
Abstract
The possible causal role of the environment in health disparities is not well understood, even though it has been a national priority for many years. Progress to investigate the relationship between genetics, environmental exposures, and health outcomes has been hampered by the lack of analytical tools to quantify the combined or cumulative effect of multiple chemical and non-chemical stressors on gene expression. The studies cited here provide a strong rationale for using epigenomic analysis to assess cumulative risk from multiple environmental exposures over the life course. The environment-specific “imprints” on the genome, coupled with transcriptomics and metabolomics, can be used to advance our understanding of the relationship between neighborhood disadvantage and health disparities.
Similar content being viewed by others
Introduction
Recent advances in cell and molecular biology have greatly enhanced our ability to elucidate the role of the environment in the etiology of common non-communicable diseases (e.g., cancer, type 2 diabetes, and cardiovascular disease). Over the past decades, the so-called revolution in genomics has transformed our understanding of the role of environmental stressors in the development of complex phenotypes. Contrary to our original expectation, we now know that genes do not determine biological fate; they establish a range of possible phenotypic outcomes through complex interaction with the environment. A second unexpected finding is that gene expression is regulated by both genetic (e.g., DNA nucleotide sequence) and epigenetic mechanisms.
To date, the three best known epigenetic mechanisms, collectively part of the epigenome, include DNA methylation, chemical modification of the histone component of chromatin, and alteration in the expression of short non-coding RNAs (e.g., miRNAs). These potentially durable and environment-specific modifications provide a plausible mechanism to explain how differential exposures associated with living in a disadvantaged neighborhood can become biologically embedded, with long-lasting and even possibly transgenerational effects on human health. This new era of science has a great potential to fundamentally change our understanding of the causes of significant common diseases.
Neighborhood Context Matters
Exactly why neighborhood or environmental context matters for human health and disease remains largely unknown. To date, the design of studies is less well suited to address this question because the appropriate tools have not been available. To increase our understanding of how neighborhood influences human health, one must expand the concept of the environment to include both chemical and non-chemical stressors. That is, environmental exposures need to be considered in the context of the community or social environment in which people actually live. Although researchers acknowledge that neighborhood-associated differences in health outcomes may have multiple determinants, most efforts still concentrate on the investigation of single risk factors.
The risk assessment practice typically focuses on one hazardous substance at a time and on one or a small number of health endpoints, and does not take into consideration the cumulative or combined impact of exposure to multiple chemical and non-chemical stressors over the life course. Of note, even though the health risk caused by a single stressor may be small, the public health consequences of cumulative exposure to multiple risk factors can be substantial in the population. Therefore, there is a need to assess the effects of cumulative or combined exposure to multiple environmental stressors and this requires the development of tools to integrate community characteristics into the risk assessment-risk management paradigm. For example, it has been suggested that epigenetic modifications can theoretically represent every chemical and non-chemical stressor to which populations are exposed from conception to death [1•, 2•]. This includes endogenous (e.g., hormones or oxygen radicals) and exogenous (e.g., diet, stress, air pollutants) agents. It has been repeatedly observed that environment-induced epigenetic influence persists even after the stressors have long gone. This observation suggests that signatures or profiles of DNA methylation, histone modification, and non-coding RNAs can serve as a long-term biomarker of past exposures, providing a powerful approach to examine the relationship between neighborhood context and health outcome [2•]. By combining epigenetic signatures with other “-omic” technologies including transcriptomics and metabolomics, one can identify metabolic pathways affected by neighborhood disadvantage.
Health Disparities and Socioeconomic Position
Health disparities remain a significant public health problem in the United States. As compared to whites, African Americans have 1.3-fold higher risk of dying from coronary disease, 1.8-fold higher for diabetes, and 2-fold higher for hypertension [3, 4]. Environmental justice advocates and researchers have long maintained that this increased prevalence of morbidity and shorter life expectancy among racial minority groups and socioeconomically “disadvantaged” versus “advantaged” populations are due to disproportionate exposure to environmental hazards [5, 6]. In fact, studies confirm that environmental risks are not uniformly distributed among the U.S. populations [7, 8]. Minority, the elderly, and socioeconomically disadvantaged communities are often disproportionately exposed to higher levels of hazardous environmental chemicals [9•]. These so-called “high-end” exposures represent levels of exposures that the general population does not encounter. Furthermore, these outliers are not captured in traditional risk management practices.
Whereas the exact causes of health disparities are unknown, they are likely due to multiple risk factors associated with poverty, including differences in access to health care and healthy foods, mental stress related to neighborhood violence, exposure to higher levels of environmental pollutants, and unhealthy behaviors [10]. Genetic variation could also play a role. However, there is very little evidence to support this view and several findings are not supportive of a significant causal role for genetics [11].
In contrast, numerous studies support the view of the disproportionate exposure to multiple extrinsic factors in “disadvantage” neighborhoods. For example, air pollution studies comparing inner city neighborhoods found that there is significant variability due to traffic patterns and siting of bus depots, incinerators, and manufacturing facilities. Socioeconomically disadvantaged communities in New York City were often disproportionately exposed to polycyclic aromatic hydrocarbons (PAHs) and particulates [12–14]. For example, six bus depots are located in Harlem and Washington Heights, predominantly African American and Hispanic communities. Similarly, smoking, malnutrition, and pesticide use are also more prevalent in these neighborhoods [15].
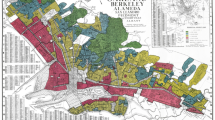
When comparing the three neighborhoods in New York City reported to have the worst health status with the three neighborhoods with the best health status (Fig. 1), sociodemographics (Fig. 1a) such as poverty and minority status were strongly associated with the worst health outcomes (Fig. 1b). Studies are underway in our laboratory to test our prediction that the epigenome of populations with such disparate health outcomes may have been uniquely programmed by neighborhood exposures (depicted schematically in Fig. 2). As such, one can use neighborhood profiles to identify gene pathways responsible for poor health outcomes of populations exposed to neighborhood disadvantage. The ability to identify neighborhoods at increased risk for developing common diseases, related to varied socioeconomic status (SES) and environmental exposures, is a public health priority.

New York City is the most diverse city in the US—a fact reflected in the distinct character of the city’s 42 neighborhoods. Here, three neighborhoods with the worst health status are compared with three best health status with respect to a sociodemographic characteristics and b adverse health outcomes. Data were extracted from “Take Care New York”, New York City Department of Health and Mental Hygiene, 2006 Report. Death rate refers to the average annual death rate in 2003–2004. Abbreviation: NYC New York City

Proposed framework that integrates neighborhood-specific epigenome profiles with cumulative risk assessment to address environmental justice issues. (Parts of the images were adapted with permission from www.motifolio.com, Biomedical PowerPoint Toolkit Suite)
Epigenome as a Biomarker for Cumulative Risk
Over the past 20 years, there has been a dramatically increasing interest in neighborhood effects on health [16]. In this paper, we further elaborate on our earlier hypothesis about how neighborhood environmental exposures interact with intrinsic biological factors (e.g., genes, age, and gender) to influence health outcomes [2•]. According to our model, advantaged and disadvantaged neighborhoods differentially “imprint” the epigenome, and these durable modifications can be detected as an exposure signature. These environment-induced epigenetic modifications can be used as a biomarker in epidemiologic studies to assess the cumulative impact of lifetime exposures to multiple chemical and non-chemical stressors. This proposed thesis lays groundwork for our understanding of how the epigenome can be used to explore the mechanism(s) by which neighborhood disadvantage is translated into health disparities.
Numerous findings are consistent with this hypothesis in that they have shown that exposure to chemical, social and behavioral factors leave their imprint on the epigenome [1•]. For example, aberrant DNA methylation, histone modifications, or altered expression of short non-coding RNAs can be induced by a variety of environmental agents, including chemical pollutants, nutrition, stress, behavior, and socioeconomic position (SEP). Furthermore, most if not all, diseases are associated with epigenetic changes, many of which alter gene expression and susceptibility to disease. Case control studies have shown that epigenetic profiles, obtained using tissue from peripheral blood, can be used to distinguish between healthy controls and individuals with various disease phenotypes [17–19]. Epigenetic changes and their influence on gene expression are best understood for tumor suppressor genes and cancer [20, 21].
Borghol et al. [22•] showed that low SEP during childhood is associated with increased morbidity later in adulthood and differential DNA methylation. For example, the number of differentially methylated genes was more than two times higher (1252 versus 545) when exposures occurred during childhood versus adulthood. This suggests that the consequences of epigenetic modifications are more pronounced if exposure to low SEP occurs during growth and development. Furthermore, by querying gene databases to gain insight on the functional relevance, the differentially methylated genes were found to be “over-represented in particular pathways and functional categories”, such as mitogen-activated protein kinase (MAPK), cytokine signaling, and DNA metabolism. Even stronger support for our hypothesis was their finding that the childhood exposed imprinted epigenome could still be detected in blood tissue extracted from individuals 45 years of age.
Similarly, Miller et al. [23] had also noted that early life exposure to extreme poverty leaves a “biological residue”, manifested by significant down regulation of genes for the glucocorticoid receptor, which regulates the secretion of cortisol and its inflammatory effect on the immune system. Low SES is also correlated with heightened expression of NF-κB and the production of the proinflammatory cytokine interleukin 6. They suggested that the chronic expression of these factors probably contribute to exaggerated adrenocortical responses which promotes aging and risk for chronic diseases. Early nurturing experiences also influence epigenetic programming of the glucocorticoid receptor gene promoter in the hippocampus of rodents and humans [24].
Likewise, cord blood derived from newborns of mothers who smoked or were depressed, has been linked to altered global DNA methylation [25]. Smoking during pregnancy was also associated with change in the expression levels of 622 genes and the methylation levels of 1024 CpG sites in DNA form the placenta [26]. Pathway analysis of differentially methylated genes showed that they played a role in oxidative phosphorylation. When the epigenetic changes were analyzed in cord blood leukocytes, it was found that 26 CpG sites in 10 genes differentially methylated in smokers were statistically significant. Similarly, Wild et al. [27] showed that individuals exposed to cigarette smoke have differential DNA methylation and gene expression profiles. Finally, McGuinness et al. [28] showed that there was an association between global methylation of DNA, socioeconomic status, lifestyle, and biomarkers for cardiovascular disease.
It is well established that a link exists between nutrition status, critical periods of development, disease pathogenesis, and epigenetic modifications [29]. Dietary restriction during pregnancy reduces methylation of the promoter of the glucocorticoid receptor genes involved in hormone-induced stress response and hypertension, with increase in expression of the glucocorticoid receptor. Food deprivation during pregnancy also resulted in low birthweight babies that were at high risk for development of type 2 diabetes and coronary heart disease in adulthood [30]. The effects of food deprivation was transgenerational in that grandchildren of the same gender as the in utero exposed individuals had increased risk for multiple diseases [31]. In summary, multiple human studies have confirmed that epigenetic programming can occur during both adulthood and embryonic development [32]. Such studies provide a mechanistic basis for the Barker hypothesis that adult diseases have their origins during embryonic development [33].
Despite improvements in the quality of the air since the passage of the Clean Air Act in 1963, adverse health effects from exposure to air pollutants, such as ozone, particulate material, PAHs, lead, and NO2 are still common occurrences. It has been estimated that air pollution contributes about 6 % of total human mortality [34]. Numerous studies have shown that exposure to high levels of air pollutants increase rates of hospital admissions and deaths from lung and cardiovascular related disorders, and that asthma prevalence was most consistently associated with socioeconomic status.
Exposure to air pollutants is also associated with epigenome modification. For example, diesel exhaust particulates altered the expression of short non-coding RNAs and histones in human airway epithelial cells; and network analysis of the 12 most altered short non-coding RNAs showed that they were part of the inflammatory response pathway associated with disease pathogenesis [35–37]. Exposure to PAHs and PM2.5 have modest effects on the global methylation of the promoter sequences of genes involved in cancer, cardiovascular, and respiratory diseases [38]. Exposure to air pollution is also associated with hypermethylation of candidate genes that play a role in the pathogenesis of asthma (e.g., the transcription factor interferon gamma) [39, 40].
Remarkable progress in epigenomic technologies has been made recently in identifying possible interactive events among exposure to air pollutants, development of cardiovascular and pulmonary dysfunctions, and epigenetic modifications. For example, both acute and chronic exposure to metal-rich PM2.5 were shown to be associated with increased risk for cardiovascular anomalies, such as variation in heart rate, systematic inflammation, and alterations in DNA methylation levels in Alu retrotransposons and long interspersed nuclear elements 1 (LINE-1) [41]. While the mechanisms by which Alu and LINE-1 methylation levels cause cardiovascular effects are largely unclear, a possible mechanism is the generation of reactive oxygen species. Exposure to PM2.5 can also induce oxidative damage to DNA and inhibit interaction between DNA and methyltransferase. Ambient air pollutants have also been shown to affect the methylation levels of genes involved in inflammation and oxidative stress [42].
Studies by Breton et al. [38, 43] showed that exposure to PM2.5 altered the methylation of several genes in the nitric oxide synthase (NOS) pathway, suggesting that these particulates may alter the production of nitric oxide. Nitric oxide homeostasis plays an important role in modulation of airway vascular smooth muscle and inflammation and therefore altered NOS expression may adversely affect lung function. To better understand how multiple pathways potentially are involved in the pathogenesis of cardiovascular disease, Bind et al. [44] developed a novel pathway analysis approach to determine whether genetic polymorphisms within relevant pathways modified the association with exposure to particulate air pollution and expression of pathway-specific biomarkers. They observed a significant association between air pollution and expression of biomarkers (e.g., C-reactive protein, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1) of these pathways and that the association was influenced by the genetic background. Also, the effects of time pattern (short and intermediate term) on biomarker expression was exposure specific [44, 45].
Conclusion
Despite substantial improvements in health care and environmental health protection over the past 50 years, large disparities in health persist in the U.S. population. Socioeconomically disadvantaged groups and minority populations (e.g., African Americans and Hispanics) tend to have worse health outcomes, with respect to higher rates of disease and reduced life expectancy, compared to the overall population in the U.S. [46, 47]. A more recent county-based survey of the U.S. population found that disparities in life expectancy can be as much as 15 years in communities that are experiencing severe poverty [8]. Most distressing is the likelihood that such devastating disparities will increase as poverty grows, and African American and Hispanic populations become a significantly larger fraction of the U.S. population. Also, the epidemic of obesity, diabetes, and cardiovascular disease among these vulnerable populations may further increase because of the transgenerational effects of epigenetic modifications. The legacy of multiple generations of exposure to higher levels of environmental stressors is likely “embedded” in the epigenome.
The premise that environmental health risks are borne disproportionately by socioeconomically disadvantaged communities has gained wider acceptance over the past 25 years [48, 49]. However, slow progress in understanding the relationship between neighborhood disadvantage and health disparities may be in part due to the lack of methodological tools to evaluate the cumulative effect of multiple environmental stressors on genetic substrates. The studies cited above provide a strong rationale to consider the link between epigenetic modifications and health disparities, with the epigenome serving as a biosensor of combined or cumulative exposure to diverse and multiple stressors over the life course. The facts that epigenetic changes are durable and often precede disease pathology make them useful as diagnostic indicators. Our prediction is that there are disease- and exposure-specific signatures or profiles with respect to DNA methylation, histone modifications, and expression of non-coding RNAs. In combination with transcriptomics and metabolomics, one can further gain an improved understanding of gene networks and metabolic pathways affected by the epigenetic modifications. However, not every modification will lead to altered gene expression; like single nucleotide polymorphisms, some will not have functional consequences.
Another interesting feature of the epigenome is that the epigenetic modifications are reversible; “durable does not mean permanent”. Unlike mutations, which alter the nucleotide sequence of DNA, the structures and mechanisms altered in the epigenome are still intact and therefore, can potentially be reversed by environmental amelioration, and by social or pharmacologic intervention. The most active and successful area of investigation involves the development of inhibitors to block histone deacetylation and DNA methylation in the treatment of cancer [50]. Moreover, some success has been achieved by changing the neighborhood environment. For example, as shown in a randomized study conducted by the U.S. Department of Housing and Urban Development, the health status of children considerably improved when moving from a high poverty neighborhood to a higher income neighborhood [51].
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Hou L, Zhang X, Wang D, Baccarelli A. Environmental chemical exposures and human epigenetics. Int J Epidemiol. 2012;41(1):79–105. This paper is an excellent comprehensive review of advances in environmental epigenetics. Data is presented to show that there is a link between environmental exposures to chemicals and alterations in epigenetic mechanisms. The authors propose a mechanism by which environmental exposures cause epigenetic changes and discuss the challenge of linking epigenetic changes with disease endpoints.
Olden K, Lin YS, Gruber D, Sonawane B. Epigenome: biosensor of cumulative exposure to chemical and nonchemical stressors related to environmental justice. Am J Public Health. 2014;104(10):1816–21. This paper is among the first pioneering attempts to adopt epigenome technology to investigate the link between cumulative effects and environmental health disparities. This work puts forth a model/hypothesis that epigenome modifications can be used as a “biosensor” of the combined or cumulative effect of environmental exposure to both chemical and non-chemical stressors over the life course.
Mensah GA. Healthy endothelium: the scientific basis for cardiovascular health promotion and chronic disease prevention. Vascul Pharmacol. 2007;46(5):310–4.
Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104(18):2158–63.
Bullard RD. Dumping in Dixie: race, class, and environmental quality. 3rd ed. Boulder: Westview Press; 2000.
Gee GC, Payne-Sturges DC. Environmental health disparities: a framework integrating psychosocial and environmental concepts. Environ Health Perspect. 2004;112(17):1645–53.
Murray CJ, Kulkarni SC, Michaud C, Tomijima N, Bulzacchelli MT, Iandiorio TJ, et al. Eight Americas: investigating mortality disparities across races, counties, and race-counties in the United States. PLoS Med. 2006;3(9):e260.
Clark LP, Millet DB, Marshall JD. National patterns in environmental injustice and inequality: outdoor NO2 air pollution in the United States. PLoS One. 2014;9(4):e94431.
Gochfeld M, Burger J. Disproportionate exposures in environmental justice and other populations: the importance of outliers. Am J Public Health. 2011;101 Suppl 1:S53–63. This paper makes a compelling case that estimating the mean population exposure is inadequate for identifying the most vulnerable or most highly exposed groups or subgroups. This may be especially true for disadvantaged populations exposed to multiple environmental chemical and non-chemical stressors.
Olden K. The complex interaction of poverty, pollution, and health status. The Scientist. 1998;1-5.
Thayer ZM, Kuzawa CW. Biological memories of past environments: epigenetic pathways to health disparities. Epigenetics. 2011;6(7):798–803.
Keeler GJ, Dvonch T, Yip FY, Parker EA, Isreal BA, Marsik FJ, et al. Assessment of personal and community-level exposures to particulate matter among children with asthma in Detroit, Michigan, as part of Community Action Against Asthma (CAAA). Environ Health Perspect. 2002;110 Suppl 2:173–81.
Gwynn RC, Thurston GD. The burden of air pollution: impacts among racial minorities. Environ Health Perspect. 2001;109 Suppl 4:501–6.
Claudio L, Tulton L, Doucette J, Landrigan PJ. Socioeconomic factors and asthma hospitalization rates in New York City. J Asthma. 1999;36(4):343–50.
Whyatt RM, Garfinkel R, Hoepner LA, Holmes D, Borjas M, Williams MK, et al. Within- and between-home variability in indoor-air insecticide levels during pregnancy among an inner-city cohort from New York City. Environ Health Perspect. 2007;115(3):383–9.
Diez-Roux AV, Nieto FJ, Muntaner C, Tyroler HA, Comstock GW, Shahar E, et al. Neighborhood environments and coronary heart disease: a multilevel analysis. Am J Epidemiol. 1997;146(1):48–63.
Chen E, Miller GE, Kobor MS, Cole SW. Maternal warmth buffers the effects of low early-life socioeconomic status on pro-inflammatory signaling in adulthood. Mol Psychiatry. 2011;16(7):729–37.
Marshall KW, Mohr S, Khettabi FE, Nossova N, Chao S, Bao W, et al. A blood-based biomarker panel for stratifying current risk for colorectal cancer. Int J Cancer. 2010;126(5):1177–86.
Kyrtopoulos SA. Making sense of OMICS data in population-based environmental health studies. Environ Mol Mutagen. 2013;54(7):468–79.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54.
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–59.
Borghol N, Suderman M, McArdle W, Racine A, Hallett M, Pembrey M, et al. Associations with early-life socio-economic position in adult DNA methylation. Int J Epidemiol. 2012;41(1):62–74. This paper reports that there is an association between early life socio-economic position and overall DNA methylation in adulthood. Analysis of the differentially methylated genes suggested plausible pathways by which SEP becomes biologically embedded.
Miller GE, Chen E, Fok AK, Walker H, Lim A, Nicholls EF, et al. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc Natl Acad Sci U S A. 2009;106(34):14716–21.
Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9(4):519–25.
Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al. 450 K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120(10):1425–31.
Suter M, Ma J, Harris A, Patterson L, Brown KA, Shope C, et al. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics. 2011;6(11):1284–94.
Wild CP, Scalbert A, Herceg Z. Measuring the exposome: a powerful basis for evaluating environmental exposures and cancer risk. Environ Mol Mutagen. 2013;54(7):480–99.
McGuinness D, McGlynn LM, Johnson PC, MacIntyre A, Batty GD, Burns H, et al. Socio-economic status is associated with epigenetic differences in the pSoBid cohort. Int J Epidemiol. 2012;41(1):151–60.
Jang H, Serra C. Nutrition, epigenetics, and diseases. Clin Nutr Res. 2014;3(1):1–8.
Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol. 2001;185(1–2):93–8.
Kaati G, Bygren LO, Pembrey M, Sjostrom M. Transgenerational response to nutrition, early life circumstances and longevity. Eur J Hum Genet. 2007;15(7):784–90.
Kuzawa CW, Sweet E. Epigenetics and the embodiment of race: developmental origins of US racial disparities in cardiovascular health. Am J Hum Biol. 2009;21(1):2–15.
Barker DJ. The fetal and infant origins of adult disease. BMJ. 1990;301(6761):1111.
Kunzli N, Kaiser R, Medina S, Studnicka M, Chanel O, Filliger P, et al. Public-health impact of outdoor and traffic-related air pollution: a European assessment. Lancet. 2000;356(9232):795–801.
Jardim MJ, Fry RC, Jaspers I, Dailey L, Diaz-Sanchez D. Disruption of microRNA expression in human airway cells by diesel exhaust particles is linked to tumorigenesis-associated pathways. Environ Health Perspect. 2009;117(11):1745–51.
Rando OJ. Combinatorial complexity in chromatin structure and function: revisiting the histone code. Curr Opin Genet Dev. 2012;22(2):148–55.
Cantone L, Nordio F, Hou L, Apostoli P, Bonzini M, Tarantini L, et al. Inhalable metal-rich air particles and histone H3K4 dimethylation and H3K9 acetylation in a cross-sectional study of steel workers. Environ Health Perspect. 2011;119(7):964–9.
Breton CV, Marutani AN. Air pollution and epigenetics: recent findings. Curr Environmen Health Rep. 2014;1(1):35–45.
States JC, Barchowsky A, Cartwright IL, Reichard JF, Futscher BW, Lantz RC. Arsenic toxicology: translating between experimental models and human pathology. Environ Health Perspect. 2011;119(10):1356–63.
Sofer T, Baccarelli A, Cantone L, Coull B, Maity A, Lin X, et al. Exposure to airborne particulate matter is associated with methylation pattern in the asthma pathway. Epigenomics. 2013;5(2):147–54.
Fan T, Fang SC, Cavallari JM, Barnett IJ, Wang Z, Su L, et al. Heart rate variability and DNA methylation levels are altered after short-term metal fume exposure among occupational welders: a repeated-measures panel study. BMC Public Health. 2014;14:1279.
Lepeule J, Bind MA, Baccarelli AA, Koutrakis P, Tarantini L, Litonjua A, et al. Epigenetic influences on associations between air pollutants and lung function in elderly men: the normative aging study. Environ Health Perspect. 2014;122(6):566–72.
Breton CV, Salam MT, Wang X, Byun HM, Siegmund KD, Gilliland FD. Particulate matter, DNA methylation in nitric oxide synthase, and childhood respiratory disease. Environ Health Perspect. 2012;120(9):1320–6.
Bind MA, Baccarelli A, Zanobetti A, Tarantini L, Suh H, Vokonas P, et al. Air pollution and markers of coagulation, inflammation, and endothelial function: associations and epigene-environment interactions in an elderly cohort. Epidemiology. 2012;23(2):332–40.
Bind MA, Coull B, Suh H, Wright R, Baccarelli A, Vokonas P, et al. A novel genetic score approach using instruments to investigate interactions between pathways and environment: application to air pollution. PLoS One. 2014;9(4):e96000.
Freeman HP. Poverty, culture, and social injustice: determinants of cancer disparities. CA Cancer J Clin. 2004;54(2):72–7.
Baquet CR, Horm JW, Gibbs T, Greenwald P. Socioeconomic factors and cancer incidence among blacks and whites. J Natl Cancer Inst. 1991;83(8):551–7.
Executive Order 12898—Federal actions to address environmental justice in minority populations and low-income populations. Federal Register. 1994:59(32). [FR Citation 59 FR 7629]. Available at: http://www.archives.gov/federal-register/executive-orders/pdf/12898.pdf (Accessed July 6, 2014).
Institute of medicine report: toward environmental justice: research, education, and health policy needs. 1999.
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92.
Goering J, Kraft J, Feins D, McInnis D, Holin MJ, Elhassan H. Moving to opportunity for fair housing demonstration program: current status and initial findings. US Dept of Housing and Urban Development: Washington, DC; 1999.
New York City Department of Health and Mental Hygiene. 2006 New York City Community Health Profiles. Available at: http://www.nyc.gov/html/doh/html/data/nyc-health-profiles.shtml (Accessed July 6, 2014).
Compliance with Ethics Guidelines
Conflict of Interest
Kenneth Olden, Heather A. Olden, and Yu-Sheng Lin declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Disclaimers
The findings and conclusions in this manuscript are those of the author(s) and do not necessarily represent the views of the U.S. Environmental Protection Agency, nor of the Henry Ford Health System. The authors declare that they have no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Environmental Epigenetics
Rights and permissions
About this article

Cite this article
Olden, K., Olden, H.A. & Lin, YS. The Role of the Epigenome in Translating Neighborhood Disadvantage Into Health Disparities. Curr Envir Health Rpt 2, 163–170 (2015). https://doi.org/10.1007/s40572-015-0048-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-015-0048-x