
Abstract
Background
The hypothalamic–pituitary–adrenal (HPA) axis is a neuroendocrine system involved in controlling stress responses in humans under physiological and pathological conditions; cortisol is the main hormone produced by the HPA axis. It is known that calorie restriction acts as a stressor and can lead to an increase in cortisol production. Renin–angiotensin–aldosterone system (RAAS) is a complex endocrine network regulating blood pressure and hydrosaline metabolism, whose final hormonal effector is aldosterone. RAAS activation is linked to cardiometabolic diseases, such as heart failure and obesity. Obesity has become a leading worldwide pandemic, associated with serious health outcomes. Calorie restriction represents a pivotal strategy to tackle obesity. On the other hand, it is well known that an increased activity of the HPA may favour visceral adipose tissue expansion, which may jeopardize a successful diet-induced weight loss. Very low-calorie ketogenic diet (VLCKD) is a normoprotein diet with a drastic reduction of the carbohydrate content and total calorie intake. Thanks to its sustained protein content, VLCKD is extremely effective to reduce adipose tissue while preserving lean body mass and resting metabolic rate.
Purpose
The purpose of this narrative review is to gain more insights on the effects of VLCKD on the HPA axis and RAAS, in different phases of weight loss and in different clinical settings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The purpose of this narrative review is to gain more insights on the effects of very low-calorie ketogenic diet (VLCKD) on the hypothalamic–pituitary–adrenal (HPA) axis and renin–angiotensin–aldosterone-system (RAAS), in different phases of weight loss and in different clinical settings.
The adrenal gland is a bilateral endocrine organ, located to the superior pool of each kidney, and is composed of two distinct anatomical areas: cortex and medulla [1, 2]. According to Kim and Choi, the adrenal cortex synthesizes steroid hormones from cholesterol through a series of biochemical metabolic pathways [1, 2]. Furthermore, the adrenal cortex consists of three different cortical zones: the glomerulosa zone, secreting mineralocorticoids such as aldosterone, the intermediate fasciculate zone, secreting glucocorticoids such as cortisol, and the innermost reticularis zone, secreting androgens [3, 4], as shown in Fig. 1, while the medullary area produces epinephrine and norepinephrine as part of the sympathetic nervous system [3, 4].

Adrenal gland and the division of the two distinct anatomical areas, with their hormone production
The HPA axis is a critical neurohormonal system that regulates cognitive, metabolic, immunological, and circadian behaviours and responds to internal and external stressors with continuous dynamic equilibration [5, 6]. Any activity, stimulus or situation that causes stress can be regarded as a stressor. Unlike homeostasis, which represents stability through consistency, allostasis is a capability to maintain stability through dynamic change. If the allostatic response is prolonged, inadequate or overstimulated, the reactive processes may lead to maladaptation and organ damage [7]. Glucocorticoids, secreted by the adrenal cortex, participate in this allostasis, control the gene expression for thousands of genes and exert multiple actions by binding to target tissues and activating mineralocorticoid (MR) and glucocorticoid receptors (GR) [8]. Upon reaching the systemic circulation, in healthy individuals, 90% of cortisol is bound to cortisol binding globulin, 5% to albumin leaving around 5–6% in an unbound, active state. The HPA axis activity also depends on glucocorticoid metabolism, clearance and plasma proteins. Glucocorticoids are metabolized irreversibly in the liver with the help of 5α and 5β reductase enzymes. Some conditions, such as obesity, alter the metabolic clearance rate causing an increased glucocorticoid secretion, while maintaining normal plasma levels [9]. Since overt hypercortisolism leads to metabolic manifestations such as visceral fat accumulation, hypertension and diabetes [10], the HPA axis was suggested as a contributor to metabolic dysregulation in obesity. Obesity is also often associated with a broad spectrum of different stressogenic factors, such as infertility, since the women seeking pregnancy are older and heavier than what was observed before [11].
GR are ubiquitously expressed in peripheral tissues and have low affinity and high sensitivity for corticosterone [12]. Some of the many effects of glucocorticoids are to oppose insulin action and stimulate energy turnover between proteins, triglycerides, glycogen and free fuel for mitochondrial oxidation [13, 14], while also having significant effects on cardiovascular tissues, vascular contractility, atherosclerotic process and angiogenesis [15].
Visceral fat represents an important target for glucocorticoid action gene expression [16]. Glucocorticoids induce differentiation of adipocytes leading to insulin resistance and an increase in adiposity [17].
Cortisol action on GR is largely under control by enzyme expression and activity: 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) that regenerates active cortisol from inactive cortisone and 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) that converts cortisol to cortisone [18]. In obesity, the activity of the adipose 11β-HSD1 is increased. This is associated with the state of chronic inflammation [19] which is a hallmark of obesity, and it was shown that anti-inflammatory treatment can reverse this effect to a certain extent [20]. In animal models, mice-overexpressing adipocyte 11β HSD-1 developed all cortisol-related comorbidities such as diabetes, hypertension, and visceral obesity [21]. On the contrary to adipose tissue, the hepatic 11β-HSD1 activity in obesity is decreased. Tissue-specific dysregulation of cortisol metabolism in human obesity and furthermore, excess liver fat increases glucocorticoid metabolite excretion in urine and can further decrease the hepatic 11β-HSD1 activity [22].
It is well known that neuronal networks that regulate food intake are tightly connected to the HPA axis, expressing a significant effect on appetite-satiety centers [23]. Every stress response is stressor-specific and can vary significantly based on its effect on the organism, how one perceives the stress and the possibility of coping [24].
Functional assessment of the HPA axis in patients with obesity can be done using the usual diagnostic procedures for evaluating patients with suspected hypercortisolism; however, the sensitivity and specificity could be significantly reduced [25]. Despite that, dynamic studies in various settings have been performed to investigate the HPA axis in obesity with regard to chronobiological changes, different types of stimulation or dexamethasone suppression [26]. Interestingly, in adult patients with obesity, there is a normal cortisol daily rhythm, normal adreno-cortico-tropic-hormone (ACTH) levels and either normal or even lower single sample of 24-h cortisol levels [27]. Furthermore, male subjects with obesity with metabolic syndrome, hypertension and/or diabetes were found not to have significant differences in urinary free cortisol, salivary cortisol and post dexamethasone cortisol levels in comparison to the subjects without these disorders [28]. Same was found when all cortisol parameters and the number of features of metabolic syndrome were compared [28].
The modulation of GR activity is influenced by different post-translational alterations such as phosphorylation, ubiquitination or gene polymorphisms [29, 30]. Many of these polymorphisms were investigated in relation to the body mass index (BMI) and other markers of metabolic syndrome [29, 30]. The N363S polymorphism is associated with an increased sensitivity to glucocorticoids, increased insulin response to overnight dexamethasone suppression testing and an increased BMI [30], while the Bcll polymorphism, also connected to enhanced sensitivity to glucocorticoids, was associated with an increased abdominal fat mass. Contrary to this, the ER22/23EK carriers had significantly better metabolic profile than noncarriers [29]. However, despite the vastly explored relationship between obesity and HPA axis, the relationship between food intake and this axis is bidirectional and the data on effects of various diets on HPA adaptation, namely hypercortisolism, and their specific differences still do not offer a unanimous conclusion.
Renin–angiotensin–aldosterone system, aldosterone, and obesity-related metabolic dysfunctions
Aldosterone is secreted by the glomerulosa zone, under the control of a complex regulatory network, namely the RAAS, potassium plasma concentrations, and, at least in part, the HPA [31].
RAAS has a pivotal role in the regulation of blood pressure, fluid and electrolyte balance and is strictly linked to the pathophysiology of several cardiometabolic diseases, such as heart failure, type 2 diabetes, and obesity [32, 33]. In this endocrine system, angiotensinogen (Agt), mainly produced by liver and adipose tissue, is enzymatically cleaved by renin, which is released into circulation by juxtaglomerular epithelioid cells located in the walls of renal afferent arterioles [34], to Ang I. Ang I is then converted by angiotensin converting enzyme (ACE) in Ang II [35]. Ang II exerts most of its physiological effects mainly through two G-protein-coupled receptors, Ang II type 1 (AT1R) and type 2 (AT2R) receptors, causing vasoconstriction and sodium/fluid retention. Hyperactivation of this pathway can lead to deleterious effects such as hypertension, fibrosis, endothelial dysfunction and inflammation [35]. On the other hand, RAAS is also characterized by a counterregulatory arm whose effects are mediated by ACE2 [extensively reviewed in [28, 36], which converts Ang-II to Ang-1,7 and Ang-1,9. These peptides elicits favourable physiologic effects through AT2R and the Mas receptor (MasR), hence counteracting the ACE1/Ang-II/AT1R arm of the RAAS, thereby determining vasodilation, increase in insulin sensitivity, and anti-inflammatory effects [37]. Aldosterone is the final hormonal effector of the RAAS, exerting its complex biological effects in almost all tissues involved in metabolic homeostasis (i.e. adipose tissue, skeletal muscle, liver, pancreas, etc.) through specific activation of MR [38].
A consistent body of evidence supports the presence of a local RAAS in adipose tissue [39], which is known to represent the major site of Agt production after liver. RAAS activation plays a major role in the regulation of adipocyte function [40]. Remarkably, adipose tissue expresses all components of RAAS necessary to generate vasoactive molecules, angiotensin peptides, and also aldosterone, although at very low concentrations [41].
The obesity state represents a condition characterized by elevated plasma aldosterone levels [42, 43] and increased RAAS activation. RAAS hyperactivation represents a major determinant of obesity complex pathophysiology [44]. Dysfunctional adipose tissue shows increased expression of MR, whose specific activation favours white adipogenesis, and inhibits the browning process, i.e. the acquisition of brown-like characteristics by white adipocytes, a process which carries great promise for protection against obesity and metabolic disorders [38]. Administration of MR antagonists prevents diet-induced obesity through induction of browning of white adipose tissue [45, 46], at least in mice models. A large body of evidence suggests that classical RAAS activation in white adipose tissue displays deleterious effect on insulin sensitivity and inflammation and plays a major role in the development of obesity-related metabolic diseases [47]. Differently, the RAAS counter-regulatory activation has shown favourable effects on adipocyte and metabolic dysfunction: in fact the administration of Ang-1,7 to high-fat diet fed mice improved insulin sensitivity through an increase in Akt phosphorylation in brown adipose tissue [48]. Hence, pharmacological, or nutritional interventions acting on the balance between the classical and counter regulatory arms of the RAAS could potentially display favourable effects in obesity-related metabolic dysfunctions.
On the other hand, weight loss and a reduction in fat mass is able to affect RAAS activation, probably due to a reduction in adipocyte factors (CTRP1, leptin, and other) capable to directly increase aldosterone synthesis by the adrenal gland, independently from renin [49]. In line with this, a recent report showed that bariatric-induced weight loss is able to lower plasma aldosterone concentration, independently of plasma renin activity and sodium excretion [50].
Diet as a stress
There are clear individual differences in humans in food intake during stressful periods. Age and sex contribute to this diverse response [51]. Even in the absence of hunger, stress tends to precipitate the intake of calorie dense foods, fast food and food rich in sugar, especially in individuals with overweight or obesity. The neural responses in brain regions of patients with obesity associated with motivation, emotion-memory and taste processing are shown to correlate with Homeostatic Model Assessment for Insulin Resistance (HOMA-IR). Alterations in insulin sensitivity can modify and suppress neural pathways associated with stress and food intake [52]. Various diets are available with a wide array of success and different metabolic outcomes. Diet induced weight loss takes place in the setting of increased energy expenditure and a net caloric deficit. Caloric restriction is one of the proven ways to reduce cardiovascular risk [53]. It is linked to a reduction in blood pressure, insulin sensitivity and leads to weight loss [53]. It is well known that the HPA axis is reactive to food intake. Glucocorticoid levels increase immediately after a meal [54]. Nevertheless, the long-term changes in total and tissue specific glucocorticoid metabolism and their relation to weight loss appear to be more complex.
Caloric intake restriction poses a stressor and can lead to cortisol output elevation as a simple manifestation of its physiological role in energy expenditure [55]. Furthermore, Purnell et al. suggest that the increased HPA activity may promote visceral weight regain following a successful diet-induced weight loss. They have shown that metabolic clearance, cortisol production rate and free cortisol do not significantly change in comparison to baseline after 24 weeks of dieting in men with obesity, but in the same group, with further weight loss, cortisol production increased, and the activity of adipose, tissue specific 11β-HSD-1 decreased [56]. In adipose tissue of patients with obesity, the inhibition of 11β-HSD-1 results in tissue-specific cortisol concentration reduction, which was suggested to improve insulin sensitivity in this group [9]. Other studies report similar findings of the unchanged circulating levels of cortisol, cortisone and urinary steroid metabolite ratios and a decreased 11β-HSD-1 activity in adipose tissue [57]. It seems that with a less significant weight loss, the expression of 11β-HSD-1 is mostly unaltered [58], but as the weight loss becomes more significant, this activity decreases. In patients with obesity that were undergoing bariatric surgery, the omental and hepatic 11β-HSD-1 were found to correlate with their BMI and the adipose 11β-HSD-1 was also significantly reduced after significant post-surgical weight loss [59].
Even though BMI is still the most used tool for stratification of obesity, obesity associated risks and the success of an individual weight loss, the body fat mass, the fat free mass and muscle gain can provide further important insight in the quality of the weight loss [60]. Various dietary approaches using low energy density, lower glycaemic index or portion controls have shown to improve body fat percentage and fat mass with no change in fat free mass [61]. In a large cohort of male patients, circulating cortisol levels were negatively associated with weight, BMI, but also waist-to-hip ratio and waist circumference. The longitudinal changes in cortisol levels were also negatively associated with these measures of adiposity [62]. Hair cortisol levels are proposed as one of the ways to measure chronic stress levels, that differ from the urinary, blood or salivary cortisol [63]; however, Larsen et al. found no association between the hair cortisol levels and weight loss maintenance in patients that had achieved a significant weight loss [64].
Manipulation of the macronutrient content is also speculated to alter glucocorticoid metabolism. In animal models, the hepatic 11β-HSD-1 was shown to be reduced in the setting of low or moderate carbohydrate diets in comparison to a high fat diet [65]. The connection between a decreased hepatic and adipose glucocorticoid regeneration and a dietary fat content has not yet been elucidated. The same group has also shown that a low-carbohydrate diet alters cortisol metabolism independently of weight loss, highlighting low-carbohydrate ketogenic diets (LCKDs) as a possible efficient tool for reversing the metabolic consequences of obesity in male patients [66]. Obesity is also an important comorbidity in various endocrinological diseases. An increasing number of patients with obesity are diagnosed with a wide spectrum of hypercortisolism ranging from (possible) autonomous cortisol secretion to overt Cushing’s syndrome and due to a long list of cardio-metabolic comorbidities associated with these conditions, treating these patients requires a personalized and a tailored approach along with lifestyle interventions [67]. Low-carb diets are known to ameliorate all metabolic complications associated with hypercortisolism, including diabetes, insulin resistance, hypertension, and obesity. In patients with both ACTH-dependent and -independent cortisol hypersecretion, it is often advised that the low-carb diet is maintained prior, during and after surgery or medical treatment of the primary disease [68]. So far, the data on the efficacy of VLCKD in treating patients with hypercortisolism are scarce; however, based on the beneficial effects that VLCKD is showing in treating all aforementioned cardio-metabolic comorbidities, Guarnotta et al. speculate that it could be successfully employed in treating these groups of patients [69].
Physical activity goes hand in hand with weight reduction programs and is almost always advised to patients with obesity within their physical limitations. However, exercise can be both a stressor itself and a modifier of stress in relation to the HPA axis [70]. Athletes who performed intense training sessions during the day had suppressed cortisol levels at night. The lowest cortisol levels were found in athletes having the most intense daytime training [70].
In animal models, exercise led to a reduction in GR expression and 11β-HSD-1 in liver and muscles, with unchanged levels of circulating cortisol [71]. However, we cannot determine the full scope of the impact of exercise on the HPA axis and its contribution to diet because of several key modulatory cofactors. Food intake prior exercise is associated with a lower postprandial cortisol elevation, while adversely, performing physical activity after a meal leads to a suppressed cortisol response [72]. Irrespective of the thermal stress in physical activity, hypohydration can significantly modify the hormonal response, manifesting with increased circulating cortisol levels, possibly due to increased internal temperature and a reduction in plasma volume [73]. Also, physical activity during different periods of a day significantly influences cortisol levels and can create more difficulty in interpreting results [74].
Weight loss in response to the low-calorie diet tends to be different in male and female patients with obesity [75, 76]. Energy balance and glucose metabolism are initially distinct between the two in part because of the effect of sex hormones, including not only circulating oestrogen and androgen levels, but also their adipose tissue production and their effect on adipose tissue distribution [77]. Postmenopausal women tend to lose lean body mass with the loss of oestrogen and show an increase in adipose tissue [78]. The number of women with obesity (BMI > 35 kg/m2) is almost double in comparison to male patients [79]. Male patients with obesity show a more significant weight loss and improvement in some cardiometabolic factors, while female patients demonstrate a lower regain of weight but have less dietary intervention effect on the cardiometabolic outcomes [80]. On a molecular level, oestrogen has a stimulatory effect on the HPA axis, both centrally and trough increasing cortisol binding globulin, and is opposed by progesterone depending on the phase of the menstrual cycle and age, while androgens down-regulate the stress induced and basal glucocorticoid levels [81]. The relationship between obesity, testosterone and steroidogenesis was investigated in various studies. Although it is well known that obesity is associated with hypogonadism [82] and weight loss promotes testosterone increase, the data on different diet regimes in relation to testosterone changes are still a matter of debate. Testosterone influences lipid, protein and carbohydrate metabolism, and lower testosterone levels favour pluripotent stem cell conversion into adipocytes while leptin itself inhibits testosterone secretion from the Leydig cells [83]. In a recent meta-analysis of 7 studies authors aimed to evaluate the potential effect of ketogenic diets (KDs) on testosterone levels. Of note, the most evident testosterone increase was found in patients with VLCKD. The authors suggest a multifactorial physiological mechanism for this, including a cholesterol intake increase, low fiber intake and alterations in glucose and insulin homeostasis [84].
Individuals with obesity often have a very hard time maintaining the newly reduced weight, even after a successful initial weight loss. Poor appetite control can sometimes be exacerbated by a diet with patients not being able to achieve the significant metabolic and psychological benefits of weight loss. Various studies have shown that dieting and food restriction can lead to anxiety, depression, or irritability [85]. For example, individual behavioural vulnerability can significantly impair the success of a low-energy diet-based weight loss program, highlighting the importance of the individualized approach in obesity management [80]. Restrained eating, a term reflecting an individual struggle to control food intake and weight [86] can pose as one of the stressogenic factors during weight loss. Elevated salivary cortisol levels were found to positively correlate with the level of self-reported dietary restraint in premenopausal women [87]. Furthermore, when taking in consideration other factors, individual differences in cognitive restraint such as body image perception, appearance beliefs and dissatisfaction also emerged as important parameters in the level of stress related to dietary restraint and cortisol levels [88]. In addition, monitoring calories increases perceived stress irrespective of calorie restriction [55]. Obesity has become a leading worldwide pandemic, associated with serious outcomes [89]. Despite all the variables mentioned in this article, the fact that cortisol levels were shown to be lower in patients with obesity, with some studies showing a U-shaped relationship with BMI across the weight spectrum [90] could mean that the subtle hypercortisolism shown in weight loss, especially VLCKD, could not only be a useful physiological consequence, but also a sign of the HPA axis reactivation or return to normal state.
Mechanisms of action of the ketogenic diet on energy metabolism and muscle mass
KD is a normoprotein diet with a drastic reduction of the carbohydrate content (approximately between 30 and 50 g/day); depending on the calorie content, KD can be defined as a high fat diet (with a fat content of approximately 60–70%), a LCKD, with a fat content > 30–40 g/day, or a VLCKD, with a fat content < 30–40 g/day [91, 92]. Thanks to their sustained protein content, KD is extremely effective to reduce adipose tissue while preserving lean mass, as reported in Fig. 2.

Short-term positive mechanisms of action of the ketogenic diet on energy metabolism and muscle mass
Importantly, β-hydroxybutyrate (βHB) has also been shown to exert anticatabolic effects on human skeletal muscle [93]. In fact, as reported by Barrea et al., KDs have a favourable impact on lean body mass preservation [94]. As reported by Basolo et al. lean body mass is the main determinant of resting metabolic rate, accounting for ~ 70% [95]. Therefore, it is extremely important, in a very low-calorie diet, to preserve lean mass to maintain resting metabolic rate. Moreover, in a recent pilot study by Camajani et al., the efficacy of a VLCKD on fat free mass, basal metabolic rate and body cell mass was evaluated: 12 patients were enrolled in the control group that underwent only VLCKD and 12 patients were instead enrolled in the experimental group that received the same diet in combination with interval training [96]. At the end of the 6-weeks study, it was seen that the experimental group preserved fat free mass, basal metabolic rate and body cell mass to a greater extent [96].
In the pilot study conducted by Merra et al., it has been demonstrated that VLCKD was highly effective in terms of body weight reduction without inducing lean body mass loss [97]. According to Barrea et al., the preservation of muscle mass, which is positively associated with muscle strength, has been included among the benefits of VLCKD due to the synergistic effects exerted by the reduction in visceral adipose tissue and obesity-related pro-inflammatory status [98]. In fact, in their study, the authors showed that at the end of a 45-days VLCKD protocol, there was an increase in muscle strength (∆ + 17.4 ± 13.2%; p = 0.001) [98].
Activation of the HPA axis during a ketogenic diet
The HPA axis is a neuroendocrine system involved in maintaining homeostasis in humans under physiological conditions and stress, and cortisol is the major hormone of the HPA axis. In obesity, calorie restriction is a good strategy to reduce visceral adipose tissue. In this respect, however, it is well known that dieting is a stress factor for the individual, resulting in negative consequences on body composition and energy metabolism reducing the free fat mass, which is an important site for glucose uptake. Furthermore, studies from the last decade have shown that the KD alters the hormonal balance through changes in the production of metabolic regulatory hormones, such as cortisol [99]. In fact, as reported by Thio, during a KD there is an increasing of serum cortisol levels, in rats [100]. In fact, in the study by Thio and colleagues, rats undergoing KD for 2 weeks had tenfold higher mid-day serum βHB levels and 30% lower glucose levels than rats undergoing standard diet. The elevated βHB levels indicated that KD produced ketosis. The reduction in glucose was expected, as low-carbohydrate diets can lower blood glucose in humans. Rats subjected to KD also had slightly increased cortisol levels (p < 0.05) [100].
In a study by Ryan et al., it was demonstrated that a nutritional manipulation characterized by a relative depletion of dietary carbohydrates, thereby inducing nutritional ketosis, acutely and chronically activated the HPA axis [101]. Male rats and mice maintained on a KD exhibited canonical markers of chronic stress, including increased basal and stress-evoked plasma corticosterone, increased adrenal sensitivity to adrenocorticotropin hormone, and thymic atrophy, an indicator of chronic glucocorticoid exposure.
In subjects with obesity, caloric restriction is a valid strategy to reduce visceral adipose tissue. A recent study was carried out by Polito et al. to assess the effects of a VLCKD for weight loss on the sympathetic nervous system and HPA axis, through evaluation of salivary cortisol and galvanic skin response levels [102]. Thirty male subjects with obesity were recruited and assessed before and after 8 weeks of VLCKD intervention to evaluate body composition and biochemical parameters. Salivary cortisol levels and galvanic skin response significantly decreased after dietary treatment; in addition, body composition and biochemical features were ameliorated. They concluded that a VLCKD had a short-term positive effect on the sympathetic nervous system and HPA axes regulating salivary cortisol levels, despite the effect observed in preclinical studies [102]. Despite VLCKD was associated with hyperactivation of the HPA axis as other nutritional protocols do and that is usually associated with fat-free muscle loss, they interestingly found that VLCKD was associated with an increase of fat free mass, probably due to the trophic effects of ketone bodies on muscle mass. Since muscle mass is important for glucose uptake in glucose metabolism, VLCKD could potentially become a promising nutritional approach, mostly in subjects with obesity and glucose metabolism derangements.
VLCKD, thanks to the dramatic reduction of the exogenous carbohydrate content and calories, determines physiological nutritional ketosis: the ketone bodies produced through lipolysis, and especially βHB, have an anti-proteolytic effect, preserving muscle mass.
In addition, there will be a decrease in adipose tissue, particularly visceral adipose tissue, with a reduction in the pro-inflammatory cytokines, corresponding reduction in the chronic low-grade inflammatory state, characteristic of subjects with obesity.
The VLCKD protocol is medicalized and well standardized; the nutritional ketosis phase, and hence the consequent reduction in carbohydrates, can only be prolonged for 12 weeks [100]. Thereafter, there will be a progressive increase in carbohydrate and calorie content, which will not activate stressogenic compensatory hormonal responses that would lead to the consequent increase in cortisol.
Potential effects of VLCKD on the RAAS
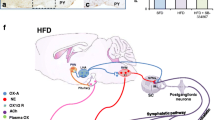
The valuable effects of VLCKD in the rapid reduction of ectopic and visceral fat displays consistent favourable effects on the major risk factors for cardiovascular diseases [22, 103]. Pioneering studies by Blackburn have demonstrated marked effects of VLCKD in the reduction of body weight, together with a significant decrease in blood pressure, fasting glucose and triglyceride plasma levels [104]. VLCKD was shown to be more effective in blood pressure lowering than a combined intervention based on a classical hypocaloric diet combined with orlistat treatment [105]. Such effect is probably linked to the increased natriuresis associated with ketone bodies urinary excretion. A recent meta-analysis of 20 studies found out a modest but significant increase in serum sodium in subject following a VLCKD [106], probably related to the important water loss occurring during the first phases of ketosis. In this context, it is important to keep in mind that a careful supplementation in minerals, including sodium, potassium, calcium, and magnesium, as well as a proper water intake, are mandatory in order to avoid potential side effects due to alteration of hydrosaline metabolism. It appears evident that VLCKD necessarily elicits rapid RAAS responses due to a different salt and water handling during nutritional ketosis. In this context, a very recent report demonstrated that a KD, with or without supplementation in ketone esters, markedly increases aldosterone plasma levels without worsening cardiometabolic risk factors [107]. Importantly, aldosterone plasma concentrations were inversely related to renin, suggesting a renin-independent activation of aldosterone production. Moreover, ketone plasma levels were positively correlated to aldosterone, suggesting a potential novel role of ketones on aldosterone production by the adrenal gland. Importantly, this substantial increase in aldosterone did not determine any adverse effect in cardiometabolic risk factors in patients following a KD, probably due to the well-known cardioprotective effects of ketone bodies [108]. However, the effects of VLCKD on all components of the RAAS still need to be clarified.
A recent study explored the differences existing in RAAS regulation in murine adipose tissue under obesogenic and ketogenic nutritional regimens [109]. The authors tested the hypothesis that the favourable vascular effects of KD were strictly linked to an increased expression in the components of the counterregulatory arm of the RAS. Interestingly, they demonstrated that KD shifted RAAS profile to the counterregulatory arm, whereas an obesogenic nutritional regimen up-regulated the expression of ACE1/Ang-II/AT1R in adipose tissue. These data suggest that VLCKD may directly affect RAAS regulation at different levels, in view of its significant impact on adipose tissue metabolism, fluid/salt regulation, appetite and thirst regulation, natriuresis, etc., potentially counteracting the adverse cardiometabolic consequences of RAAS dysregulation in obesity. The mechanisms underlying these effects are still unclear. Ketone bodies could display powerful effects both on aldosterone secretion by adrenal cells, both on the expression and function of RAAS peptides. Caloric restriction, which shares with RAAS blockade similar effects on longevity [45], could also play a substantial role in the RAAS adaptation to a completely different dietary regimen. However, this hypothesis needs further studies, both in preclinical models exposed to KD, and in patients with obesity undergoing a VLCKD.
Conclusions
There is a strong relationship between obesity, stress, responses to low calorie diets, weight loss, the HPA axis and the RAAS, which indeed play a key role in short- and long-term metabolic adaptation to a very low-calorie diet. VLCKD represents a valuable nutritional strategy to tackle obesity, inducing a rapid and effective loss of adipose tissue. Its potential impact in the adaptation of the HPA axis and RAAS to a novel metabolic, hormonal, cardiovascular and psychological status, has been poorly addressed so far, and requires ad hoc studies, to understand the effects of VLCKD on adrenal function. VLCKD could display favourable effects against stress-induced hypercortisolism and has been shown to directly increase aldosterone production by the adrenal glands, without any detrimental effect on cardiovascular risk. Importantly, due to a significant loss of visceral and subcutaneous fat, VLCKD may strongly affects the peripheral metabolism of steroid hormones by adipose tissue, with subsequent important impact on cortisol effects on central and peripheral tissues. More studies are deemed necessary in this regard, in order to better define precision nutrition strategies, optimally adapting to the hormonal changes related to weight loss, to maintain the novel metabolic status and avoid weight regain.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Abbreviations
- HPA:
-
Hypothalamic–pituitary–adrenal
- MR:
-
Mineralocorticoid receptor
- GR:
-
Glucocorticoid receptors
- 11β-HSD1:
-
11β-Hydroxysteroid dehydrogenase type 1
- 11β-HSD2:
-
11β-Hydroxysteroid dehydrogenase type 2
- ACTH:
-
Adreno-cortico-tropic-hormone
- BMI:
-
Body mass index
- HOMA-IR:
-
Homeostatic model assessment for insulin resistance
- KD:
-
Ketogenic diet
- BHB:
-
β-Hydroxybutyrate
- RMR:
-
Resting metabolic rate
- VLCKD:
-
Very low-calorie ketogenic diet
- Ang:
-
Angiotensin
- Agt:
-
Angiotensinogen
- ACE:
-
Angiotensin-converting enzyme
- AT1R:
-
Angiotensin II type 1 receptor
- AT2R:
-
Angiotensin II type 2 receptor
References
Guthrie HD, Barber JA, Leighton JK, Hammond JM (1994) Steroidogenic cytochrome P450 enzyme messenger ribonucleic acids and follicular fluid steroids in individual follicles during preovulatory maturation in the pig. Biol Reprod 51(3):465–471
Kim JH, Choi MH (2020) Embryonic development and adult regeneration of the adrenal gland. Endocrinol Metab (Seoul) 35(4):765–773
Niemeyer CS, Mescher T, Griggs R, Orlicky DJ, Wilkerson GK, Bubak AN et al (2021) Histopathological analysis of adrenal glands after simian varicella virus infection. Viruses 13(7):1245
Wolfraim LA, Langis R, Tyson H, Dhindsa RS (1993) cDNA sequence, expression, and transcript stability of a cold acclimation-specific gene, cas18, of alfalfa (Medicago falcata) cells. Plant Physiol 101(4):1275–1282
Lightman SL, Birnie MT, Conway-Campbell BL (2020) Dynamics of ACTH and cortisol secretion and implications for disease. Endocr Rev. https://doi.org/10.1210/endrev/bnaa002
Lightman SL, Conway-Campbell BL (2010) The crucial role of pulsatile activity of the HPA axis for continuous dynamic equilibration. Nat Rev Neurosci 11(10):710–718
McEwen BS (2006) Protective and damaging effects of stress mediators: central role of the brain. Dialogues Clin Neurosci 8(4):367–381
Oakley RH, Cidlowski JA (2013) The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132(5):1033–1044
Stewart PM, Boulton A, Kumar S, Clark PM, Shackleton CH (1999) Cortisol metabolism in human obesity: impaired cortisone–>cortisol conversion in subjects with central adiposity. J Clin Endocrinol Metab 84(3):1022–1027
Chanson P, Salenave S (2010) Metabolic syndrome in Cushing’s syndrome. Neuroendocrinology 92(Suppl 1):96–101
Medenica S, Spoltore ME, Ormazabal P, Marina LV, Sojat AS, Faggiano A et al (2023) Female infertility in the era of obesity: the clash of two pandemics or inevitable consequence? Clin Endocrinol (Oxf) 98(2):141–152
De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M (1998) Brain corticosteroid receptor balance in health and disease. Endocr Rev 19(3):269–301
Dallman MF, Strack AM, Akana SF, Bradbury MJ, Hanson ES, Scribner KA et al (1993) Feast and famine: critical role of glucocorticoids with insulin in daily energy flow. Front Neuroendocrinol 14(4):303–347
Walker BR (2007) Extra-adrenal regeneration of glucocorticoids by 11beta-hydroxysteroid dehydrogenase type 1: physiological regulator and pharmacological target for energy partitioning. Proc Nutr Soc 66(1):1–8
Hadoke PW, Macdonald L, Logie JJ, Small GR, Dover AR, Walker BR (2006) Intra-vascular glucocorticoid metabolism as a modulator of vascular structure and function. Cell Mol Life Sci 63(5):565–578
Veilleux A, Laberge PY, Morency J, Noel S, Luu-The V, Tchernof A (2010) Expression of genes related to glucocorticoid action in human subcutaneous and omental adipose tissue. J Steroid Biochem Mol Biol 122(1–3):28–34
Gathercole LL, Morgan SA, Bujalska IJ, Hauton D, Stewart PM, Tomlinson JW (2011) Regulation of lipogenesis by glucocorticoids and insulin in human adipose tissue. PLoS ONE 6(10):e26223
Walker BR (2007) Glucocorticoids and cardiovascular disease. Eur J Endocrinol 157(5):545–559
Sutinen J, Kannisto K, Korsheninnikova E, Nyman T, Ehrenborg E, Andrew R et al (2004) In the lipodystrophy associated with highly active antiretroviral therapy, pseudo-Cushing’s syndrome is associated with increased regeneration of cortisol by 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue. Diabetologia 47(10):1668–1671
Nixon M, Wake DJ, Livingstone DE, Stimson RH, Esteves CL, Seckl JR et al (2012) Salicylate downregulates 11beta-HSD1 expression in adipose tissue in obese mice and in humans, mediating insulin sensitization. Diabetes 61(4):790–796
Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR et al (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294(5549):2166–2170
Ahmed A, Rabbitt E, Brady T, Brown C, Guest P, Bujalska IJ et al (2012) A switch in hepatic cortisol metabolism across the spectrum of non-alcoholic fatty liver disease. PLoS ONE 7(2):e29531
Kyrou I, Chrousos GP, Tsigos C (2006) Stress, visceral obesity, and metabolic complications. Ann N Y Acad Sci 1083:77–110
Chrousos GP, Gold PW (1992) The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA 267(9):1244–1252
Baid SK, Rubino D, Sinaii N, Ramsey S, Frank A, Nieman LK (2009) Specificity of screening tests for Cushing’s syndrome in an overweight and obese population. J Clin Endocrinol Metab 94(10):3857–3864
Pasquali R, Vicennati V, Cacciari M, Pagotto U (2006) The hypothalamic-pituitary-adrenal axis activity in obesity and the metabolic syndrome. Ann N Y Acad Sci 1083:111–128
Chalew S, Nagel H, Shore S (1995) The hypothalamic-pituitary-adrenal axis in obesity. Obes Res 3(4):371–382
Abraham SB, Rubino D, Sinaii N, Ramsey S, Nieman LK (2013) Cortisol, obesity, and the metabolic syndrome: a cross-sectional study of obese subjects and review of the literature. Obesity (Silver Spring) 21(1):E105–E117
Manenschijn L, van den Akker EL, Lamberts SW, van Rossum EF (2009) Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Ann N Y Acad Sci 1179:179–198
van Rossum EF, Lamberts SW (2004) Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Horm Res 59:333–357
Chong C, Hamid A, Yao T, Garza AE, Pojoga LH, Adler GK et al (2017) Regulation of aldosterone secretion by mineralocorticoid receptor-mediated signaling. J Endocrinol 232(3):525–534
Feraco A, Armani A, Mammi C, Fabbri A, Rosano GM, Caprio M (2013) Role of mineralocorticoid receptor and renin-angiotensin-aldosterone system in adipocyte dysfunction and obesity. J Steroid Biochem Mol Biol 137:99–106
Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM (2007) Renin-angiotensin system and cardiovascular risk. Lancet 369(9568):1208–1219
Schweda F, Friis U, Wagner C, Skott O, Kurtz A (2007) Renin release. Physiology (Bethesda) 22:310–319
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM (2014) Classical Renin-Angiotensin system in kidney physiology. Compr Physiol 4(3):1201–1228
Paz Ocaranza M, Riquelme JA, Garcia L, Jalil JE, Chiong M, Santos RAS et al (2020) Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat Rev Cardiol 17(2):116–129
Frigolet ME, Torres N, Tovar AR (2013) The renin-angiotensin system in adipose tissue and its metabolic consequences during obesity. J Nutr Biochem 24(12):2003–2015
Armani A, Marzolla V, Fabbri A, Caprio M (2015) Cellular mechanisms of MR regulation of adipose tissue physiology and pathophysiology. J Mol Endocrinol 55(2):R1-10
Menikdiwela KR, Ramalingam L, Rasha F, Wang S, Dufour JM, Kalupahana NS et al (2020) Autophagy in metabolic syndrome: breaking the wheel by targeting the renin-angiotensin system. Cell Death Dis 11(2):87
Frantz EDC, Prodel E, Braz ID, Giori IG, Bargut TCL, Magliano DC et al (2018) Modulation of the renin-angiotensin system in white adipose tissue and skeletal muscle: focus on exercise training. Clin Sci (Lond) 132(14):1487–1507
Briones AM, Nguyen Dinh Cat A, Callera GE, Yogi A, Burger D, He Y et al (2012) Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension 59(5):1069–1078
Dudenbostel T, Ghazi L, Liu M, Li P, Oparil S, Calhoun DA (2016) Body mass index predicts 24-hour urinary aldosterone levels in patients with resistant hypertension. Hypertension 68(4):995–1003
Kawarazaki W, Fujita T (2016) The role of aldosterone in obesity-related hypertension. Am J Hypertens 29(4):415–423
Parasiliti-Caprino M, Bollati M, Merlo FD, Ghigo E, Maccario M, Bo S (2022) Adipose tissue dysfunction in obesity: role of mineralocorticoid receptor. Nutrients 14(22):4735
Armani A, Cinti F, Marzolla V, Morgan J, Cranston GA, Antelmi A et al (2014) Mineralocorticoid receptor antagonism induces browning of white adipose tissue through impairment of autophagy and prevents adipocyte dysfunction in high-fat-diet-fed mice. FASEB J 28(8):3745–3757
Hirata A, Maeda N, Hiuge A, Hibuse T, Fujita K, Okada T et al (2009) Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice. Cardiovasc Res 84(1):164–172
Kalupahana NS, Massiera F, Quignard-Boulange A, Ailhaud G, Voy BH, Wasserman DH et al (2012) Overproduction of angiotensinogen from adipose tissue induces adipose inflammation, glucose intolerance, and insulin resistance. Obesity (Silver Spring) 20(1):48–56
Morimoto H, Mori J, Nakajima H, Kawabe Y, Tsuma Y, Fukuhara S et al (2018) Angiotensin 1–7 stimulates brown adipose tissue and reduces diet-induced obesity. Am J Physiol Endocrinol Metab 314(2):E131–E138
Infante M, Armani A, Mammi C, Fabbri A, Caprio M (2017) Impact of adrenal steroids on regulation of adipose tissue. Compr Physiol 7(4):1425–1447
Berney M, Vakilzadeh N, Maillard M, Faouzi M, Grouzmann E, Bonny O et al (2021) Bariatric surgery induces a differential effect on plasma aldosterone in comparison to dietary advice alone. Front Endocrinol (Lausanne) 12:745045
Pasquali R (2012) The hypothalamic-pituitary-adrenal axis and sex hormones in chronic stress and obesity: pathophysiological and clinical aspects. Ann N Y Acad Sci 1264(1):20–35
Jastreboff AM, Sinha R, Lacadie C, Small DM, Sherwin RS, Potenza MN (2013) Neural correlates of stress- and food cue-induced food craving in obesity: association with insulin levels. Diabetes Care 36(2):394–402
Bales CW, Kraus WE (2013) Caloric restriction: implications for human cardiometabolic health. J Cardiopulm Rehabil Prev 33(4):201–208
Benedict C, Hallschmid M, Scheibner J, Niemeyer D, Schultes B, Merl V et al (2005) Gut protein uptake and mechanisms of meal-induced cortisol release. J Clin Endocrinol Metab 90(3):1692–1696
Tomiyama AJ, Mann T, Vinas D, Hunger JM, Dejager J, Taylor SE (2010) Low calorie dieting increases cortisol. Psychosom Med 72(4):357–364
Purnell JQ, Kahn SE, Samuels MH, Brandon D, Loriaux DL, Brunzell JD (2009) Enhanced cortisol production rates, free cortisol, and 11beta-HSD-1 expression correlate with visceral fat and insulin resistance in men: effect of weight loss. Am J Physiol Endocrinol Metab 296(2):E351–E357
Stomby A, Simonyte K, Mellberg C, Ryberg M, Stimson RH, Larsson C et al (2015) Diet-induced weight loss has chronic tissue-specific effects on glucocorticoid metabolism in overweight postmenopausal women. Int J Obes (Lond) 39(5):814–819
Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Heintze U, Janke J et al (2004) Regulation of 11beta-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes Res 12(1):9–17
Woods CP, Corrigan M, Gathercole L, Taylor A, Hughes B, Gaoatswe G et al (2015) Tissue specific regulation of glucocorticoids in severe obesity and the response to significant weight loss following bariatric surgery (BARICORT). J Clin Endocrinol Metab 100(4):1434–1444
Ghachem A, Paquin J, Brochu M, Dionne IJ (2018) Should waist circumference cutoffs in the context of cardiometabolic risk factor assessment be specific to sex, age, and BMI? Metab Syndr Relat Disord 16(7):366–374
Melanson KJ, Summers A, Nguyen V, Brosnahan J, Lowndes J, Angelopoulos TJ et al (2012) Body composition, dietary composition, and components of metabolic syndrome in overweight and obese adults after a 12-week trial on dietary treatments focused on portion control, energy density, or glycemic index. Nutr J 11:57
Travison TG, O’Donnell AB, Araujo AB, Matsumoto AM, McKinlay JB (2007) Cortisol levels and measures of body composition in middle-aged and older men. Clin Endocrinol (Oxf) 67(1):71–77
Short SJ, Stalder T, Marceau K, Entringer S, Moog NK, Shirtcliff EA et al (2016) Correspondence between hair cortisol concentrations and 30-day integrated daily salivary and weekly urinary cortisol measures. Psychoneuroendocrinology 71:12–18
Larsen SC, Turicchi J, Christensen GL, Larsen CS, Jorgensen NR, Mikkelsen MK et al (2021) Hair cortisol concentration, weight loss maintenance and body weight variability: a prospective study based on data from the European NoHoW trial. Front Endocrinol (Lausanne) 12:655197
Stimson RH, Lobley GE, Maraki I, Morton NM, Andrew R, Walker BR (2010) Effects of proportions of dietary macronutrients on glucocorticoid metabolism in diet-induced obesity in rats. PLoS ONE 5(1):e8779
Stimson RH, Johnstone AM, Homer NZ, Wake DJ, Morton NM, Andrew R et al (2007) Dietary macronutrient content alters cortisol metabolism independently of body weight changes in obese men. J Clin Endocrinol Metab 92(11):4480–4484
Miomira Ivović LVM, Antoan S. Šojat, Milina Tančić-Gajić, Zorana Arizanovic, Aleksandra Kendereškie Svetlana Vujovic. Approach to the patient with subclinical Cushing's syndrome
Mary Kimberly Dugandzic E-CP-MaTK. The ketogenic diet initially masks the symptoms of hypercortisolism in Cushing's disease
Valentina Guarnotta FE, Roberta Amedei and Carlo Giordano Very low-calorie ketogenic diet: a potential application in the treatment of comorbidities of hypercortisolism
Hackney AC (2006) Stress and the neuroendocrine system: the role of exercise as a stressor and modifier of stress. Expert Rev Endocrinol Metab 1(6):783–792
Coutinho AE, Campbell JE, Fediuc S, Riddell MC (2006) Effect of voluntary exercise on peripheral tissue glucocorticoid receptor content and the expression and activity of 11beta-HSD1 in the Syrian hamster. J Appl Physiol 100(5):1483–1488
Brandenberger G, Follenius M, Hietter B (1982) Feedback from meal-related peaks determines diurnal changes in cortisol response to exercise. J Clin Endocrinol Metab 54(3):592–596
Judelson DA, Maresh CM, Yamamoto LM, Farrell MJ, Armstrong LE, Kraemer WJ et al (2008) Effect of hydration state on resistance exercise-induced endocrine markers of anabolism, catabolism, and metabolism. J Appl Physiol 105(3):816–824
Kanaley JA, Weltman JY, Pieper KS, Weltman A, Hartman ML (2001) Cortisol and growth hormone responses to exercise at different times of day. J Clin Endocrinol Metab 86(6):2881–2889
Bray GA, Fruhbeck G, Ryan DH, Wilding JP (2016) Management of obesity. Lancet 387(10031):1947–1956
Meyer A, Montastier E, Hager J, Saris WHM, Astrup A, Viguerie N et al (2018) Plasma metabolites and lipids predict insulin sensitivity improvement in obese, nondiabetic individuals after a 2-phase dietary intervention. Am J Clin Nutr 108(1):13–23
Geer EB, Shen W (2009) Gender differences in insulin resistance, body composition, and energy balance. Gend Med 6(Suppl 1):60–75
Leeners B, Geary N, Tobler PN, Asarian L (2017) Ovarian hormones and obesity. Hum Reprod Update 23(3):300–321
Collaboration NCDRF (2016) Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 387(10026):1377–1396
Trouwborst I, Goossens GH, Astrup A, Saris WHM, Blaak EE (2021) Sexual dimorphism in body weight loss, improvements in cardiometabolic risk factors and maintenance of beneficial effects 6 months after a low-calorie diet: results from the randomized controlled DiOGenes trial. Nutrients 13(5):1588
Moisan MP (2021) Sexual Dimorphism in Glucocorticoid Stress Response. Int J Mol Sci 22(6):3139
Tancic-Gajic M, Vukcevic M, Ivovic M, Marina LV, Arizanovic Z, Soldatovic I et al (2021) Obstructive sleep apnea is associated with low testosterone levels in severely obese men. Front Endocrinol (Lausanne) 12:622496
Kelly DM, Jones TH (2013) Testosterone: a metabolic hormone in health and disease. J Endocrinol 217(3):R25-45
Furini C, Spaggiari G, Simoni M, Greco C, Santi D (2023) Ketogenic state improves testosterone serum levels-results from a systematic review and meta-analysis. Endocrine 79(2):273–282
French SA, Jeffery RW (1994) Consequences of dieting to lose weight: effects on physical and mental health. Health Psychol 13(3):195–212
Heatherton TF, Herman CP, Polivy J, King GA, McGree ST (1988) The (mis)measurement of restraint: an analysis of conceptual and psychometric issues. J Abnorm Psychol 97(1):19–28
Anderson DA, Shapiro JR, Lundgren JD, Spataro LE, Frye CA (2002) Self-reported dietary restraint is associated with elevated levels of salivary cortisol. Appetite 38(1):13–17
Putterman E, Linden W (2006) Cognitive dietary restraint and cortisol: importance of pervasive concerns with appearance. Appetite 47(1):64–76
Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C et al (2014) Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384(9945):766–781
Schorr M, Lawson EA, Dichtel LE, Klibanski A, Miller KK (2015) Cortisol measures across the weight spectrum. J Clin Endocrinol Metab 100(9):3313–3321
Caprio M, Infante M, Moriconi E, Armani A, Fabbri A, Mantovani G et al (2019) Very-low-calorie ketogenic diet (VLCKD) in the management of metabolic diseases: systematic review and consensus statement from the Italian Society of Endocrinology (SIE). J Endocrinol Invest 42(11):1365–1386
Trimboli P, Castellana M, Bellido D, Casanueva FF (2020) Confusion in the nomenclature of ketogenic diets blurs evidence. Rev Endocr Metab Disord 21(1):1–3
Thomsen HH, Rittig N, Johannsen M, Moller AB, Jorgensen JO, Jessen N et al (2018) Effects of 3-hydroxybutyrate and free fatty acids on muscle protein kinetics and signaling during LPS-induced inflammation in humans: anticatabolic impact of ketone bodies. Am J Clin Nutr 108(4):857–867
Barrea L, Vetrani C, Caprio M, Cataldi M, Ghoch ME, Elce A et al (2022) From the ketogenic diet to the Mediterranean diet: the potential dietary therapy in patients with obesity after CoVID-19 infection (Post CoVID syndrome). Curr Obes Rep 11(3):144–165
Basolo A, Magno S, Santini F, Ceccarini G (2022) Ketogenic diet and weight loss: is there an effect on energy expenditure? Nutrients 14(9):1814
Camajani E, Feraco A, Proietti S, Basciani S, Barrea L, Armani A et al (2022) Very low calorie ketogenic diet combined with physical interval training for preserving muscle mass during weight loss in sarcopenic obesity: a pilot study. Front Nutr 9:955024
Merra G, Miranda R, Barrucco S, Gualtieri P, Mazza M, Moriconi E et al (2016) Very-low-calorie ketogenic diet with aminoacid supplement versus very low restricted-calorie diet for preserving muscle mass during weight loss: a pilot double-blind study. Eur Rev Med Pharmacol Sci 20(12):2613–2621
Barrea L, de Alteriis G, Muscogiuri G, Vetrani C, Verde L, Camajani E et al (2022) Impact of a very low-calorie ketogenic diet (VLCKD) on changes in handgrip strength in women with obesity. Nutrients 14(19):4213
Thio LL (2012) Hypothalamic hormones and metabolism. Epilepsy Res 100(3):245–251
Thio LL, Erbayat-Altay E, Rensing N, Yamada KA (2006) Leptin contributes to slower weight gain in juvenile rodents on a ketogenic diet. Pediatr Res 60(4):413–417
Ryan KK, Packard AEB, Larson KR, Stout J, Fourman SM, Thompson AMK et al (2018) Dietary manipulations that induce ketosis activate the HPA axis in male rats and mice: a potential role for fibroblast growth factor-21. Endocrinology 159(1):400–413
Polito R, Messina G, Valenzano A, Scarinci A, Villano I, Monda M et al (2021) The role of very low calorie ketogenic diet in sympathetic activation through cortisol secretion in male obese population. J Clin Med 10(18):4230
Parretti HM, Jebb SA, Johns DJ, Lewis AL, Christian-Brown AM, Aveyard P (2016) Clinical effectiveness of very-low-energy diets in the management of weight loss: a systematic review and meta-analysis of randomized controlled trials. Obes Rev 17(3):225–234
Palgi A, Read JL, Greenberg I, Hoefer MA, Bistrian BR, Blackburn GL (1985) Multidisciplinary treatment of obesity with a protein-sparing modified fast: results in 668 outpatients. Am J Public Health 75(10):1190–1194
Yancy WS Jr, Westman EC, McDuffie JR, Grambow SC, Jeffreys AS, Bolton J et al (2010) A randomized trial of a low-carbohydrate diet vs orlistat plus a low-fat diet for weight loss. Arch Intern Med 170(2):136–145
Castellana M, Conte E, Cignarelli A, Perrini S, Giustina A, Giovanella L et al (2020) Efficacy and safety of very low calorie ketogenic diet (VLCKD) in patients with overweight and obesity: a systematic review and meta-analysis. Rev Endocr Metab Disord 21(1):5–16
Belany P, Kackley ML, Zhao S, Kluwe B, Buga A, Crabtree C et al (2023) Effects of hypocaloric low-fat, ketogenic and ketogenic & ketone supplement diets on aldosterone and renin. J Clin Endocrinol Metab. https://doi.org/10.1210/clinem/dgad009
Makievskaya CI, Popkov VA, Andrianova NV, Liao X, Zorov DB, Plotnikov EY (2023) Ketogenic diet and ketone bodies against ischemic injury: targets, mechanisms, and therapeutic potential. Int J Mol Sci 24(3):2576
Da Eira D, Jani S, Stefanovic M, Ceddia RB (2023) Obesogenic versus ketogenic diets in the regulation of the renin-angiotensin system in rat white and brown adipose tissues. Nutrition 105:111862
Funding
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
LB and GM were responsible for designing the review protocol, writing the protocol and report, conducting the search and screening potentially eligible studies. LV, EC, ASS and LM were responsible for designing the review protocol, screening potentially eligible studies and writing the original draft. AC and MC provided feedback on the review. GM and LB and LV provided to reviewing and editing the final manuscript. All authors contributed to and agreed on the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Ljiljana Marina and Massimiliano Caprio are members of the editorial board of the Journal of Endocrinological Investigation.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, formal consent is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barrea, L., Verde, L., Camajani, E. et al. Effects of very low-calorie ketogenic diet on hypothalamic–pituitary–adrenal axis and renin–angiotensin–aldosterone system. J Endocrinol Invest 46, 1509–1520 (2023). https://doi.org/10.1007/s40618-023-02068-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-023-02068-6