
Abstract
Background
The Coronavirus disease 2019 (COVID-19) and type 2 diabetes (T2D) are two pandemics that share the dramatic impact on global mortality and economic resources. COVID-19 largely exhibits mild to moderate clinical manifestations. However, severe pneumonia with high fatality rate may occur, especially in the elderly and in patients with underlying conditions, such as diabetes and cardiovascular disease. SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) binds to the angiotensin-converting enzyme 2 (ACE2), a ubiquitous trans-membrane carboxypeptidase, to enter the cells.
Aims
This short review discusses some open questions about the link between COVID-19 and diabetes, principally focusing on the possible effects of commonly used drugs in patients with diabetes.
Results
Preclinical studies have reported that angiotensin receptor blockers (ARBs) and ACE inhibitors might increase ACE2 expression in several cell types. Hence, it has been speculated that the treatment with these agents might influence the course of the infection, and both harmful and beneficial effects have been supposed. Other pharmacological agents are thought to increase ACE2 expression, including statins and proliferator-activated receptor gamma (PPAR-γ) agonists. All these drug classes are broadly adopted in T2D. Besides ACE2, other unknown co-factors might be involved in cell infection. It has been recently observed that dipeptidyl peptidase-4 (DPP4), the receptor for MERS-CoV (Middle East respiratory syndrome-related coronavirus) and ACE2 have similar expression profiles in the lung. DPP4 has important metabolic and immune functions and is a target for commonly used therapies in T2D.
Conclusions
Although clinical data supporting an influence of all these drugs on the course of the disease are limited, this is an interesting background for further research that might help unravel the complex mechanisms underlying the link between COVID-19 and diabetes.
Similar content being viewed by others
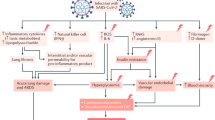


The burden of COVID-19 and diabetes
The Coronavirus disease 2019 (COVID-19), caused by the novel coronavirus SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), has abruptly reached pandemic proportions. SARS-CoV-2 is a member of the Betacoronavirus family, as well as SARS-CoV (severe acute respiratory syndrome coronavirus) and MERS-CoV (Middle East respiratory syndrome-related coronavirus). Since the beginning of 2020, the number of confirmed cases of COVID-19 has dramatically increased worldwide, leaping to more than a million by March 2020 [1]. Although the majority of COVID-19 patients develop mild to moderate clinical features [2], severe pneumonia, acute respiratory distress syndrome (ARDS) and multi-organ failure, leading to high death rate, may develop. Notably, marked sex differences have emerged in COVID-19 prevalence and prognosis. In particular, the age-specific risk of disease is reported to be significantly higher in males than in females, except under the age of 50 years. Moreover, the age-specific risks of death and hospitalization is at least two fold higher in males than in females among all age ranges [3]. Furthermore, patients with advanced age and underlying pathologies, mainly hypertension, diabetes and cardiovascular disease (CVD), are more prone to experience severe form of the disease [4]. Even though the overall mortality considerably varies among countries, the fatality-rate by age groups displays very similar patterns, rising consistently in the 60–69-year age group [5]. Importantly, the presence of comorbidities further increases mortality. In a sample of 355 patients who died of COVID-19 in Italy, the prevalence of diabetes was 35.5% [5]. Not so differently from COVID-19, the spread of diabetes has known no boundaries, and the number of affected people has reached nearly half a billion worldwide [6]. The number of deaths attributed to diabetes and its complications was around 4.2 million in 2019 [6]. Notably, more than 65% of diabetic patients are over 65 years old [7]. Thus, although with extremely different connotations, COVID-19 and diabetes are two pandemics that share the burden of a wide diffusion in the elderly population and a dramatic impact on global mortality and economic health resources.
This short review is focused on some open questions emerging from the effort to fully understand the link between COVID-19 and diabetes, mainly in the context of possible harmful or beneficial effects of commonly used drugs in patients with diabetes on the course of COVID-19 infection.
Diabetes as a risk factor for COVID-19 severity
Diabetes is reportedly a major cause of mortality and morbidity worldwide. A conceivable link between diabetes and infectious diseases has been postulated. In particular, lower respiratory tract infections are known to be rather common and severe in the elderly with type 2 diabetes (T2D) [8]. Although evidence suggests that diabetes is unlikely to significantly increase the susceptibility to SARS-CoV-2 infection, a higher risk of worse COVID-19 progression and outcomes has been observed [9]. The mechanisms underlying this association are not completely clear yet, but the exacerbated pro-inflammatory cascade and the impaired immune response in diabetic patients with COVID-19 are suspected to be crucially involved. In particular, increased levels of pro-inflammatory markers, such as leukocyte and neutrophil count, pro-calcitonin, C-reactive protein, ferritin, and circulating cytokines that trigger the cytokine storm, namely IL-6, IL-8, IL-2 receptor, TNF-α, have been detected in diabetic patients with severe COVID-19 compared to patients without diabetes [10]. Furthermore, an altered immune response in diabetic patients, possibly due to impaired lymphocyte, neutrophil and monocyte/macrophage function, has been long hypothesised, especially in the presence of poorly controlled glycaemia [11, 12]. Higher concentrations of D-dimer and fibrinogen, hallmarks of severe forms of COVID-19, have been also reported in patients with diabetes compared to patients without diabetes, suggesting that these patients might be more prone to develop coagulation abnormalities in the course of the infection, known to be significantly associated with worse prognosis [10, 13].
In a recent retrospective study involving Chinese patients (n = 904) with moderate, severe or critical disease, the major risk factors for mortality and poor outcomes were evaluated. Among the enrolled patients 15% (n = 136) had diabetes, including T2D, type 1 diabetes and gestational diabetes. Overall, diabetes increased the odds of in-hospital death and poor prognosis. Diabetic patients were older and had significant alterations of several laboratory parameters associated with poor prognosis compared to patients without diabetes. In particular, they had higher levels of D-dimer, while female diabetic patients had increased LDH and neutrophil count, compared to non-diabetic patients. CVD was more common in men with diabetes than in non-diabetic men. Notably, in multivariable regression analysis, insulin therapy was independently associated with poor prognosis. C-reactive protein was the only independent risk factor for either mortality or poor prognosis in patients with COVID-19 and diabetes [14]. Besides this, further research is also needed to clarify whether hypoglycaemia might also contribute to adverse outcomes and increased mortality in COVID-19 patients with diabetes [15].
ACE2 modulation in lung injury
Patients with diabetes have high prevalence of high blood pressure and are at increased risk of CVD [16]. Hence, the treatment of hypertension largely includes drug classes that have demonstrated to reduce cardiovascular events, such as angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) [17]. Since ACE converts angiotensin I into angiotensin II, a potent vasoconstrictor and sodium-retentive agent, the blockage of the renin–angiotensin–aldosterone system (RAAS) leads to an effective reduction in blood pressure. ACE shows homology with angiotensin-converting enzyme 2 (ACE2), the receptor to which the transmembrane Spike glycoprotein on the surface of SARS-CoV and SARS-CoV-2 binds, allowing the internalization of the virus by the alveolar cells and other cell types [18]. ACE2, which is clearly not a direct target of ACE inhibitors, is a ubiquitous mono-carboxypeptidase that converts angiotensin II into the anti-inflammatory and anti-fibrotic angiotensin-(1–7), contrasting its potent vasoconstrictor, sodium-retentive, pro-inflammatory and pro-fibrotic effects. Indeed, angiotensin-(1–7) binding to the Mas receptor induces nitric oxide release, protecting against tissue injury [19]. Another known substrate of ACE2 is angiotensin I, which is converted into angiotensin-(1–9) [20, 21]. The latter is in turn converted into angiotensin-(1–7) by ACE, therefore competing with angiotensin II for its binding [22].
Limited animal and in vitro studies have reported an upregulation of ACE2, induced by ARBs and, to a far lesser extent, by ACE inhibitors, predominantly in the heart and in the kidney. The effect is partly explained by an increase in local levels of angiotensin II, induced by ARBs, but not by ACE inhibitors [20]. Indeed, ARBs block angiotensin II type 1 (AT1) receptors and, consequently, increase the availability of the substrate angiotensin II, which upregulates ACE2 [21]. Based on this assumption, it has been speculated that the treatment with ARBs and ACE inhibitors might favour the entry of SARS-CoV-2 into the lung cells, therefore increasing the risk for adverse outcomes and severe forms of COVID-19 infection.
Conversely, a beneficial effect of a background therapy with ARB on the course of the disease has been supposed. Two complex mechanisms might support this hypothesis. Firstly, angiotensin II, by binding to its receptor AT1, might enhance the shedding of trans-membrane ACE2, promoted by ADAM17, a disintegrin and metalloprotease, therefore releasing soluble ACE2 locally. The soluble form of ACE2 is unable to mediate SARS-Cov-2 internalization, as it is no longer annexed to the cell membrane [21]. Secondly, the up-regulation of ACE2, probably induced by ARBs might not necessarily have negative effects. As SARS-CoV-2 internalization in the lung cells inevitably induces the downregulation of ACE2, the increase in its substrate, angiotensin II, a potent vasoconstrictor and pro-inflammatory agent, might contribute to an exacerbation of lung injury. Indeed, in animal models of lung injury, a clear association between the activation of the angiotensin II-AT1 signalling and the severity of lung damage has been observed, while the detrimental effects of this cascade are counterbalanced by the activation of the ACE2-angiotensin-(1–7)-Mas receptor axis [23]. Specifically, in models of sepsis or acid aspiration, which frequently cause acute lung injury and ARDS, ACE2 knockout mice experienced a more severe tissue damage, characterized by marked inflammatory cell infiltration, increased vascular permeability, lung oedema and bleeding, compared to wild-type mice. Similarly, angiotensin II type 2 (AT2) receptors knockout mice had a more marked lung damage. In the same models, the injection of recombinant human ACE2 protein into ACE2 knockout mice considerably decreased the severity of lung injury [24]. In line with these findings, the pre-treatment with recombinant human ACE2 was able to reduce inflammatory cell infiltration and tissue injury in a mouse model of respiratory syncytial virus infection [25]. Evidence from clinical studies is still rather limited. In patients with ARDS, recombinant human ACE2 increased angiotensin-(1–7) and decreased angiotensin-II concentrations, even though the study was prematurely discontinued for the lack of clinical improvement [26]. Overall, evidence from animal models suggests a conceivable pneumo-protective role of both ACE2 and AT2 receptor in the context of lung injury. A loss of pulmonary ACE2 might therefore have harmful effects, exacerbating inflammation and promoting respiratory distress and fibrosis. In the same way, the activation of ACE, angiotensin II and AT1 receptor axis might exert detrimental effects, worsening lung damage. The rationale of RAAS inhibition is therefore counterbalancing these effects, by reducing angiotensin II and increasing ACE2. Notably, ARB and ACE inhibitors attenuated LPS-induced lung injury in several preclinical studies [27,28,29,30,31]. Recently, Milne et al. analysed gene expression of ACE2, ACE, AGTR1 (encoding for AT1), TMPRSS2 (transmembrane protease serine 2, requested cofactor for the internalization of SARS-CoV-2) and ADAM17 in a large number of healthy lung tissue samples collected from patients undergoing lung resection for cancer. A significant association between use of ACE inhibitors and reduced ACE2 gene expression was observed. Differently, the use of ARBs was not linked to relevant changes in ACE2 expression, but was significantly correlated to increased ACE and decreased AGTR1 expression [32]. Although these findings are quite in contrast with data from animal studies, it should be highlighted that gene expression rather than protein concentration was analysed, and that the collected tissue samples did not exhibit lung injury or ARDS. COVID-19 results in severe lung damage and an imbalance of the RAAS system in this context cannot be excluded. Recently, in a pilot study, COVID-19 patients were found to have higher levels of circulating angiotensin II compared to healthy subjects, and a correlation between angiotensin II concentration and the severity of the disease was observed as well [33]. Thus, the effects of ACE inhibitors and ARBs might be very different in extremely damaged context, and should be therefore explored in animal models of COVID-19.
Impact of ACE inhibitors and ARBs on COVID-19 infection: clinical evidence
Mounting evidence from recent clinical studies suggests that RAAS inhibitor use is not related to an increased risk of COVID-19 infection or poor outcomes. In a retrospective cohort study of 18,472 patients screened for COVID-19, test positivity was not significantly associated with ACE inhibitor or ARB therapy [34]. Similarly, in a US study no relationship emerged between prior ACE inhibitor or ARB use and the increased likelihood of a positive SARS-CoV-2 test result, adjusting for relevant confounding factors [35]. In an Italian community-based case–control study, no association was found between ACE inhibitor or ARB treatment and COVID-19 infection, although the use of these drugs was rather frequent, due to high prevalence of CVD [36]. Overall, these findings excluded an increased risk of COVID-19 infection in patients already treated with RAAS inhibitors.
Other studies have evaluated the link between COVID-19 progression and RAAS inhibitor therapy. In a recent Korean population-based cohort study, involving 5179 COVID-19 patients, the use of ACE inhibitors or ARBs was not associated with a higher mortality risk, after adjustment for confounders [37]. In a recent systematic review, including a total of n = 23,565 COVID-19 patients from different countries, ACE inhibitor or ARB use was not associated with increased severity of COVID-19 illness [38]. Interestingly, in a meta-analysis of more than 19,000 COVID-19 patients from seven cohort studies and five case–control studies, the use of RAAS inhibitors was not linked to a higher risk of COVID-19 infection, severity or mortality, whereas mortality risk was significantly lower in patients treated with ACE inhibitors or ARBs compared to those treated with other antihypertensive drugs [39].
Taking together, data from observational studies indicates that RAAS inhibitor therapy is unlikely to have a negative impact on the course of the infection. However, whether patients with COVID-19 might take advantage of initiating these drugs or not in terms of disease progression and outcomes should be explored in clinical trials, which are still ongoing.
ACE2 modulation in myocardial injury
CVD is a well-known major complications of diabetes. The risk of developing CVD is 2- to fivefold higher in patients with T2D than in non-diabetic patients, mainly due to diabetes-related coronary atherosclerosis [40]. In addition, diabetic cardiomyopathy may occur independently of coronary artery disease, likely developing from T helper 1 driven responses within the cardiomyocyte [41, 42]. Besides the common clinical features of COVID-19, cardiovascular damage may occur in nearly 12% of patients [43], being a cause of major concern, due to a high related mortality. The mechanisms underlying this complication are largely unknown. The direct viral damage and the destabilization of coronary plaques, reportedly, are suspected to play a consistent role. Notably, in a sample of 187 patients with COVID-19, Guo et al. observed that the presence of background coronary heart disease or cardiomyopathy was associated with frequent occurrence of myocardial damage, highlighted by a marked increase in serum Troponin T in the course of the disease [44]. Accordingly, mortality was considerably higher among patients with myocardial injury and concomitant CVD (69.4%) than in patients without CVD (37.5%). Remarkably, a considerable percentage of patients displaying myocardial injury had diabetes (30%), compared to patients without myocardial injury (8.9%). Myocardial injury may therefore contribute to the poorer outcomes observed in diabetic patients with COVID-19 [45]. Of note, patients with myocardial injury were more prone to develop malignant arrhythmias, ARDS, coagulopathy and acute kidney injury, which explained high mortality [44]. Importantly, ACE inhibitors and ARBs were broadly used in patients with myocardial injury, but mortality did not significantly differ between treated and non-treated patients. Although consistent evidence excluding harms or suggesting benefits of RAAS inhibitors on COVID-19 patients with hearth injury is lacking, most ARBs were able to increase ACE2 expression in the hearts of animal models of arterial hypertension [46]. Even in this pathological context, the possible effects of these drugs should be further elucidated, as the down-regulation of ACE2 in cardiomyocytes and in blood vessels, induced by SARS-CoV2 internalization, might exacerbate myocardial injury and endothelial dysfunction, compromising heart function in patients with diabetes and underlying CVD [20, 47].
In addition, patients with T2D have high rate of lipid alterations and statins are the first line therapy for dyslipidaemia. Remarkably, evidence from preclinical studies suggests that statins might modulate ACE2 expression in several tissues, including the heart. It has been observed that atorvastatin increased ACE2 protein expression in the heart and in the kidney of rabbits with atherosclerosis [48]. In another study, diabetic rats displayed significantly lower expression of ACE2 and impaired systolic and diastolic function compared to control rats, and the combined treatment with insulin and fluvastatin was associated with higher levels of ACE2, compared to non-treated diabetic rats [49].
ACE2 is a multi-substrate enzyme and is a targeted by several molecules. Although extremely limited, there is evidence in animal studies that peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists, which are insulin-sensitizer drugs for the treatment of type 2 diabetes (T2D), might increase the expression of ACE2 in different tissues. In an animal model of hypertension, the insulin-sensitizer Rosiglitazone modulated the ACE2/angiotensin-(1–7)/angiotensin II axis in aortic tissue and lowered blood pressure by increasing the expression of ACE2 [50]. Similarly, in high fat diet fed rats, an animal model of obesity, pioglitazone increased the expression of ACE2 in insulin-sensitive tissues (liver, adipose tissue and skeletal muscle) likely through the modulation of renin-angiotensin system [51].
COVID-19 and diabetes: beyond ACE2 modulation
ACE2 is widely distributed in human tissues and, remarkably, its expression was found to be even lower in the lung than in other sites [4]. Hence, other mediators are thought to play a role in facilitating SARS-CoV-2 infection. The priming by the serine protease TMPRSS2 is requested for the binding of ACE2 and the internalization of SARS-CoV-2. TMPRSS2 cleaves the S protein in S1 and S2 subunits, a necessary step for membrane fusion and viral internalization, which contextually drags ACE2 into the alveolar cells [18]. In light of this, both antibodies targeting S-protein and TMPRSS2 inhibitors might prevent SARS-CoV-2 entry in lung cell [18]. Individuating other possible candidate co-receptors is, therefore, of great importance for the development of new therapeutic strategies to contrast the infection.
DPP4 is a ubiquitously expressed cell surface exopeptidase with important metabolic functions. It mainly regulates the bioactivity of gut-derived hormones, such as glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). They stimulate insulin secretion in response to oral glucose administration, known as the incretin effect [52]. The DPP4 inhibitors (DPP4i) are commonly used drugs for the treatment of T2D. They prevent the degradation of incretins by inhibiting the catalytic activity of DPP4. As a result, ß-cell insulin secretion is potentiated and glycaemia is significantly reduced. Independently of its catalytic function, DPP4 displays inflammatory and immune functions [53]. Indeed, it is expressed mainly by T-lymphocytes and is involved in their activation and proliferation [54]. Interestingly, pro-inflammatory factors significantly increase DPP4 expression in circulating immune cells [55]. Unlike SARS-CoV and SARS-CoV-2, which binds to ACE2 to entry into lung cells, DPP4 is the functional receptor for MERS-CoV [56]. Hence, several mouse and human monoclonal antibodies preventing protein S binding to DPP4 have given promising results for the treatment of MERS [57]. Of note, it has been recently reported that DPP4 shares patterns of expression with ACE2 in the lung, precisely in alveolar type 2 cells, the main target of Sars-CoV-2 [58]. Vankadari et al. have recently set up a docking model of DPP4 and the SARS-CoV-2 Spike glycoprotein [59]. They predicted several DPP4 residues that might be involved in the interaction with SARS-CoV-2 S1 domain of the Spike protein, also targeted by other coronaviruses that enter the host cells through the functional receptor DPP4. However, further validation is needed to draw conclusions and to establish whether the possible adhesion between DPP4 and Spike protein may play a role in SARS-CoV-2 infection. Consequently, there are some important issues to be addressed before claiming possible beneficial effects of DPP4 modulation on COVID-19 [60]. According to data from Chen et al., in patients with diabetes and moderate-severe COVID-19 the use of DPP4i did not significantly impact on mortality and clinical outcomes [14].
Conclusion
The presence of diabetes considerably influences the course of COVID-19 infection, being a risk factor for poor outcomes. In limited preclinical studies, ACE2 expression was found to be modulated by several players in different tissues. Most of them are largely employed in the treatment of diabetes, such as antihypertensive and, to a considerably lesser extent, insulin-sensitizers and statins. In light of this, the potential harmful or protective effects of these treatments in COVID-19 infection might be an interesting soil for further research. To date, data supporting that these drugs clearly interfere with SARS-Cov-2 access in lung cells or in other tissues are completely lacking. It should be stressed that in the absence of evidence from consistent clinical studies, the discontinuation or the adoption, of these therapies in the attempt to influence the course of the infection is strongly discouraged.
References
World Health Organization (2018) Global report on diabetes. https://www.who.int/news-room/fact-sheets/detail/diabetes. Accessed 17 Mar 2020
Xu XW, Wu XX, Jiang XG, Xu KJ, Ying LJ, Ma CL, Li SB, Wang HY, Zhang S, Gao HN, Sheng JF, Cai HL, Qiu YQ, Li LJ (2020) Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: retrospective case series. BMJ 368:m606. https://doi.org/10.1136/bmj.m606
Giorgi Rossi P, Marino M, Formisano D, Venturelli F, Vicentini M, Grilli R, The Reggio Emilia COVID-19 Working Group (2020) Characteristics and outcomes of a cohort of SARS-CoV-2 patients in the Province of Reggio Emilia, Italy. https://www.medrxiv.org/content/10.1101/2020.04.13.20063545v1. Accessed May 25 2020
Zou X, Chen K, Zou J, Han P, Hao J, Han Z (2020) Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. https://doi.org/10.1007/s11684-020-0754-0
Onder G, Rezza G, Brusaferro S (2020) Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA. https://doi.org/10.1001/jama.2020.4683
International Diabetes Federation (2019) IDF Diabetes Atlas. https://www.diabetesatlas.org/en/. Accessed 17 Mar 2020
Pinchevsky Y, Butkow N, Raal FJ, Chirwa T, Rothberg A (2020) Demographic and clinical factors associated with development of type 2 diabetes: a review of the literature. Int J Gen Med 13:121–129. https://doi.org/10.2147/IJGM.S226010
Pearson-Stuttard J, Blundell S, Harris T, Cook DG, Critchley J (2016) Diabetes and infection: assessing the association with glycaemic control in population-based studies. Lancet Diabetes Endocrinol 4(2):148–158. https://doi.org/10.1016/S2213-8587(15)00379-4
Fadini GP, Morieri ML, Longato E, Avogaro A (2020) Prevalence and impact of diabetes among people infected with SARS-CoV-2. J Endocrinol Invest 43(6):867–869. https://doi.org/10.1007/s40618-020-01236-2
Yan Y, Yang Y, Wang F, Ren H, Zhang S, Shi X, Yu X, Dong K (2020) Clinical characteristics and outcomes of patients with severe covid-19 with diabetes. BMJ Open Diabetes Res Care. https://doi.org/10.1136/bmjdrc-2020-001343
Moutschen MP, Scheen AJ, Lefebvre PJ (1992) Impaired immune responses in diabetes mellitus: analysis of the factors and mechanisms involved. Relevance to the increased susceptibility of diabetic patients to specific infections. Diabete Metab 18(3):187–201
Geerlings SE, Hoepelman AI (1999) Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol Med Microbiol 26(3–4):259–265. https://doi.org/10.1111/j.1574-695X.1999.tb01397.x
Spiezia L, Boscolo A, Poletto F, Cerruti L, Tiberio I, Campello E, Navalesi P, Simioni P (2020) COVID-19-Related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb Haemost. https://doi.org/10.1055/s-0040-1710018
Chen Y, Yang D, Cheng B, Chen J, Peng A, Yang C, Liu C, Xiong M, Deng A, Zhang Y, Zheng L, Huang K (2020) Clinical characteristics and outcomes of patients with diabetes and COVID-19 in association with glucose-lowering medication. Diabetes Care. https://doi.org/10.2337/dc20-0660
Zhou J, Tan J (2020) Diabetes patients with COVID-19 need better blood glucose management in Wuhan China. Metabolism 107:154216. https://doi.org/10.1016/j.metabol.2020.154216
Solini A, Zoppini G, Orsi E, Fondelli C, Trevisan R, Vedovato M, Cavalot F, Lamacchia O, Arosio M, Baroni MG, Penno G, Pugliese G, Renal I, Cardiovascular Events Study G (2014) Resistant hypertension in patients with type 2 diabetes: clinical correlates and association with complications. J Hypertens 32(12):2401–2410. https://doi.org/10.1097/HJH.0000000000000350
Saglimbene V, Palmer SC, Ruospo M, Natale P, Maione A, Nicolucci A, Vecchio M, Tognoni G, Craig JC, Pellegrini F, Lucisano G, Hegbrant J, Ariano R, Lamacchia O, Sasso A, Morano S, Filardi T, De Cosmo S, Pugliese G, Procaccini DA, Gesualdo L, Palasciano G, Johnson DW, Tonelli M, Strippoli GFM, Long-Term Impact of RASIoCOI (2018) The long-term impact of renin-angiotensin system (RAS) inhibition on cardiorenal outcomes (LIRICO): a randomized controlled trial. J Am Soc Nephrol 29(12):2890–2899. https://doi.org/10.1681/ASN.2018040443
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Muller MA, Drosten C, Pohlmann S (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. https://doi.org/10.1016/j.cell.2020.02.052
Romero CA, Orias M, Weir MR (2015) Novel RAAS agonists and antagonists: clinical applications and controversies. Nat Rev Endocrinol 11(4):242–252. https://doi.org/10.1038/nrendo.2015.6
Sahara M, Ikutomi M, Morita T, Minami Y, Nakajima T, Hirata Y, Nagai R, Sata M (2014) Deletion of angiotensin-converting enzyme 2 promotes the development of atherosclerosis and arterial neointima formation. Cardiovasc Res 101(2):236–246. https://doi.org/10.1093/cvr/cvt245
Danser AHJ, Epstein M, Batlle D (2020) Renin-angiotensin system blockers and the COVID-19 pandemic: at present there is no evidence to abandon renin-angiotensin system blockers. Hypertension. https://doi.org/10.1161/HYPERTENSIONAHA.120.15082
Arendse LB, Danser AHJ, Poglitsch M, Touyz RM, Burnett JC Jr, Llorens-Cortes C, Ehlers MR, Sturrock ED (2019) Novel therapeutic approaches targeting the renin-angiotensin system and associated peptides in hypertension and heart failure. Pharmacol Rev 71(4):539–570. https://doi.org/10.1124/pr.118.017129
Imai Y, Kuba K, Penninger JM (2008) The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol 93(5):543–548. https://doi.org/10.1113/expphysiol.2007.040048
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM (2005) Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436(7047):112–116. https://doi.org/10.1038/nature03712
Gu H, Xie Z, Li T, Zhang S, Lai C, Zhu P, Wang K, Han L, Duan Y, Zhao Z, Yang X, Xing L, Zhang P, Wang Z, Li R, Yu JJ, Wang X, Yang P (2016) Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci Rep 6:19840. https://doi.org/10.1038/srep19840
Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, Hall R, Poirier G, Ronco JJ, Tidswell M, Hardes K, Powley WM, Wright TJ, Siederer SK, Fairman DA, Lipson DA, Bayliffe AI, Lazaar AL (2017) A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care 21(1):234. https://doi.org/10.1186/s13054-017-1823-x
Wosten-van Asperen RM, Lutter R, Specht PA, Moll GN, van Woensel JB, van der Loos CM, van Goor H, Kamilic J, Florquin S, Bos AP (2011) Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1–7) or an angiotensin II receptor antagonist. J Pathol 225(4):618–627. https://doi.org/10.1002/path.2987
Liu L, Qiu HB, Yang Y, Wang L, Ding HM, Li HP (2009) Losartan, an antagonist of AT1 receptor for angiotensin II, attenuates lipopolysaccharide-induced acute lung injury in rat. Arch Biochem Biophys 481(1):131–136. https://doi.org/10.1016/j.abb.2008.09.019
Li Y, Zeng Z, Li Y, Huang W, Zhou M, Zhang X, Jiang W (2015) Angiotensin-converting enzyme inhibition attenuates lipopolysaccharide-induced lung injury by regulating the balance between angiotensin-converting enzyme and angiotensin-converting enzyme 2 and inhibiting mitogen-activated protein kinase activation. Shock 43(4):395–404. https://doi.org/10.1097/SHK.0000000000000302
Deng W, Deng Y, Deng J, Wang DX, Zhang T (2015) Losartan attenuated lipopolysaccharide-induced lung injury by suppression of lectin-like oxidized low-density lipoprotein receptor-1. Int J Clin Exp Pathol 8(12):15670–15676
Boskabadi J, Askari VR, Hosseini M, Boskabady MH (2019) Immunomodulatory properties of captopril, an ACE inhibitor, on LPS-induced lung inflammation and fibrosis as well as oxidative stress. Inflammopharmacology 27(3):639–647. https://doi.org/10.1007/s10787-018-0535-4
Minle S, Yang CY, Timens W, Bossè Y, Sin DD (2020) SARS-CoV-2 receptor ACE2 gene expression and RAAS inhibitors. Lancet Resp. Med. https://www.thelancet.com/journals/lanres/article/PIIS2213-2600(20)30224-1/fulltext - %2520. Accessed May 25 2020
Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, Wang Z, Li J, Li J, Feng C, Zhang Z, Wang L, Peng L, Chen L, Qin Y, Zhao D, Tan S, Yin L, Xu J, Zhou C, Jiang C, Liu L (2020) Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci 63(3):364–374. https://doi.org/10.1007/s11427-020-1643-8
Mehta N, Kalra A, Nowacki AS, Anjewierden S, Han Z, Bhat P, Carmona-Rubio AE, Jacob M, Procop GW, Harrington S, Milinovich A, Svensson LG, Jehi L, Young JB, Chung MK (2020) Association of use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.1855
Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB, Hausvater A, Newman JD, Berger JS, Bangalore S, Katz SD, Fishman GI, Kunichoff D, Chen Y, Ogedegbe G, Hochman JS (2020) Renin-angiotensin-aldosterone system inhibitors and risk of Covid-19. N Engl J Med. https://doi.org/10.1056/NEJMoa2008975
Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G (2020) Renin-angiotensin-aldosterone system blockers and the risk of Covid-19. N Engl J Med. https://doi.org/10.1056/NEJMoa2006923
Jung SY, Choi JC, You SH, Kim WY (2020) Association of renin-angiotensin-aldosterone system inhibitors with COVID-19-related outcomes in Korea: a nationwide population-based cohort study. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa624
Mackey K, King VJ, Gurley S, Kiefer M, Liederbauer E, Vela K, Sonnen P, Kansagara D (2020) Risks and impact of angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers on SARS-CoV-2 infection in adults. Ann Intern Med. https://doi.org/10.7326/M20-1515
Zhang X, Yu J, Pan LY, Jiang HY (2020) ACEI/ARB use and risk of infection or severity or mortality of COVID-19: a systematic review and meta-analysis. Pharmacol Res. https://doi.org/10.1016/j.phrs.2020.104927
Mizamtsidi M, Paschou SA, Grapsa J, Vryonidou A (2016) Diabetic cardiomyopathy: a clinical entity or a cluster of molecular heart changes? Eur J Clin Invest 46(11):947–953. https://doi.org/10.1111/eci.12673
Giannattasio S, Corinaldesi C, Colletti M, Di Luigi L, Antinozzi C, Filardi T, Scolletta S, Basili S, Lenzi A, Morano S, Crescioli C (2019) The phosphodiesterase 5 inhibitor sildenafil decreases the proinflammatory chemokine IL-8 in diabetic cardiomyopathy: in vivo and in vitro evidence. J Endocrinol Invest 42(6):715–725. https://doi.org/10.1007/s40618-018-0977-y
Filardi T, Ghinassi B, Di Baldassarre A, Tanzilli G, Morano S, Lenzi A, Basili S, Crescioli C (2019) Cardiomyopathy associated with diabetes: the central role of the cardiomyocyte. Int J Mol Sci. https://doi.org/10.3390/ijms20133299
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. Lancet 395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, Wang H, Wan J, Wang X, Lu Z (2020) Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.1017
Guo W, Li M, Dong Y, Zhou H, Zhang Z, Tian C, Qin R, Wang H, Shen Y, Du K, Zhao L, Fan H, Luo S, Hu D (2020) Diabetes is a risk factor for the progression and prognosis of COVID-19. Diabetes Metab Res Rev. https://doi.org/10.1002/dmrr.3319
Wang X, Ye Y, Gong H, Wu J, Yuan J, Wang S, Yin P, Ding Z, Kang L, Jiang Q, Zhang W, Li Y, Ge J, Zou Y (2016) The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang(1–7)-Mas axes in pressure overload-induced cardiac remodeling in male mice. J Mol Cell Cardiol 97:180–190. https://doi.org/10.1016/j.yjmcc.2016.05.012
Yousif MH, Dhaunsi GS, Makki BM, Qabazard BA, Akhtar S, Benter IF (2012) Characterization of Angiotensin-(1–7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacol Res 66(3):269–275. https://doi.org/10.1016/j.phrs.2012.05.001
Tikoo K, Patel G, Kumar S, Karpe PA, Sanghavi M, Malek V, Srinivasan K (2015) Tissue specific up regulation of ACE2 in rabbit model of atherosclerosis by atorvastatin: role of epigenetic histone modifications. Biochem Pharmacol 93(3):343–351. https://doi.org/10.1016/j.bcp.2014.11.013
Shin YH, Min JJ, Lee JH, Kim EH, Kim GE, Kim MH, Lee JJ, Ahn HJ (2017) The effect of fluvastatin on cardiac fibrosis and angiotensin-converting enzyme-2 expression in glucose-controlled diabetic rat hearts. Heart Vessels 32(5):618–627. https://doi.org/10.1007/s00380-016-0936-5
Sanchez-Aguilar M, Ibarra-Lara L, Del Valle-Mondragon L, Rubio-Ruiz ME, Aguilar-Navarro AG, Zamorano-Carrillo A, Ramirez-Ortega MDC, Pastelin-Hernandez G, Sanchez-Mendoza A (2019) Rosiglitazone, a ligand to PPARgamma, improves blood pressure and vascular function through renin-angiotensin system regulation. PPAR Res 2019:1371758. https://doi.org/10.1155/2019/1371758
Zhang W, Xu YZ, Liu B, Wu R, Yang YY, Xiao XQ, Zhang X (2014) Pioglitazone upregulates angiotensin converting enzyme 2 expression in insulin-sensitive tissues in rats with high-fat diet-induced nonalcoholic steatohepatitis. ScientificWorldJournal. https://doi.org/10.1155/2014/603409
Nauck MA, Meier JJ (2016) The incretin effect in healthy individuals and those with type 2 diabetes: physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol 4(6):525–536. https://doi.org/10.1016/S2213-8587(15)00482-9
Trzaskalski NA, Fadzeyeva E, Mulvihill EE (2020) Dipeptidyl peptidase-4 at the interface between inflammation and metabolism. Clin Med Insights Endocrinol Diabetes 13:1179551420912972. https://doi.org/10.1177/1179551420912972
Yu DM, Slaitini L, Gysbers V, Riekhoff AG, Kahne T, Knott HM, De Meester I, Abbott CA, McCaughan GW, Gorrell MD (2011) Soluble CD26 / dipeptidyl peptidase IV enhances human lymphocyte proliferation in vitro independent of dipeptidyl peptidase enzyme activity and adenosine deaminase binding. Scand J Immunol 73(2):102–111. https://doi.org/10.1111/j.1365-3083.2010.02488.x
Zhong J, Rao X, Deiuliis J, Braunstein Z, Narula V, Hazey J, Mikami D, Needleman B, Satoskar AR, Rajagopalan S (2013) A potential role for dendritic cell/macrophage-expressing DPP4 in obesity-induced visceral inflammation. Diabetes 62(1):149–157. https://doi.org/10.2337/db12-0230
Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, Muth D, Demmers JA, Zaki A, Fouchier RA, Thiel V, Drosten C, Rottier PJ, Osterhaus AD, Bosch BJ, Haagmans BL (2013) Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495(7440):251–254. https://doi.org/10.1038/nature12005
Du L, Yang Y, Zhou Y, Lu L, Li F, Jiang S (2017) MERS-CoV spike protein: a key target for antivirals. Expert Opin Ther Targets 21(2):131–143. https://doi.org/10.1080/14728222.2017.1271415
Qi F, Qian S, Zhang S, Zhang Z (2020) Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2020.03.044
Vankadari N, Wilce JA (2020) Emerging Wuhan (COVID-19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect 9(1):601–604. https://doi.org/10.1080/22221751.2020.1739565
Barchetta I, Cavallo MG, Baroni MG (2020) COVID-19 and diabetes: is this association driven by the DPP4 receptor? Potential clinical and therapeutic implications. Diabetes Res Clin Pract 163:108165. https://doi.org/10.1016/j.diabres.2020.108165
Acknowledgments
This review did not receive any specific grant from funding agencies in the public, commercial, or no-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study formal consent in not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Filardi, T., Morano, S. COVID-19: is there a link between the course of infection and pharmacological agents in diabetes?. J Endocrinol Invest 43, 1053–1060 (2020). https://doi.org/10.1007/s40618-020-01318-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-020-01318-1