the PMS reports, data from the tofacitinib clinical development program found that nasopharyngitis and upper respiratory tract infections were the most common AEs experienced by patients receiving tofacitinib for PsA or RA [39]. These differences may relate to methodology; in clinical trials, all potential AEs are captured in a tightly controlled setting (including nonsevere respiratory illnesses), whereas for spontaneous reporting, AEs that are suspected to be treatment-related may be prioritized.
In this analysis, off-label use was coded as an AE in line with its inclusion as a MedDRA PT. Off-label use was the second most frequently reported AE for PsA, which might be attributable to use as monotherapy (tofacitinib is approved for PsA in combination with methotrexate or nonbiologic DMARDs, depending on the country). A recent analysis of US claims data found that 62.6% of patients treated with tofacitinib for PsA were receiving monotherapy [52]. Alternatively, off-label use may represent utilization of a higher dose, such as 10 mg BID, which is not approved for PsA, and is approved for RA in Russia and Botswana (and formerly Switzerland, until 2020).
Our study provides important insight into the safety profile of tofacitinib in PsA, which was similar overall to the safety profile in RA. Higher RRs for AEs were observed for PsA than for RA in the present study over the full period of data collection, which covered 9 years for RA and 4 years for PsA. However, when restricting the analysis to the first 4 years post-approval for each indication, to align the duration of data collection, the RRs were higher for RA versus PsA. RRs are typically highest in the first 2 years post-approval (a phenomenon referred to as the Weber effect) [53], so these results were not unexpected, given that tofacitinib was approved for RA before PsA, and had a novel mechanism of action at the time of its first approval. Also, RA has been associated with a generally higher comorbidity burden than PsA [8], which may increase patients’ likelihood of experiencing AEs [54]. In pooled analyses of the tofacitinib clinical development program, incidence rates of SAEs and AEs leading to discontinuation were higher in RA than in PsA, while rates of AESIs were generally comparable, with the exception of numerically lower rates of serious infections and HZ in PsA versus RA [39]. These differences were suggested to relate to patient characteristics including older age and higher corticosteroid use in the RA cohort versus the PsA cohort [39]. In these PMS data, we did not observe noticeable differences in frequency or RR of AESIs between PsA and RA. Previous registry studies have found higher exposure-adjusted incidence rates and/or risk ratios of infections [9], MACE [13, 14], malignancies [10], and VTE [11] in RA versus PsA. In this analysis, the most frequent AEs reported by PT were similar between PsA and RA, with the exception of differences in off-label use, as discussed above. To our knowledge, no other PMS data are available comparing rates of AESIs in PsA versus RA.
Overall, the AESIs observed in this study were aligned with those observed in other PMS studies of bDMARDs in PsA and RA [55,56,57,58,59], but differences in geographical regions, patient and disease characteristics, and data-collection methodology make it difficult to compare AE rates across studies. In previous real-world safety studies, rates of AESIs were generally comparable between tofacitinib and bDMARDs in overall populations of patients with RA, with the exception of HZ, which occurred more frequently with tofacitinib than with bDMARDs [45, 46, 60]. No comparable large studies have been published in PsA, but an analysis comparing tofacitinib clinical data with US Truven MarketScan registry data for bDMARDs (with patient exclusion criteria similar to those of the tofacitinib clinical trials applied) showed generally similar incidence rates for most AESIs in PsA, except for rates of HZ which, as expected, were higher in the tofacitinib clinical data than in the bDMARD observational data [61]. In patients with RA and elevated cardiovascular risk, differences in AESI rates between tofacitinib and bDMARDs have been noted in the ORAL Surveillance and STAR-RA studies [35, 45, 46].
A trend towards higher reporting of AEs and SAEs with the MR formulation than with the IR formulation was observed across indications, although RRs for AESIs and fatal cases were similar. There are several factors that might explain the higher volume of reports, and resulting RR, for MR relative to IR. Notably, SAEs occurred with similar frequency (in terms of percentage) between the MR and IR formulations, possibly indicating that the difference could be due to reporting trends rather than a reflection of differing safety profiles. The total exposure time for the MR formulation was lower than for the IR formulation, which may have impacted the resulting RR. Almost all of the case reports received for the MR formulation originated from North America, potentially indicating a regional trend in reporting frequency. The MR formulation was first approved in North America, and the US contributes the highest number of AE reports globally to large Individual Case Safety Reports databases such as VigiBase and the FDA Event Reporting System (FAERS) [62, 63]. In addition, specialists may be more likely than general practitioners to report AEs [64], and the US has a higher density of rheumatologists than other countries with large populations [65]. Furthermore, the MR QD formulation might be preferred by patients receiving multiple treatments per day, who may have comorbidities and may potentially experience a greater number of AEs [54]. Notably, approximately twice as many patients receiving the MR formulation reported multiple indications as those receiving the IR formulation. In a randomized clinical trial setting, the safety profile of the IR and MR formulations was comparable [40]. The two formulations have been shown to have equivalent pharmacokinetic profiles based on areas under the curve [66], and similar effectiveness in the real-world CorEvitas (formerly Corrona) RA Registry [67].
External factors such as regulatory safety alerts, or changes to the approval status of a drug, have been shown to lead to increases in AE reporting [68], which is termed notoriety bias [69]. In 2019, an ad hoc safety analysis of the ORAL Surveillance study revealed increases in rates of pulmonary embolism with tofacitinib 10 mg BID versus TNFi, and all-cause mortality with tofacitinib 10 mg BID versus tofacitinib 5 mg BID and TNFi. Additionally, in the final results, non-inferiority was not shown for the combined tofacitinib doses (5 and 10 mg BID) versus TNFi for the co-primary endpoints of MACE and malignancy excluding NMSC [35]. After the results of ORAL Surveillance, signal detection studies of JAK inhibitors using FAERS and VigiBase have revealed increased reporting odds ratios for VTE or thromboembolic events [70, 71]. In another analysis of VigiBase directly comparing JAK inhibitors and TNFi, no increased reporting odds ratio was found for MACE, although an increased risk of VTE was observed [47]. In our analysis of PMS data collected from the Pfizer safety database, an increase in event reporting over time was not consistently observed across AEs, which might suggest that differences in reporter identity or training can influence post-marketing data collection.
There was a higher proportion of reports received for females than for males; this trend is expected for RA, given the epidemiological rates of disease prevalence (approximately 2–5 times higher in females than males, depending on age [72]); however, this was unexpected for PsA (approximately 1:1 female:male ratio [2]), suggesting a bias for reporting AEs in females compared with males. It is not surprising that a higher RR is observed in female patients versus male patients, given the higher volume of reports for females. It will be important to further explore whether these trends are based on a higher likelihood of the attending HCP, or the consumer, reporting an event occurring in a female, or if the rate of AEs occurring is truly higher in females. When considering SAEs, AESIs, and fatal cases, the RRs were similar between sexes. Higher RRs were also observed in patients < 65 versus ≥ 65 years of age. In this case, it is possible that AEs in patients ≥ 65 years of age are more likely to be attributed to older age than to the treatment and are therefore less likely to be reported. Alternatively, these trends in RRs could point to differences in clinical care between subgroups. In the VigiBase analysis of MACE and VTE comparing JAK inhibitors and TNFi, age and sex did not significantly influence RRs [47].
Limitations of this analysis include the potential for reporting bias (e.g., favoring female and/or younger patients), varied reporter identity/training (e.g., consumer versus physician; specialists may be more likely to report than general practitioners), and exposure estimation from commercial sales data (covering only 61 countries and one region, with indication-share derived from even fewer countries [n = 18] where prescription data were available). When interpreting the AE data, the use of mixed data sources should be noted; the RRs in this analysis were calculated using the estimated exposure data, while the number and frequency (percentage) are solely based on data from case reports. Also, more data were available for RA than for PsA, and for IR than for MR tofacitinib, which should be considered when interpreting differences across indications and formulations. Furthermore, causality of AEs, considering tofacitinib compared with other concomitant medications, was not robustly collected. Most patients would have received concomitant methotrexate or other csDMARDs per regulatory labeling, so the role of these medications in contributing to AE risk cannot be ruled out. Other limitations of PMS data in general include under-reporting of nonserious AEs, difficulty identifying events with low frequency, and difficulty quantifying risks (owing to the lack of a reliable denominator) [73].
Conclusions
This PMS study using data ascertained from submitted AE reports found that safety findings for overall AEs and AESIs with tofacitinib were consistent between PsA and RA, and were aligned with the known safety profile of tofacitinib. Frequencies of SAEs, AESIs, and fatal cases (as a proportion of total AEs or total cases) were similar between tofacitinib formulations, while RRs were higher with the MR formulation versus the IR formulation. This difference in RRs may relate to differences in cumulative exposure, regional reporting trends, or different patient populations. Potential trends in reporting by sex and age require further assessment, with higher RRs observed in females than in males, and in younger than in older patients. While these results should be interpreted in the context of the limitations of PMS studies and spontaneous AE reporting, this study provides important insight into the global real-world safety profile of tofacitinib, reported here for the first time in PsA.
Change history
02 December 2023
A Correction to this paper has been published: https://doi.org/10.1007/s40744-023-00615-4
References
Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. 2017;376:957–70.
Scotti L, Franchi M, Marchesoni A, Corrao G. Prevalence and incidence of psoriatic arthritis: a systematic review and meta-analysis. Semin Arthritis Rheum. 2018;48:28–34.
Hagberg KW, Li L, Peng M, Paris M, Shah K, Jick SS. Rates of cancers and opportunistic infections in patients with psoriatic arthritis compared with patients without psoriatic arthritis. J Clin Rheumatol. 2016;22:241–7.
Smolen JS, Aletaha D, Barton A, et al. Rheumatoid arthritis. Nat Rev Dis Primers. 2018;4:18001.
Almutairi K, Nossent J, Preen D, Keen H, Inderjeeth C. The global prevalence of rheumatoid arthritis: a meta-analysis based on a systematic review. Rheumatol Int. 2021;41:863–77.
Roubille C, Richer V, Starnino T, et al. Evidence-based recommendations for the management of comorbidities in rheumatoid arthritis, psoriasis, and psoriatic arthritis: expert opinion of the Canadian Dermatology-Rheumatology Comorbidity Initiative. J Rheumatol. 2015;42:1767–80.
Taylor PC, Atzeni F, Balsa A, Gossec L, Müller-Ladner U, Pope J. The key comorbidities in patients with rheumatoid arthritis: a narrative review. J Clin Med. 2021;10:509.
Merola JF, Espinoza LR, Fleischmann R. Distinguishing rheumatoid arthritis from psoriatic arthritis. RMD Open. 2018;4: e000656.
Krabbe S, Grøn KL, Glintborg B, et al. Risk of serious infections in arthritis patients treated with biological drugs: a matched cohort study and development of prediction model. Rheumatology (Oxford). 2021;60:3834–44.
Dreyer L, Mellemkjaer L, Andersen AR, et al. Incidences of overall and site specific cancers in TNFα inhibitor treated patients with rheumatoid arthritis and other arthritides—a follow-up study from the DANBIO Registry. Ann Rheum Dis. 2013;72:79–82.
Ogdie A, Kay McGill N, Shin DB, et al. Risk of venous thromboembolism in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a general population-based cohort study. Eur Heart J. 2018;39:3608–14.
Ferguson LD, Siebert S, McInnes IB, Sattar N. Cardiometabolic comorbidities in RA and PsA: lessons learned and future directions. Nat Rev Rheumatol. 2019;15:461–74.
Ogdie A, Yu Y, Haynes K, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2015;74:326–32.
Lauper K, Courvoisier DS, Chevallier P, Finckh A, Gabay C. Incidence and prevalence of major adverse cardiovascular events in rheumatoid arthritis, psoriatic arthritis, and axial spondyloarthritis. Arthritis Care Res (Hoboken). 2018;70:1756–63.
Fraenkel L, Bathon JM, England BR, et al. 2021 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2021;73:1108–23.
Smolen JS, Landewé RBM, Bergstra SA, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. 2022;82:3–18.
Gossec L, Baraliakos X, Kerschbaumer A, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis. 2020;79:700–12.
Singh JA, Guyatt G, Ogdie A, et al. Special article: 2018 American College of Rheumatology/National Psoriasis Foundation guideline for the treatment of psoriatic arthritis. Arthritis Rhematol. 2019;71:5–32.
Coates LC, Soriano ER, Corp N, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA): updated treatment recommendations for psoriatic arthritis 2021. Nat Rev Rheumatol. 2022;18:465–79.
Haynes K, Beukelman T, Curtis JR, et al. Tumor necrosis factor α inhibitor therapy and cancer risk in chronic immune-mediated diseases. Arthritis Rheum. 2013;65:48–58.
Ozen G, Pedro S, England BR, Mehta B, Wolfe F, Michaud K. Risk of serious infection in patients with rheumatoid arthritis treated with biologic versus nonbiologic disease-modifying antirheumatic drugs. ACR Open Rheumatol. 2019;1:424–32.
Curtis JR, Patkar N, Xie A, et al. Risk of serious bacterial infections among rheumatoid arthritis patients exposed to tumor necrosis factor alpha antagonists. Arthritis Rheum. 2007;56:1125–33.
Gladman D, Rigby W, Azevedo VF, et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med. 2017;377:1525–36.
Mease P, Hall S, FitzGerald O, et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med. 2017;377:1537–50.
Leng X, Lin W, Liu S, et al. Efficacy and safety of tofacitinib in Chinese patients with active psoriatic arthritis: a phase 3, randomised, double-blind, placebo-controlled study. RMD Open. 2023;9: e002559.
Nash P, Coates LC, Fleishaker D, et al. Safety and efficacy of tofacitinib up to 48 months in patients with active psoriatic arthritis: final analysis of the OPAL Balance long-term extension study. Lancet Rheumatol. 2021;3:e270–83.
Burmester GR, Blanco R, Charles-Schoeman C, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451–60.
Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507.
Lee EB, Fleischmann R, Hall S, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370:2377–86.
Kremer J, Li Z-G, Hall S, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159:253–61.
van der Heijde D, Tanaka Y, Fleischmann R, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013;65:559–70.
van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–19.
van der Heijde D, Strand V, Tanaka Y, et al. Tofacitinib in combination with methotrexate in patients with rheumatoid arthritis: clinical efficacy, radiographic, and safety outcomes from a twenty-four-month, phase III study. Arthritis Rheumatol. 2019;71:878–91.
Fleischmann R, Mysler E, Hall S, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet. 2017;390:457–68.
Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386:316–26.
Wollenhaupt J, Lee EB, Curtis JR, et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: final results of a global, open-label, long-term extension study. Arthritis Res Ther. 2019;21:89.
Wollenhaupt J, Silverfield J, Lee EB, et al. Safety and efficacy of tofacitinib, an oral Janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J Rheumatol. 2014;41:837–52.
Yamanaka H, Tanaka Y, Takeuchi T, et al. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background methotrexate, in Japanese patients with rheumatoid arthritis: an open-label, long-term extension study. Arthritis Res Ther. 2016;18:34.
Burmester GR, Nash P, Sands BE, et al. Adverse events of special interest in clinical trials of rheumatoid arthritis, psoriatic arthritis, ulcerative colitis and psoriasis with 37 066 patient-years of tofacitinib exposure. RMD Open. 2021;7: e001595.
Tanaka Y, Sugiyama N, Toyoizumi S, et al. Modified- versus immediate-release tofacitinib in Japanese rheumatoid arthritis patients: a randomized, phase III, non-inferiority study. Rheumatology (Oxford). 2019;58:70–9.
Cohen SB, Pope J, Haraoui B, et al. Methotrexate withdrawal in patients with rheumatoid arthritis who achieve low disease activity with tofacitinib modified-release 11 mg once daily plus methotrexate (ORAL Shift): a randomised, phase 3b/4, non-inferiority trial. Lancet Rheumatol. 2019;1:E23–34.
Charles-Schoeman C, Fleischmann R, Davignon J, et al. Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheumatol. 2015;67:616–25.
Charles-Schoeman C, Gonzalez-Gay MA, Kaplan I, et al. Effects of tofacitinib and other DMARDs on lipid profiles in rheumatoid arthritis: implications for the rheumatologist. Semin Arthritis Rheum. 2016;46:71–80.
Pfizer Inc. Xeljanz® (tofacitinib): highlights of prescribing information. 2020. http://labeling.pfizer.com/ShowLabeling.aspx?id=959. Accessed Oct 14, 2021.
Khosrow-Khavar F, Desai RJ, Lee H, Lee SB, Kim SC. Tofacitinib and risk of malignancy: results from the Safety of TofAcitinib in Routine care patients with Rheumatoid Arthritis (STAR-RA) study. Arthritis Rheumatol. 2022;74:1648–59.
Khosrow-Khavar F, Kim SC, Lee H, Lee SB, Desai RJ. Tofacitinib and risk of cardiovascular outcomes: results from the Safety of TofAcitinib in Routine care patients with Rheumatoid Arthritis (STAR-RA) study. Ann Rheum Dis. 2022;81:798–804.
Montastruc F, Flumian C, Degboe Y, Constantin A, Ruyssen-Witrand A. OP0268 Comparison of major cardiovascular and thromboembolic events in safety reports between rheumatoid arthritis patients treated with JAK inhibitors versus anti-TNF: results from VigiBase [abstract]. Ann Rheum Dis. 2022;81(Suppl 1):OP0268.
Ramiro S, Nikiphorou E, Sepriano A, et al. ASAS-EULAR recommendations for the management of axial spondyloarthritis: 2022 update [abstract]. Arthritis Rheumatol. 2022;74(Suppl 9):0542.
Cohen S, Curtis JR, DeMasi R, et al. Worldwide, 3-year, post-marketing surveillance experience with tofacitinib in rheumatoid arthritis. Rheumatol Ther. 2018;5:283–91.
Rubin DT, Modesto I, Vermeire S, et al. Worldwide post-marketing safety surveillance experience with tofacitinib in ulcerative colitis. Aliment Pharmacol Ther. 2021;55:302–10.
Galíndez-Agirregoikoa E, Prieto-Peña D, Martín-Varillas JL, et al. Treatment with tofacitinib in refractory psoriatic arthritis: a national multicenter study of the first 87 patients in clinical practice. J Rheumatol. 2021;48:1552–8.
Mease PJ, Young P, Gruben D, Fallon L, Germino R, Kavanaugh A. Early real-world experience of tofacitinib for psoriatic arthritis: data from a United States Healthcare Claims Database. Adv Ther. 2022;39:2932–45.
Hartnell NR, Wilson JP. Replication of the Weber effect using postmarketing adverse event reports voluntarily submitted to the United States Food and Drug Administration. Pharmacotherapy. 2004;24:743–9.
Bechman K, Clarke BD, Rutherford AI, et al. Polypharmacy is associated with treatment response and serious adverse events: results from the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis. Rheumatology. 2019;58:1767–76.
Koike T, Harigai M, Inokuma S, et al. Effectiveness and safety of tocilizumab: postmarketing surveillance of 7901 patients with rheumatoid arthritis in Japan. J Rheumatol. 2014;41:15–23.
Kanbori M, Suzuka H, Yajima T, et al. Postmarketing surveillance evaluating the safety and effectiveness of golimumab in Japanese patients with rheumatoid arthritis. Mod Rheumatol. 2018;28:66–75.
Koike T, Harigai M, Inokuma S, et al. Postmarketing surveillance of safety and effectiveness of etanercept in Japanese patients with rheumatoid arthritis. Mod Rheumatol. 2011;21:343–51.
Gottlieb AB, Deodhar A, McInnes IB, et al. Long-term safety of secukinumab over five years in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis and ankylosing spondylitis: update on integrated pooled clinical trial and post-marketing surveillance data. Acta Derm Venereol. 2022;102:adv00698.
Deodhar A, Mease PJ, McInnes IB, et al. Long-term safety of secukinumab in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: integrated pooled clinical trial and post-marketing surveillance data. Arthritis Res Ther. 2019;21:111.
Kremer JM, Bingham CO, Cappelli LC, et al. Postapproval comparative safety study of tofacitinib and biological disease-modifying antirheumatic drugs: 5-year results from a United States-based rheumatoid arthritis registry. ACR Open Rheumatol. 2021;3:173–84.
Burmester GR, Curtis JR, Yun H, et al. An integrated analysis of the safety of tofacitinib in psoriatic arthritis across phase III and long-term extension studies with comparison to real-world observational data. Drug Saf. 2020;43:379–92.
U.S. Food & Drug Administration. FAERS domestic and foreign reports by year. 2015. https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/faers-domestic-and-foreign-reports-year. Accessed Dec 5, 2022.
Watson S, Caster O, Rochon PA, den Ruijter H. Reported adverse drug reactions in women and men: aggregated evidence from globally collected individual case reports during half a century. EClinicalMedicine. 2019;17: 100188.
Thiessard F, Roux E, Miremont-Salamé G, et al. Trends in spontaneous adverse drug reaction reports to the French pharmacovigilance system (1986–2001). Drug Saf. 2005;28:731–40.
Al Maini M, Adelowo F, Al Saleh J, et al. The global challenges and opportunities in the practice of rheumatology: white paper by the World Forum on Rheumatic and Musculoskeletal Diseases. Clin Rheumatol. 2015;34:819–29.
Lamba M, Wang R, Fletcher T, Alvey C, Kushner J, Stock TC. Extended-release once-daily formulation of tofacitinib: evaluation of pharmacokinetics compared with immediate-release tofacitinib and impact of food. J Clin Pharmacol. 2016;56:1362–71.
Cohen S, Litman HJ, Chen C, et al. Clinical effectiveness of tofacitinib 11 mg once daily (QD) versus tofacitinib 5 mg twice daily (BID) in the Corrona US RA registry [abstract]. Arthritis Rheumatol. 2018;70(Suppl 10):580.
Motola D, Vargiu A, Leone R, et al. Influence of regulatory measures on the rate of spontaneous adverse drug reaction reporting in Italy. Drug Saf. 2008;31:609–16.
Pariente A, Gregoire F, Fourrier-Reglat A, Haramburu F, Moore N. Impact of safety alerts on measures of disproportionality in spontaneous reporting databases: the notoriety bias. Drug Saf. 2007;30:891–8.
Setyawan J, Azimi N, Strand V, Yarur A, Fridman M. Reporting of thromboembolic events with JAK inhibitors: analysis of the FAERS database 2010–2019. Drug Saf. 2021;44:889–97.
Vallejo-Yagüe E, Weiler S, Micheroli R, Burden AM. Thromboembolic safety reporting of tofacitinib and baricitinib: an analysis of the WHO VigiBase. Drug Saf. 2020;43:881–91.
Kvien TK, Uhlig T, Ødegård S, Heiberg MS. Epidemiological aspects of rheumatoid arthritis: the sex ratio. Ann N Y Acad Sci. 2006;1069:212–22.
Goldman SA. Limitations and strengths of spontaneous reports data. Clin Ther. 1998;20 Suppl C:C40–4.
Acknowledgements
The authors would like to acknowledge Rob Julg, an employee and shareholder of Pfizer Inc, for important contributions to this work.
Funding
This study was sponsored by Pfizer. The sponsor funded the journal’s Rapid Service Fee and medical writing support, as described below.
Medical Writing, Editorial, and Other Assistance
Medical writing support, under the direction of the authors, was provided by Julia King, PhD, and Justine Juana, BScH, CMC Connect, a division of IPG Health Medical Communications, and was funded by Pfizer, New York, NY, USA, in accordance with Good Publication Practice (GPP2022) guidelines (Ann Intern Med. 2022;175:1298–304).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Gerd R. Burmester, Stanley B. Cohen, Ivana Vranic, Edward Nagy, Kenneth Kwok, Lara Fallon, and Cassandra Kinch made substantial contributions to the study conception and design. Gerd R. Burmester, Ivana Vranic, Edward Nagy, Irina Lazariciu, and Cassandra Kinch made substantial contributions to the acquisition of data. Gerd R. Burmester, Laura C. Coates, Stanley B. Cohen, Yoshiya Tanaka, Ivana Vranic, Edward Nagy, Irina Lazariciu, All-shine Chen, Kenneth Kwok, Lara Fallon, and Cassandra Kinch made substantial contributions to the analysis and interpretation of data. All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be submitted for publication.
Prior Presentation
Parts of the data in this manuscript were presented at EULAR 2023 (European Congress of Rheumatology; May 31–June 3, 2023; Milan, Italy).
Disclosures
Gerd R. Burmester has received honoraria for lectures and consulting for AbbVie, Amgen, Bristol Myers Squibb, Eli Lilly, Galapagos, Janssen, Novartis, Pfizer Inc, and Sanofi. Laura C. Coates has been paid as a speaker for AbbVie, Amgen, Biogen, Celgene, Eli Lilly, Galapagos, Gilead Sciences, GSK, Janssen, Medac, Novartis, Pfizer Inc, and UCB; has worked as a paid consultant for AbbVie, Amgen, Bristol Myers Squibb, Celgene, Eli Lilly, Gilead, Galapagos, Janssen, MoonLake, Novartis, Pfizer Inc, and UCB; and has received grants/research support from AbbVie, Amgen, Celgene, Eli Lilly, Janssen, Novartis, Pfizer Inc, and UCB. Stanley B. Cohen has received consulting fees or other remuneration from AbbVie, Amgen, Boehringer Ingelheim, Gilead Sciences, Merck, and Pfizer Inc. Yoshiya Tanaka has received speaking fees and/or honoraria from AbbVie, AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Chugai, Daiichi Sankyo, Eisai, Eli Lilly, Gilead Sciences, GSK, Mitsubishi Tanabe, and Pfizer Inc; and has received research grants from AbbVie, Asahi Kasei, Boehringer Ingelheim, Chugai, Daiichi Sankyo, Eisai, and Takeda. Ivana Vranic, Edward Nagy, All-shine Chen, Kenneth Kwok, Lara Fallon, and Cassandra Kinch are employees and shareholders of Pfizer Inc. Irina Lazariciu is an employee of IQVIA, who were paid contractors to Pfizer Inc in the development of this manuscript and in providing statistical support.
Compliance with Ethics Guidelines
The spontaneously reported PMS AE data in this analysis were not collected as part of a clinical study and were non-interventional; therefore, no ethics approval was required. All data were reported in aggregate form in summary reports; no individual case-level data were evaluated or reported.
Data Availability
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original online version of this article was revised to correct few incorrect values in Table 5.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Burmester, G.R., Coates, L.C., Cohen, S.B. et al. Post-Marketing Safety Surveillance of Tofacitinib over 9 Years in Patients with Psoriatic Arthritis and Rheumatoid Arthritis. Rheumatol Ther 10, 1255–1276 (2023). https://doi.org/10.1007/s40744-023-00576-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-023-00576-8