
Abstract
Obesity and diabetes have overtaken smoking as the number 1 preventable health determinate in the United States. In its basic form, obesity is due to disruptions of the endocrine systems that control food intake, satiety, and metabolic rate. Recent studies have identified a subclass of endocrine disrupting chemicals that interfere with hormonally regulated metabolic processes, especially during early development. These chemicals, called “obesogens,” may predispose individuals to gain weight despite efforts to limit caloric intake and increase physical activity. Evidence suggests that chemical exposures early in life can predispose individuals to weight gain through programming changes, which may enhance dysfunctional eating behaviors later in life. This review examines the latest research on the obesogen hypothesis and its underpinnings in the Developmental Origins of Heath and Disease model. We provide examples of known and suspected obesogens, and evidence of their general mechanisms of action. The research reviewed here provides a solid foundation of knowledge from which health scientists may draw from and build upon to inform their research and decision-making.
Similar content being viewed by others


Introduction
Humans in both developed and developing countries are gaining weight faster than they have in the past [1]. It is widely agreed upon that this obesity epidemic is the product of poor nutrition and lack of exercise. However, obesity is a complex endocrine disease caused by disruption of many control systems involving interaction between genetic and environmental cues such as nutrition, exercise, drugs, behavior, alterations in the microbiome, and exposure to environmental chemicals. Genome wide association studies using large populations have uncovered some 40 novel single nucleotide polymorphisms that are associated with increased body mass index (BMI), but together these genetic identifiers account for less than 5 % of the incidence of obesity [2]. Indeed, there are no known genetic mechanisms that can explain the remarkable changes in body composition over the past 20 years. These facts provide real biological plausibility to the notion that environmental exposures interfere with endocrine signaling, disrupt hormonally regulated metabolic processes, and are contributing to increased rates of obesity.
Recent studies have identified a subclass of endocrine disrupting chemicals (EDCs), which can disrupt hormonally regulated metabolic processes, especially during early development [3]. These chemicals, called “obesogens,” may predispose individuals to gain weight despite efforts to limit caloric intake and increase physical activity [4]. Plausible evidence also suggests that chemical exposures early in life can predispose individuals to weight gain through changes in metabolic ‘set-points’, and enhance dysfunctional eating behaviors later in life. This chapter reviews the latest research on the obesogen concept, including discussions of susceptibility and the Developmental Origins of Heath and Disease (DOHaD) model.
Conventional wisdom posits that obesity is strictly an energy balance disorder caused by excess calorie intake coupled with a sedentary lifestyle. It is now clear that this explanation is far too simplistic to explain all cases of obesity as there are data showing that in situations where there is equal access to food, only a subset of people become overweight [5]. Obese people tend to eat more high fat and high sugar foods and continue to eat even when they are not hungry, suggesting differences in brain chemistry, which may predispose some individuals to gain weight [6]. Recent studies show that the number of overweight infants under 6 months old increased more than 70 % from 1980 to 2001 [7]. The puzzling rise in infant weight and body mass appears to begin at birth and cannot be explained by changes in diet and exercise habits among the infants. In the US 12.4 % of kindergarten children are obese and another 14.9 % are overweight. Indeed, data show that 87 % of obese 8th graders had a BMI above the 50th percentile in kindergarten and 75 % had been above the 70th percentile, whereas only 13 % of children that were normal weight were overweight in kindergarten [8]. These data point to infancy or early childhood as an important time in establishing body weight regulation and increased susceptibility for individuals to gain weight later in life.
Early development is a time period in which an organism is critically sensitive to perturbations such as alterations in hormone levels that can lead to changes in gene expression and protein levels, which persist as tissues and organs develop [9]. This increased sensitivity is also a consequence of incomplete development or partial function of protective mechanisms such as DNA repair, immunity, xenobiotic metabolism, and the blood brain barrier in the fetus or newborn compared with older individuals. Adverse perturbations in the metabolic system of the developing organism translate to a higher risk of metabolic and hormonal disorders later in life [10]. Thus, the DOHaD hypothesis provides a framework to assess the effects of exposure to diverse factors, such as nutrition and obesogenic chemicals, on long-term health. Many disease patterns linked to poor nutrition, such as elevated risks for cardiovascular disease, diabetes, and obesity, have also been linked to maternal chemical exposure [11••], suggesting a common mechanism for chemical and nutritional stress that ultimately leads to long-term obesity in the offspring. Altogether a confluence of factors including altered developmental programming (in utero and the first few years of life), followed by overnutrition, decreased activity, and additional environmental exposures over the lifespan creates an important combination that is likely driving the obesity epidemic [12••, 13]. Childhood and adolescence are also marked by continued maturation of key endocrine systems, including the major metabolic organs, and are, therefore, also likely susceptible to chemical exposures that may alter developmental programming.
At least part of the developmental programming of disease and metabolic dysfunction is the result of alterations in the epigenetic control of gene expression during development. Epigenetic modifications, such as DNA methylation and histone methylation and acetylation, regulate gene expression during development and are, thus, responsible for normal tissue and organ development [14, 15•]. Indeed, it is now clear that the epigenetic system is responsive to environmental stimuli, such as drugs of abuse, diet or chemical exposures [16]. Animal studies indicate that many changes to our epigenetic landscape are likely to be permanent and can be manifested in multiple generations, even if the original chemical insult is no longer present (Fig. 1) [17].

Nutrition and environmental exposures share common pathway to increased disease risk. The developmental disruption effects associated with nutrients and environmental chemicals are likely 2 sides of the same coin. Developmental nutritional imbalance or environmental chemical exposures can lead to disorders such as obesity and many other diseases over the lifespan.
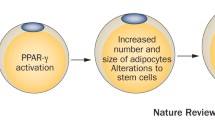
There are now a variety of environmental chemicals, including those that activate peroxisome proliferator-activated receptor gamma (PPARγ), such as tributyltin (TBT), estrogenic chemicals such as bisphenol A (BPA), and chemicals acting through other mechanisms such as lead, perfluoroctanoic acid, phthalates and tobacco smoke (a highly complex mixture of chemicals), that have been shown to lead to weight gain later in life [18–21]. New advances relative to the obesogen hypotheses include the delineation of emerging chemical obesogens, the development of nuclear receptor-activating screens to detect new obesogens, and mechanisms of action of obesogens including epigenetic and transgenerational effects, plus new data linking exposures to obesity in human populations. Recent general reviews related to the obesogen hypothesis have reviewed potential mechanisms by which environmental chemicals can cause obesity (Fig. 2) [13, 21–24].

A potential mechanism by which environmental chemicals can cause obesity in animals and humans. Only a fraction of known obesogens have a defined mechanism of action. However, several chemicals such as organotins, phthalates, phenols, and organochlorines are known to activate PPAR, the master regulator of fat development.
Chemical Obesogens
Bisphenol A
There has been a great deal of interest in BPA because of its high production volume and widespread commercial use. Numerous animal studies have shown a link between BPA exposure during development and increased body weight and adiposity across the lifespan and it is presumed, although not yet demonstrated, that these effects of BPA are mediated through estrogen receptors [25, 26]. However, several other studies have failed to find direct associations between BPA and weight gain [27–29], suggesting a need for more research to delineate the effects of BPA on metabolic systems including the importance of diet, animal species and strain, doses, and timing of exposures in the BPA effect on weight gain. Epidemiologic studies linking BPA exposure to weight gain are inconsistent overall, with the most significant associations observed in cross sectional analyses of NHANES data in the US [30]. Large prospective birth cohort studies are needed to confirm and validate findings from cross sectional human studies as well as findings in laboratory animals.
Several laboratory animal studies have demonstrated that BPA exposure can not only lead to increased body weight but also can alter other aspects of the complex endocrine system controlling weight gain and metabolism [31–33]. For instance, Macay et al demonstrated that BPA exposed female mice on a high fat diet consumed more food and gained more weight than control animals on the same diet. Their study demonstrated that early-life exposure leads to sexually dimorphic alterations in the structure of hypothalamic energy balance circuitry, leading to increased vulnerability for developing diet-induced obesity and metabolic impairments, such as glucose intolerance [33]. Similarly, a recent report showed that in vitro BPA increased neural progenitor cell proliferation and altered neurogenesis, with some of the changes in gene expression similar to those occurring in the offspring of obese dams, which have been related to hyperphagia [34].
Flame Retardants
Flame retardants are chemicals applied to a variety of materials including furniture, electronics, and construction materials and intended to reduce their flammability or delay their combustion. Polybrominated biphenyls (PBBs) and polybrominated diphenylethers (PBDEs) are widely used as flame retardant. Although a subset of these are now banned, PBDEs have been detected at biologically active levels in the majority of the people tested in the US and have been associated with a variety of adverse health outcomes including obesity and reduced thyroid function [35–39]. Prenatal or neonatal exposure to PBDEs is associated with low birth weight and thyroid function in offspring [40, 41], which are confounders of obesity later in life, however, these populations have not been followed up to actually show if changes in body weight would occur. One specific PBDE, BDE-47, is used in several commercial mixtures of flame retardants throughout the world. It has recently been shown to stimulate adipogenesis in the 3T3-L1 adipogenic screen [42] and to increased weight gain in rats exposed in utero [43].
The flame retardant mixture known as “Firemaster 550” came to market as the use of chemical flame retardants containing PBDEs were being phased out. It is now the second most commonly detected flame retardant sold in the United States. Patisaul et al [44] evaluated developmental toxicity of Firemaster 550 in pregnant rats and discovered phenotypic biomarkers associated with metabolic syndrome in the offspring. Effects noted included advanced female puberty, weight gain, which became evident prior to puberty and continued into adulthood, male cardiac hypertrophy, glucose intolerance, and increased serum thyroxine levels and reduced hepatic carboxylesterase activity in the dams. This study is the first to implicate FM 550 as an endocrine disruptor and an obesogen at environmentally relevant levels [44].
Air Pollution
Polycyclic aromatic hydrocarbons (PAHs) are a family of environmental chemicals that occur in oil, coal, and tar deposits, and are produced as byproducts of fuel burning (whether fossil fuel or biomass), including cigarette smoke and diesel exhaust. Benzo(a)pyrene is a PAH compound that has been shown to inhibit lipolysis and to cause increased fat in adult mice [45]. A recent epidemiologic study using of pregnant women who wore personal air monitors during the second trimester showed that higher prenatal exposures to PAHs was associated with increased body size of their children at age 5 and 7 [46]. Animals exposed to diesel exhaust during development and a high fat diet as adults showed increased susceptibility to diet-induced weight gain and neuroinflammation later in life [47]. Similarly early-life exposure to air pollution particulates (PM 2.5), (beginning at week 3 and continuing for 10 weeks) in mice lead to increased visceral obesity, insulin resistance, and inflammation [48]. Also, mice fed a high fat diet for 10 weeks then exposed to particulate matter (PM 2.5) as adults for 24 weeks showed increased visceral obesity, insulin resistance, and inflammation [49]. These results indicate an interaction between air pollution, diet, and metabolic programming, particularly during developmental periods. Although these select studies indicate that exposure to various components of air pollution may play a role in susceptibility to obesity, more research is needed in both animals models and humans to establish a causal relationship.
PCBs
Polychlorinated biphenyls (PCBs) are organochlorine compounds once widely used as dielectric and coolant fluids in transformers, capacitors, and electric motors. Because of their proven environmental toxicity and classification as a persistent organic pollutant, use of PCB chemicals was banned in the United States in 1979, yet they remain persistent environmental contaminants. PCBs measured in serum have been associated with increased incidence of diabetes and visceral obesity in humans studies, mostly cross sectional in nature. Recent prospective cohort study of children in the Faroe Islands showed that prenatal exposure to PCBs and dichlorodiphenyl-dichloroethylene (DDE) as a result of eating seafood can result in increased body weight at age 5–7 in girls with overweight mothers [50]. In some cases, the effects of PCB exposure on weight gain appear to be nonmonotonic, with lower doses causing weight gain, whereas higher doses result in weight loss. These results may be attributed to PCB interaction with the endocrine system at lower doses and general toxicity at higher doses. Currently, there are no animal studies reporting PCB exposure and weight gain. However, studies have shown PCB effects on brain development, including areas that could impact satiety and metabolism [51, 52].
High Fructose Corn Syrup
Sugars have been increasingly linked with obesity in Western societies that consume large amounts of sweetened soda. There is growing evidence that the obesogenic effect of dietary sugars is due mainly to the fructose content [53]. Table sugar (sucrose) is 50 % glucose and 50 % fructose and it is the fructose component that is most problematic. In addition, fructose is commercially derived from sugar cane, sugar beets, and maize and processed mainly into high fructose corn syrup. Fructose is commonly added to foods and drinks for palatability and taste enhancement, and for browning of some foods, such as baked goods. The glycemic index of fructose is significantly lower than glucose. The majority of fructose quickly metabolized in the liver [54] without inducing insulin secretion [55]. For this reason, low doses of fructose are thought to help regulate glucose homeostasis [56], but at the higher doses ingested by many Americans compared with other countries [57], as a result of the addition of fructose to many products and the general increased consumption of sugar, fructose has pathologic consequences. Unlike glucose, which is stored as glycogen, fructose metabolites are stored as triglycerides in the liver [58], and excess fat is secreted in the form of very low density lipoprotein (VLDL), which is highly associated with type 2 diabetes [59]. Fructose results in de novo lipogenesis in the liver, and thus, is a unique obesogen by directly stimulating inappropriate storage of fat in the liver, as opposed to the adipocyte. Increasing the palatability of food by addition of sucrose undermines normal satiety signals and motivates energy intake independent of energy need.
Fructose, and thus, high fructose corn syrup, exert specific biochemical effects beyond their caloric equivalent, thus, a calorie is not a calorie when it comes to comparing glucose and fructose. Human and animal data have shown that fructose acts on the satiety centers of the brain and blunts blood levels of the satiety signal Peptide YY3-36 and causes leptin resistance, which is consistent with increased caloric intake [60•]. While glucose is metabolized into carbon dioxide, water, and ATP, fructose is metabolized into different intermediary metabolites, which tend to overwhelm hepatic mitochondrial capacity. This, in turn, promotes de novo lipogenesis, hepatic insulin resistance, which drives chronic metabolic disease. Aberrant fructose metabolism also leads to inflammation because of fructosylation of proteins with resultant superoxide formation putting increasing strain on the pancreatic beta cells. There are now some animal studies [61] showing that the offspring of fructose-fed rats (10 % solution in addition to chow) caused increased body weight and fat tissue, increased food intake, and decreased sensitivity to leptin and insulin resistance throughout their lifetime. Thus, like other obesogens, developmental exposures to fructose may increase susceptibility to obesity later in life.
Transgenerational Obesogenic Transmission
In the last decade a considerable body of evidence has shown that chemicals administered during precise time periods of fetal development in animal models have been shown to generate phenotypes not just in the first generation, but in the second, or third generation [62]. These phenotypes are likely nongenetically determined since low dose exposure to EDCs typically do not damage DNA but rather cause changes to the epigenetic profile to create a permanent change in the germline [63]. Certain diseases or gene expression changes can be found in the F3 generation and beyond, presumably resulting from chemical exposures that affect DNA loci, which escape reprogramming mechanisms during gametogenesis that normally erase epigenetic marks acquired by the previous generation [15•, 18, 64]. For example, exposure to certain fungicides, pesticides, plastics, and air pollution is linked to ovarian diseases in the F3 generation [65]. Maternal exposure to BPA has been linked to social behavioral changes in F4 through epigenetic changes in imprinted genes [66].
A preponderance of data surrounding transgenerational inheritance of obesity is beginning to appear in the literature. Prenatal exposure of pregnant mice to TBT led to increased adipose depot weight, larger adipocyte size and biased cell fate in the mesenchymal stem cell compartment to favor the adipocyte formation: effects that were transmitted in the F1, F2, and F3 generations [18]. Moreover, prenatal TBT exposure led to fatty liver in all 3 generations [18]. Recent data also show the transgenerational inheritance of obesity from exposure to DDT, a mixture of BPA and Di(2-ethylhexyl)phthalate (DEHP) and a hydrocarbon mixture (jet fuel JP-8) [67–69]. There was no transgenerational transmission of obesity after exposure to dioxin, vinclozolin, or a mixture of permethrin and DEET, showing selectivity to metabolic processes [63]. These results demonstrate that the effects of early-life exposure to at least some obesogens are permanent and transgenerational, increasing the risk of future generations to develop obesity and related disorders.
Obesogen Screens
In order to expand and further validate the obesogen hypothesis it is critical to develop robust, reproducible, and sensitive assays to screen large numbers of chemicals for obesogenic properties. During the past few years several screens have been developed that meet these criteria.
In Vitro Assays
In vitro cell models allow for rapid and systematic targeting of signaling pathways linked to obesity in humans. For example, a common obesogen target is PPARγ, which is strongly expressed in adipose tissue where it is essential for adipose tissue differentiation [12••]. Pereira-Fernandes et al developed a standardized and reproducible protocol using 3T3-L1 cells and a direct measurement of Nile red staining [70]. This adipocyte differentiation assay proved very sensitive to the positive controls TBT and rosiglitazone and showed positive results for BPA, parabens, several musks, and phthalate compounds. This assay, used in combination with the CALUX assay (chemical-activated Luciferase gene expression) (BioDetection Systems; Amsterdam) which measures gene expression in cells, has proven to be reliable and efficient tools to study the influence of EDCs on transcriptional regulations of genes involved in fat metabolism [71].
In Vivo Assays
Whole animal assays with fish and other organisms allow for high throughput examinations of multiple endpoints for multiple hormones, and multiple mechanisms of action. New compounds, thus, can be screened without prior information on suspected activity or mechanisms of action. Zebra fish are a good model in which to study metabolism because they possess all the key organs required for metabolic control in humans, from the appetite circuits that are present in the hypothalamus, through to the pancreas and insulin-sensitive tissues [liver, muscle. and white adipose tissue (WAT)] [72]. Zebra fish also express orthologues of key genes such as PPARg, fatty acid binding protein 4 (FABP4), Glucose transporter type 4 (GLUt4), leptin and adiponectin and one can measure relative amounts of subcutaneous and visceral white adipose tissue during development in the presence or absence of chemical challenges.
Similar to humans, Zebra fish store fat in the form of large unilocular lipid droplets in white adipocytes, suggesting that the Zebra fish could serve as a useful model in which to study the biology of the adipose depot itself. Adipocytes can by visualized and quantified by staining with the neutral dye Oil Red O or with Nile red, a lipophilic stain that fluoresces in lipid-rich environments [73]. The advantage of fluorescent staining is that it can be used in live fish, allowing real-time imaging of the formation of adipocytes and their expansion under conditions of nutrient excess. Development of transgenic and reporter lines for adipose tissue will provide an invaluable tool for obesity research, and will be an excellent resource for high-throughput screening of potential drugs for the treatment of obesity [72].
Conclusions
Metabolism is a complex process involving many hormones such as estrogens, androgens, glucocorticoids, insulin and thyroid hormones. These hormones play important roles in controlling adipose tissue development, metabolism and satiety, and body weight. The metabolic system is also sensitive to environmental chemicals and drugs that can affect these hormonal pathways. It is well known that many pharmaceuticals have adverse side effects of increasing weight and reducing energy levels [74]. While the targets of these drugs that result in weight gain is not known in many cases, rosiglitazone, which regulates glucose levels, also targets pathways/genes involved in obesity such as PPAR gamma/RXR (18). Therefore, it is likely that environmental chemicals that act through similar biological mechanisms are capable of altering weight gain and metabolism in humans.
Certainly overeating and lack of exercise play an important role in the etiology of obesity. However, questions remain concerning the biological forces driving humans toward this behavior. Data now show that exposures to obesogens during development can permanently alter metabolic programming and alter the “set point” for adipose tissue formation, and regulation of satiety. In addition, obesogen exposure can trigger changes in the hypothalamus that play a particularly important role in feeding behaviors. Thus, improper hypothalamic programming may help explain differences between the eating behavior of lean and obese individuals.
The obesogen hypothesis proposes a mechanism for the increased epidemic of obesity and a possible solution. If obesity is due in part to developmental exposures to obesogens, then there are clear preventative strategies including reducing exposures to environmental chemicals during development that we can take to stem propagation of this disease. Indeed, incorporation of strategies to prevent early life exposures to obesogenic chemicals should be the first line of defense to prevent the obesity epidemic.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Hebert JR et al. Scientific decision making, policy decisions, and the obesity pandemic. Mayo Clin Proc. 2013;88:593–604. doi:10.1016/j.mayocp.2013.04.005.
Speliotes EK et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–48. doi:10.1038/ng.686. Epub 2010 Oct 10.
Diamanti-Kandarakis E et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342.
Grun F, Blumberg B. Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology. 2006;147(Suppl):S50–5.
Speakman JR et al. Set points, settling points and some alternative models: theoretical options to understand how genes and environments combine to regulate body adiposity. Dis Model Mech. 2011;4:733–45. doi:10.1242/dmm.008698.
Corsica JA, Pelchat ML. Food addiction: true or false? Curr Opin Gastroenterol. 2010;26:165–9. doi:10.1097/MOG.0b013e328336528d.
Kim J et al. Trends in overweight from 1980 through 2001 among preschool-aged children enrolled in a health maintenance organization. Obesity (Silver Spring). 2006;14:1107–12.
Cunningham SA, Kramer MR, Narayan K. Incidence of childhood obesity in the United States. N Engl J Med. 2014;370:403–11. doi:10.1056/NEJMoa1309753.
Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol. 2007;23:297–307.
Newbold RR, Padilla-Banks E, Jefferson WN. Environmental estrogens and obesity. Mol Cell Endocrinol. 2009;304:84–9.
Barouki R et al. Developmental origins of non-communicable disease: implications for research and public health. Environ Health. 2012;11:42. Important overview of DOHaD.
Janesick A, Blumberg B. Obesogens, stem cells and the developmental programming of obesity. Int J Androl. 2012;35:437–48. doi:10.1111/j.1365-2605.2012.01247.x. Important early review of obesogens.
Heindel JJ, Schug TT. The perfect storm for obesity. Obesity (Silver Spring). 2013;21:1079–80. doi:10.1002/oby.20222.
Bernal AJ, Jirtle RL. Epigenomic disruption: the effects of early developmental exposures. Birth Defects Res A Clin Mol Teratol. 2010;88:938–44.
Skinner MK, Manikkam M, Guerrero-Bosagna C. Epigenetic transgenerational actions of endocrine disruptors. Reprod Toxicol. 2011;31:337–43. Overview of transgenerational effects.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2011;13:97–109.
Skinner MK et al. Environmentally induced transgenerational epigenetic reprogramming of primordial germ cells and the subsequent germ line. PLoS One. 2013;8:e66318.
Chamorro-Garcia R, et al. Transgenerational inheritance of prenatal obesogen exposure. Environ Health Perspect. 2012.
Kirchner S et al. Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol Endocrinol. 2010;24:526–39.
Grun F et al. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20:2141–55.
Thayer KA et al. Role of environmental chemicals in diabetes and obesity: a National Toxicology Program workshop review. Environ Health Perspect. 2012;120:779–89. doi:10.1289/ehp.1104597.
Kelishadi R et al. Childhood obesity: today and tomorrow's health challenge. J Obes. 2013;2013:208392. doi:10.1155/2013/208392.
Tang-Peronard JL et al. Endocrine-disrupting chemicals and obesity development in humans: a review. Obes Rev. 2011;12:622–36. doi:10.1111/j.1467-789X.2011.00871.x.
Regnier SM, Sargis RM. Adipocytes under assault: environmental disruption of adipose physiology. Biochim Biophys Acta. 1842;2014:520–33. doi:10.1016/j.bbadis.2013.05.028.
Olea N et al. Estrogenicity of resin-based composites and sealants used in dentistry. Environ Health Perspect. 1996;104:298–305.
Rubin BS. Bisphenol A: an endocrine disruptor with widespread exposure and multiple effects. J Steroid Biochem Mol Biol. 2011;127:27–34.
Anderson OS et al. Perinatal bisphenol A exposure promotes hyperactivity, lean body composition, and hormonal responses across the murine life course. Faseb J. 2013;27:1784–92. doi:10.1096/fj.12-223545.
Kendig EL et al. Estrogen-like disruptive effects of dietary exposure to bisphenol A or 17alpha-ethinyl estradiol in CD1 mice. Int J Toxicol. 2012;31:537–50. doi:10.1177/1091581812463254.
Mirmira P, Evans-Molina C. Bisphenol A, obesity, and type 2 diabetes mellitus: genuine concern or unnecessary preoccupation? Transl Res. 2014;13:00087–5.
LaKind JS, Goodman M, Mattison DR. Bisphenol A and indicators of obesity, glucose metabolism/type 2 diabetes and cardiovascular disease: a systematic review of epidemiologic research. Crit Rev Toxicol. 0:1–30.
Masuno H et al. Bisphenol A in combination with insulin can accelerate the conversion of 3T3-L1 fibroblasts to adipocytes. J Lipid Res. 2002;43:676–84.
Sakurai K et al. Bisphenol A affects glucose transport in mouse 3T3-F442A adipocytes. Br J Pharmacol. 2004;141:209–14.
Mackay H et al. Organizational effects of perinatal exposure to bisphenol-A and diethylstilbestrol on arcuate nucleus circuitry controlling food intake and energy expenditure in male and female CD-1 mice. Endocrinology. 2013;154:1465–75. doi:10.1210/en.2012-2044.
Desai M. et al. Bisphenol A increases neural progenitor cell proliferation and alters neurogenesis. Repro Sci. 2013;20(Suppl 3):204A
Allgood EL. The effect of diet and polybrominated diphenyl ether (PBDE) exposure on adipocyte and whole body metabolism in male Wistar rats. Molecular, Cellular, & Biomedical Sciences, UNH. 2009.
Hallgren S et al. Effects of polybrominated diphenyl ethers (PBDEs) and polychlorinated biphenyls (PCBs) on thyroid hormone and vitamin A levels in rats and mice. Arch Toxicol. 2001;75:200–8.
Hoppe AA, Carey GB. Polybrominated diphenyl ethers as endocrine disruptors of adipocyte metabolism. Obesity (Silver Spring). 2007;15:2942–50.
van der Ven LT et al. A 28-day oral dose toxicity study enhanced to detect endocrine effects of hexabromocyclododecane in Wistar rats. Toxicol Sci. 2006;94:281–92.
van der Ven LT et al. A 28-day oral dose toxicity study enhanced to detect endocrine effects of a purified technical pentabromodiphenyl ether (pentaBDE) mixture in Wistar rats. Toxicology. 2008;245:109–22.
Chao HR et al. Levels of polybrominated diphenyl ethers (PBDEs) in breast milk from central Taiwan and their relation to infant birth outcome and maternal menstruation effects. Environ Int. 2007;33:239–45.
Herbstman JB et al. Birth delivery mode modifies the associations between prenatal polychlorinated biphenyl (PCB) and polybrominated diphenyl ether (PBDE) and neonatal thyroid hormone levels. Environ Health Perspect. 2008;116:1376–82.
Bastos Sales L et al. Effects of endocrine disrupting chemicals on in vitro global DNA methylation and adipocyte differentiation. Toxicol in Vitro. 2013;27:1634–43. doi:10.1016/j.tiv.2013.04.005.
Suvorov A, Battista MC, Takser L. Perinatal exposure to low-dose 2,2′,4,4′-tetrabromodiphenyl ether affects growth in rat offspring: what is the role of IGF-1? Toxicology. 2009;260:126–31. doi:10.1016/j.tox.2009.03.018.
Patisaul HB et al. Accumulation and endocrine disrupting effects of the flame retardant mixture Firemaster(R) 550 in rats: an exploratory assessment. J Biochem Mol Toxicol. 2013;27:124–36. doi:10.1002/jbt.21439.
Irigaray P et al. Benzo[a]pyrene impairs beta-adrenergic stimulation of adipose tissue lipolysis and causes weight gain in mice. A novel molecular mechanism of toxicity for a common food pollutant. FEBS J. 2006;273:1362–72.
Rundle A et al. Association of childhood obesity with maternal exposure to ambient air polycyclic aromatic hydrocarbons during pregnancy. Am J Epidemiol. 2012;175:1163–72. doi:10.1093/aje/kwr455.
Bolton JL et al. Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J. 2012;26:4743–54. doi:10.1096/fj.12-210989.
Zheng Z et al. Exposure to ambient particulate matter induces a NASH-like phenotype and impairs hepatic glucose metabolism in an animal model. J Hepatol. 2013;58:148–54. doi:10.1016/j.jhep.2012.08.009.
Sun Q et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation. 2009;119:538–46. doi:10.1161/CIRCULATIONAHA.108.799015.
Tang-Peronard JL et al. Association between prenatal polychlorinated biphenyl exposure and obesity development at ages 5 and 7 years: a prospective cohort study of 656 children from the Faroe Islands. Am J Clin Nutr. 2014;99:5–13. doi:10.3945/ajcn.113.066720.
Tan SW, Zoeller RT. Integrating basic research on thyroid hormone action into screening and testing programs for thyroid disruptors. Crit Rev Toxicol. 2007;37:5–10.
Dickerson SM, Cunningham SL, Gore AC. Prenatal PCBs disrupt early neuroendocrine development of the rat hypothalamus. Toxicol Appl Pharmacol. 2011;252:36–46. doi:10.1016/j.taap.2011.01.012.
Lustig RH. Fructose: metabolic, hedonic, and societal parallels with ethanol. J Am Diet Assoc. 2010;110:1307–21. doi:10.1016/j.jada.2010.06.008.
Bjorkman O, Crump M, Phillips RW. Intestinal metabolism of orally administered glucose and fructose in Yucatan miniature swine. J Nutr. 1984;114:1413–20.
Bruckdorfer KR, Kang SS, Yudkin J. Plasma concentrations of insulin, corticosterone, lipids and sugars in rats fed on meals with glucose and fructose. Proc Nutr Soc. 1973;32:12A–3A.
Sievenpiper JL et al. ‘Catalytic’ doses of fructose may benefit glycaemic control without harming cardiometabolic risk factors: a small meta-analysis of randomised controlled feeding trials. Br J Nutr. 2012;108:418–23. doi:10.1017/S000711451200013X.
Lustig RH, Schmidt LA, Brindis CD. Public health: the toxic truth about sugar. Nature. 2012;482:27–9.
Ouyang X et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol. 2008;48:993–9.
Adiels M et al. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–36.
Goran MI et al. The obesogenic effect of high fructose exposure during early development. Nat Rev Endocrinol. 2013;9:494–500. doi:10.1038/nrendo.2013.108. Overview of effects of fructose.
Goran MI et al. The obesogenic effect of high fructose exposure during early development. Nat Rev Endocrinol. 2013;9:494–500.
Guerrero-Bosagna C, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of phenotype and disease. Mol Cell Endocrinol. 2012;354:3–8. doi:10.1016/j.mce.2011.10.004.
Manikkam M et al. Pesticide and insect repellent mixture (permethrin and DEET) induces epigenetic transgenerational inheritance of disease and sperm epimutations. Reprod Toxicol. 2012;34:708–19. doi:10.1016/j.reprotox.2012.08.010.
Anway MD, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors. Endocrinology. 2006;147(Suppl):S43–9.
Nilsson E et al. Environmentally induced epigenetic transgenerational inheritance of ovarian disease. PLoS One. 2012;7:e36129.
Wolstenholme JT, et al. Gestational Exposure to Bisphenol A produces transgenerational changes in behaviors and gene expression. Endocrinology. 2012;153:3828-38
Tracey R et al. Hydrocarbons (jet fuel JP-8) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. Reprod Toxicol. 2013;36:104–16.
Skinner MK et al. Ancestral dichlorodiphenyltrichloroethane (DDT) exposure promotes epigenetic transgenerational inheritance of obesity. BMC Med. 2013;11:228.
Manikkam M et al. Plastics derived endocrine disruptors (BPA, DEHP, and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS One. 2013;8:e55387.
Pereira-Fernandes A et al. Evaluation of a screening system for obesogenic compounds: screening of endocrine disrupting compounds and evaluation of the PPAR dependency of the effect. PLoS One. 2013;8:e77481. doi:10.1371/journal.pone.0077481.
Punzon I et al. Towards a humanized PPARgamma reporter system for in vivo screening of obesogens. Mol Cell Endocrinol. 2013;374:1–9. doi:10.1016/j.mce.2013.04.004.
Seth A, Stemple DL, Barroso I. The emerging use of Zebra fish to model metabolic disease. Dis Model Mech. 2013;6:1080–8. doi:10.1242/dmm.011346.
Flynn III EJ, Trent CM, Rawls JF. Ontogeny and nutritional control of adipogenesis in Zebra fish (Danio rerio). J Lipid Res. 2009;50:1641–52. doi:10.1194/jlr.M800590-JLR200.
Schug TT et al. Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol. 2011;127:204–15.
Compliance with Ethics Guidelines
Conflict of Interest
Dr. Jerrold J. Heindel and Dr. Thaddeus T. Schug each declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article may be the work of an employee of the National Institute of Environmental Health Sciences (NIEHS), NIH. However, the statements, opinions or conclusions contained therein do not necessarily represent those of NIEHS, NIH or the U.S. Government.
Rights and permissions
About this article

Cite this article
Heindel, J.J., Schug, T.T. The Obesogen Hypothesis: Current Status and Implications for Human Health. Curr Envir Health Rpt 1, 333–340 (2014). https://doi.org/10.1007/s40572-014-0026-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-014-0026-8