
Abstract
Purpose
In 2015, an interdisciplinary project was started to fill the gap of knowledge on the toxicokinetics of aluminium (Al) after exposure from adjuvanted products for subcutaneous immunotherapy (SCIT).
Methods
Two complementary initiatives of the project are explained. The results of two studies are reviewed and put in connection with the overarching goal. An estimate is given which steps have been reached and which are still needed.
Results
Recent in vivo data provided evidence of systemically available Al from SCIT products in rats (Weisser et al. 2020 [1]). The data are highly valuable for further development of the physiology-based toxicokinetic (PBTK) model for Al exposure which has been established in parallel (Hethey et al. 2021 [2]).
Conclusion
The Hethey model is an important step towards prediction of Al exposure in man from various sources. For use in risk assessment of Al exposure from SCIT products further extension of the model is warranted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Allergen-specific immunotherapy (AIT) is a well-established treatment for allergies with gradually increasing doses of allergen extracts over a period of 2–3 years. Most of the subcutaneous allergen immunotherapy (SCIT) products marketed in Europe are adsorbed to aluminium hydroxide (AH) [3]. Production is based on either adsorption to commercial AH gel-like suspensions (e.g., Alhydrogel®, Croda International, Snaith, UK) or in situ coprecipitation of AH with the candidate antigens [4]. According to the European Pharmacopoeia the Al content in SCIT products is limited to 1.25 mg per dose and has to be labelled in the product information [5].
With regard to the cumulative Al amount administered per year (up to 15 mg Al following 4‑weekly maximum doses), SCIT constitutes an important source of Al in humans considering the provisional tolerable weekly intake (PTWI) for Al from all dietary sources (2 mg/kg body weight) and an oral bioavailability of aluminium from food of 0.1% [6].
Up to 2014, knowledge of aluminium toxicokinetics after subcutaneous (SC) injection of Al-adjuvanted SCIT products was sparse. While Al bioavailability after parenteral administration is supposed to be 100%, the rate of absorption and thus the potential Al increase in plasma and tissues over time in humans was unknown. Due to the insolubility of the Al complexes absorption rate was expected to be slow. Investigations at SC [7] or intramuscular (IM) [8] injection sites in animals suggested absorption time periods of several months for an AH adjuvant.
On the one hand, a physiology-based toxicokinetic (PBTK) model was urgently needed for extrapolation of animal data to humans [9]. On the other hand, relevant animal data on Al absorption and distribution after administration of Al-adjuvanted products to inform such a model were lacking [10, 11]. In 2015, we therefore started an ambitious interdisciplinary toxicokinetic project. The main objectives were on the one hand to generate new in vivo data on the kinetics of Al adjuvants and on the other hand to develop a PBTK model suitable for prediction of Al exposure in rats and humans. The ultimate goal was to get a valid tool for prediction of Al tissue accumulation after Al adjuvant exposure from SCIT and vaccine products in all age groups. Such predictions were expected to substantially improve regulatory risk assessment.
Methods
Two main studies have been completed so far and are comparatively reviewed in the following.
The kinetic study in rats was conducted as described in [1] (full methods see Appendix). Figure 1 depicts a summary of the study design: In brief, groups of Wistar rats (approximately 2 months of age; mean body weight 350 g) received a single SC injection (1 mL) of either plain AH adjuvant (AH: Alhydrogel®, 1.25 mg Al/mL; n = 6), each of two AH-adjuvanted SCIT products at highest marketed strength which differ in adjuvant manufacture (P1: Alhydrogel®-adjuvanted mixed grass pollen allergen, 1 mg Al/mL; P2: in situ coprecipitated AH-adjuvanted mixed grass pollen allergen, 1.13 mg Al/mL; n = 6 each), or vehicle (saline; n = 3). Al in plasma (over 80 days) and tissues (at day 80; whole humerus bone, right brain hemisphere, and injection site subcutis) was determined by atomic absorption spectrometric (AAS) procedures.

Design of the rat study on the kinetics of AH adjuvant and AH-adjuvanted allergen products P1 and P2 [1]
In parallel, a comprehensive collection of study data on 26Al kinetics in rat and man has been established and utilised to build a PBTK model [2]. The final curated dataset contained 319 human and 620 rat 26Al observations in various tissues (Table 2 in [2]). All data resulted from single PO (peroral) or IV (intravenous) dose administration of 26Al citrate or chloride, except for a single human, which received a second dose after 2 years. The model performance was investigated by use of an external 27Al dataset on the plasma kinetics of IV Al citrate in rats [12], a full-body retention dataset after IV 26Al citrate in humans [13], and a data set of Al plasma data in humans published by de Ligt et al. (2018, [14]) comprising 14 women who IV received small amounts of 26Al as citrate. All datasets and calculations were provided in the Supplementary Material to [2].
Results
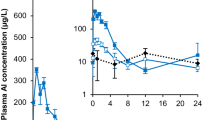
Time courses of Al in plasma did not exhibit profiles distinctive from that of the vehicle group, partly due to a relatively high value of 11 µg/L in the vehicle group (see Fig. 1 in [1]). However, after P2, but not after P1 and AH, a significant increase in Al plasma AUC (area under the curve) over 80 days [AUC(0–80d)] compared to vehicle was observed (1431 ± 314 vs. 909 ± 115 µg/L⋅d; mean ± standard deviation [SD]; Fig. 2).

Area under the curve over 80 days [AUC(0–80d), +SD] of Al plasma concentrations after administration of plain adjuvant (AH), SCIT products P1 or P2, or vehicle (saline). *p < 0.05 (Wilcoxon–Mann–Whitney test on difference to vehicle)
Bone Al content was significantly increased in all groups, again strongest in the P2 group [mean (standard deviation): 1.28 (0.26), 0.64 (0.12), 0.72 (0.10), and 0.40 (0.11) µg/g wet weight (ww) for P2, P1, AH, and vehicle, respectively]. No significant difference in brain Al concentration between any treatment and vehicle was observed (Fig. 2 and Table S2 in [1]).
A significant decrease (%) of the total Al amount injected was observed in samples of the injection site subcutis at day 80 for P1 (11.2%) and P2 (56.9%), but not for plain AH (1.7%) (Figure S3 in [1]). Figure 3 shows the calculated absolute amounts of Al released from the injection sites. The ranking of the bars of Al amounts released (Fig. 3) was similar to the ranking of Al plasma AUC (Fig. 1) and Al in bone after 80 days (Fig. 2 in [1]). This was corroborated by the close relationship between the Al amounts released with Al plasma AUC and bone Al on the individual rat level (Fig. S4 in [1]).

Total Al amount (+SD) lost/released from the injection site 80 days after administration of plain adjuvant (AH), or SCIT products (P1, P2)
In parallel, a basic PBTK model for the PO and IV exposure of soluble 26Al salts in rats and humans was successfully implemented and validated [2]. The model differentiates between administration of Al as chloride and as citrate reflecting the difference in renal filterability of Al citrate compared to Al chloride [15]. Higher plasma clearance for Al citrate compared to other salts was confirmed in our recent kinetic study after IV injection of Al citrate in rats [12].
The final model parameters predict a half-life of Al from bone of 198 (162) weeks in male (female) humans [2]. This means that each accumulated amount of Al in bone is expected to be released again but in a very slow manner taking many years for complete elimination. In contrast, none of the experimental data available in brain gave an indication of release from the tissue over time. Therefore, and in line with [16] this organ was (from a toxicological point of view) conservatively modelled as a sink.
Discussion
Our adjuvant study in rats constituted the first-time proof of systemically available Al from marketed SCIT products in vivo [1]. Al increase was mainly visible in bone, less in plasma, but not in brain. Shortly before, we could demonstrate similar evidence for Al adjuvanted vaccines administered IM in rats. It is more than two decades ago that Flarend et al. [17] investigated short-term plasma and various tissue Al concentrations in two rabbits; however, they did not evaluate Al levels in bone and injected plain self-prepared 26Al-adjuvants. More recently, McDougall et al. (2016 [7]) failed to detect changes in Al levels in plasma, kidney or brain after SC administration of an Alhydrogel® adjuvanted formulation in rats [18].
Furthermore, our data indicated that Al-adjuvanted SCIT products do not behave uniformly. We observed a remarkable difference in the degree of Al bioavailability on day 80 after injection between two marketed AH-adjuvanted SCIT products. The rate of SC absorption of Al appeared higher from a coprecipitated product than from a product adjuvanted with commercial Alhydrogel®. We are aware that tissue determination on day 80 is only a cross-sectional view and that different bone levels might only reflect different rates of absorption. This would imply that, once Al absorption is completed, two products containing the same Al dose will reach comparable cumulative Al levels in bone, however at different time points. Linear extrapolation from the loss of Al from the injection site indicated that Al absorption might have been completed after 120 (coprecipitated product) and 350 (Alhydrogel® product) days, respectively. This suggests that each single SC injection sets a depot of AH which continuously releases Al over a period of 5 months up to 1 year.
The difference in SC absorption rate observed is most probably related to the difference in physicochemical properties of the adjuvant preparation such as chemical composition, surface area, electric charge, morphological structure, and particle size [4, 19]. Between preformed commercial AH products like Alhydrogel® and coprecipitated AH adjuvants mainly the degree of crystallinity and the chemical composition appears to be different: The former consist of crystalline aluminium oxyhydroxide (AlO(OH)) identified as boehmite, whereas the latter is an amorphous aluminium hydroxyl-(buffer ion)-sulphate. Buffer and sulphate anions substitute for hydroxyls leading to varying composition and properties dependent on the precipitation conditions. For example, Al adjuvants precipitated in the presence of a phosphate buffer are essentially the same as aluminium phosphate (AP) adjuvants [4, 19]. Of note, the differences seen between P2 and P1 on day 80 resemble those between AP and AH adjuvant indicated in the early Flarend study [17] and confirmed by our recent vaccine study [18]. Thus, both amorphous structure and higher solubility might have contributed to the higher rate of systemic Al availability observed for the coprecipitated product.
The coprecipitated product P2 only showed a significant increase in total Al plasma AUC(0–80d) being a robust quantitative measure of plasma exposure. Unfortunately, the lack of a distinct plasma profile did not allow model-based evaluation of Al absorption kinetics. However, a shape without a clear Cmax might be more in accordance with zero order than with first order input kinetics.
In line with the increase in plasma AUC the P2 group also showed the highest absolute increase of Al concentration in bone (0.87 µg/g ww). A more visible increase in bone rather than plasma exposure is not surprising: rapid renal Al plasma clearance prevents a sharp rise of plasma levels above a relatively high baseline level, whereas elimination of Al from bone is very slow. The skeleton is the major storage compartment for Al in the body of both rats and humans [16]. Thus, Al amounts reaching bone build a long-term deposit which facilitates detection.
The highest total bone Al concentration measured in our rats (1.3 µg/g ww) is far below levels of toxicological concern. Studies conducted by Sun et al. [20, 21] indicated that rats with bone Al concentrations up to 15 µg/g (ww) were without abnormal findings, whereas above 20 µg/g (ww) bone formation markers decreased and oxidative stress markers increased, and in groups > 30 µg/g (ww) bone mineral density (BMD) decreased significantly. Similarly, bone Al levels below 10–15 µg/g (ww) in humans are not associated with “Al-overload” or any signs of bone toxicity [22,23,24].
We did not find a significant increase in brain Al concentration in all groups. This is not unexpected as from 26Al kinetic data in rats it is known that in contrast to bone only a very small fraction of dose retains in brain [25, 26]. As we determined Al concentration in a whole brain hemisphere also the potential presence of Al clusters due to focal accumulation reported for human brain tissues [27] could not have been missed. Furthermore, as determination by AAS comprises dissolved Al3+ ions as well as insoluble/particulate Al species, our measurements would also have captured any Al particles hypothetically transported into the brain by macrophages [28]. Thus, our results show that contribution of such particulate Al amounts, if any, are marginal.
The positive relationship found between absolute Al amount released from the injection site and both plasma AUC(0-80d) and bone Al concentration at the individual rat level confirm that the Al loss measured can be interpreted as bioavailable amount and increase in bone and plasma exposure are fairly proportional to this amount. However, we cannot exclude overestimation of Al bioavailability as we have not considered potential alternative pathways such as phagocytosis and transport of insoluble AH to the draining lymph node by antigen-presenting immune cells.
Increases of Al exposure in plasma and bone observed in rats cannot be translated one-to-one to humans; this is especially true for bone allometry with interspecies differences in bone architecture and remodelling [29]. Valid extrapolation to humans (especially children) requires physiologically based modelling. In this respect, the successful development of the Hethey-model was an important step paving the way towards a more quantitative risk assessment in humans. It has been built on the most extensive and diverse dataset of IV and PO administered 26Al exposure to date. It is a validated tool for predicting Al tissue concentrations after PO or IV Al exposure of soluble Al salts (Al citrate or Al chloride) in adult rats and humans.
However, due to those limitations (PO or IV, soluble Al salts, adulthood) further extension of the model is needed in order to enable predictions of Al exposure resulting from SC or IM applications of adjuvanted products, especially in children.
Firstly, parameters characterizing the SC or IM input kinetics of Al from adjuvants have to be implemented. Injection site data from various adjuvant studies could be helpful in this respect. Secondly, inclusion of dynamic physiological growth and maturation from neonatal age to adulthood is needed. This comprises growth of organ volumes and blood flows as well as the maturation of kidney function (GFR, glomerular filtration rate) during the first year of life which is of high importance for the elimination of Al. Thirdly, as bone is toxicologically and quantitatively highly relevant some refinement of the bone model is crucial. The model should be able to reflect changes in bone constitution, bone density and bone remodelling between species and ages. Some guidance is available from similar models for other bone-seeking elements like Sr or Pb [30, 31]. In a complementary manner the adjuvant studies in rats reviewed here can serve as valuable external data sets to test the performance of such an extended model.
Abbreviations
- AAS:
-
Atomic absorption spectrometric
- AH:
-
Aluminium hydroxide
- AIT:
-
Allergen-specific immunotherapy
- Al:
-
Aluminium
- AlO(OH):
-
Aluminium oxyhydroxide
- AP:
-
Aluminium phosphate
- AUC:
-
Area under the curve
- BMD:
-
Bone mineral density
- GFR:
-
Glomerular filtration rate
- IM:
-
Intramuscular
- PBTK:
-
Physiology-based toxicokinetic
- PTWI:
-
Provisional tolerable weekly intake
- SC:
-
Subcutaneous
- SCIT:
-
Subcutaneous immunotherapy
- ww:
-
Wet weight
References
Weisser K, Göen T, Oduro JD, Wangorsch G, Hanschmann KMO, Keller-Stanislawski B. Aluminium from adjuvanted subcutaneous allergen immunotherapeutics is mainly detected in bone. Allergy. 2020;75:215–7.
Hethey C, Hartung N, Wangorsch G, Weisser K, Huisinga W. Physiology-based toxicokinetic modelling of aluminium in rat and man. Arch Toxicol. 2021;95:2977–3000.
Mahler V, Esch RE, Kleine-Tebbe J, Lavery WJ, Plunkett G, Vieths S, et al. Understanding Differences in Allergen Immunotherapy Products and Practices in North America 13and Europe. J Allergy Clin Immunol. 2019;143:813–28.
Hem SL, HogenEsch H. Relationship between physical and chemical properties of aluminum-containing adjuvants and immunopotentiation. Expert Rev Vaccines. 2007;6:685–98.
Ph. Eur. 9.6, Monograph 1063: Allergen products (01/2019)
JECFA (Joint FAO/WHO Expert Committee on Food Additives): Safety evaluation of certain food additives and contaminants. WHO Food Additives. Series, Vol. 65. Geneva: World Health Organization; 2012.
McDougall SA, Heath MD, Kramer MF, Skinner MA. Analysis of aluminium in rat following administration of allergen immunotherapy using either aluminium or microcrystalline-tyrosine-based adjuvants. Bioanalysis. 2016;8:547–56.
Verdier F, Burnett R, Michelet-Habchi C, Moretto P, Fievet-Groyne F, Sauzeat E. Aluminium assay and evaluation of the local reaction at several time points after intramuscular administration of aluminium containing vaccines in the Cynomolgus monkey. Vaccine. 2005;23:1359–67.
Krewski D, Yokel RA, Nieboer E, Borchelt D, Cohen J, Harry J, et al. Human Health Risk Assessment for Aluminium, Aluminium Oxide, and Aluminium Hydroxide. J Toxicol Environ Health B Crit Rev. 2007;10(Suppl 1):1–269.
Weisser K, Stübler S, Matheis W, Huisinga W. Towards toxicokinetic modelling of aluminium exposure from adjuvants in medicinal products. Regul Toxicol Pharmacol. 2017;88:310–21.
Masson JD, Crépeaux G, Authier FJ, Exley C, Gherardi RK. Critical analysis of reference studies on the toxicokinetics of aluminum-based adjuvants. J Inorg Biochem. 2018;181:87–95.
Weisser K, Göen T, Oduro JD, Wangorsch G, Hanschmann KMO, Keller-Stanislawski B. Aluminium toxicokinetics after intramuscular, subcutaneous, and intravenous injection of Al citrate solution in rats. Arch Toxicol. 2019;93:37–47.
Newton D, Talbot R. Long-term retention of injected aluminium-26. Human Exp Toxicol. 2012;1:1195–8.
de Ligt R, van Duijn E, Grossouw D, Bosgra S, Burggraaf J, Windhorst A, et al. Assessment of dermal absorption of aluminum from a representative antiperspirant formulation using a 26Al microtracer approach. Clin Transl Sci. 2018;11:573–81.
Shirley DG, Lote CJ. Renal handling of aluminium. Nephron Physiol. 2005;101:p99–103.
Priest ND. The biological behaviour and bioavailability of aluminium in man, with special reference to studies employing aluminium-26 as a tracer: review and study update. J Environ Monit. 2004;6:375–403.
Flarend RE, Hem SL, White JL, Elmore D, Suckow MA, Rudy AC, et al. In vivo absorption of aluminium-containing vaccine adjuvants using 26Al. Vaccine. 1997;15:1314–8.
Weisser K, Göen T, Oduro JD, Wangorsch G, Hanschmann KMO, Keller-Stanislawski B. Aluminium in plasma and tissues after intramuscular injection of adjuvanted human vaccines in rats. Arch Toxicol. 2019;93:2787–2796
HogenEsch H, O’Hagan DT, Fox CB. Optimizing the utilization of aluminum adjuvants in vaccines: you might just get what you want. Npj Vaccines. 2018;3:51.
Sun X, Cao Z, Zhang Q, Liu S, Xu F, Che J, et al. Aluminum trichloride impairs bone and downregulates Wnt/β-catenin signaling pathway in young growing rats. Food Chem Toxicol. 2015;86:154–62.
Sun X, Liu J, Zhuang C, Yang X, Han Y, Shao B, et al. Aluminum trichloride induces bone impairment through TGF-β1/Smad signaling pathway. Toxicology. 2016;371:49–57.
Klein GL. Aluminum toxicity to bone: A multisystem effect? Osteoporos Sarcopenia. 2019;5:2–5.
Hellström HO, Mjöberg B, Mallmin H, Michaëlsson K. No association between the aluminium content of trabecular bone and bone density, mass or size of the proximal femur in elderly men and women. BMC Musculoskelet Disord. 2006;7:69.
Van Landeghem GF, D’Haese PC, Lamberts LV, Djukanovic L, Pejanovic S, Goodman WG, et al. Low serum aluminum values in dialysis patients with increased bone aluminum levels. Clin Nephrol. 1998;50:69–76.
Walker VR, Sutton RA, Meirav O, Sossi V, Johnson R, Klein J, et al. Tissue disposition of 26aluminium in rats measured by accelerator mass spectrometry. Clin Invest Med. 1994;17:420–5.
Yumoto S, Nagai H, Imamura M, Matsuzaki H, Hayashi K, Masuda A, et al. 26Al uptake and accumulation in the rat brain. Nucl Instrum Methods Phys Res B. 1997;123:279–82.
House E, Esiri M, Forster G, Ince PG, Exley C. Aluminium, iron and copper in human brain tissues donated to the Medical Research Council’s Cognitive Function and Ageing Study. Metallomics. 2012;4:56–65.
Gherardi RK, Eidi H, Crépeaux G, Authier FJ, Cadusseau J. Biopersistence and brain translocation of aluminum adjuvants of vaccines. Front Neurol. 2015;6:4.
Barak MM, Lieberman DE, Hublin JJ. Of mice, rats and men: trabecular bone architecture in mammals scales to body mass with negative allometry. J Struct Biol. 2013;183:123–31.
Pertinez H, Chenel M, Aarons L. A physiologically based pharmacokinetic model for strontium exposure in rat. Pharm Res. 2013;30:1536–52. https://doi.org/10.1007/s11095-013-0991-x.
O’Flaherty EJ. Physiologically based models for bone-seeking elements. I. Rat skeletal and bone growth. Toxicol Appl Pharmacol. 1991;111:299–312.
Funding
This work was funded by the German Ministry of Health (ZMVI1-2515-FSB-772).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
K. Weisser declares that she has no competing interests.
Ethical standards
All applicable international, national institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution (preclinics GmbH, Germany) at which the studies were conducted.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weisser, K. Toxicokinetics of aluminium—novel insights in an old adjuvant. Allergo J Int (2024). https://doi.org/10.1007/s40629-024-00288-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40629-024-00288-7