
Abstract
Aging is an important risk factor for several human diseases such as cancer, cardiovascular disease and neurodegenerative disorders, resulting from a combination of genetic and environmental factors (e.g., diet, smoking, obesity and stress), which, at molecular level, cause changes in gene expression underlying the decline of physiological function. Epigenetics, which include mechanisms regulating gene expression independently of changes to DNA sequence, regulate gene expression by modulating the structure of chromatin or by regulating the binding of transcriptional machinery to DNA. Several studies showed that an impairment of epigenetic mechanisms promotes alteration of gene expression underlying several aging-related diseases. Alteration of these mechanisms is also linked with changes of gene expression that occurs during aging processes of different tissues. In this review, we will outline the potential role of epigenetics in the onset of two age-related pathologies, cancer and cardiovascular diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aging is associated with a progressive decline of numerous physiological processes, which is accompanied by an increase of risk for development of severe diseases such as cancer, cardiovascular disease, senile dementias and type-II diabetes in the elderly. Interestingly, the incidence of many cancers increases after 50 years of age, and diagnoses of heart failure (HF) increases from the age of 60 years, with most patients being over 70 years old. This, coupled with the fact that the average life span has increased, is resulting in aging having a considerable importance at both economic and social levels [1]. Despite this, the mechanisms that link aging with the onset of aging-related diseases remain largely unknown.
The aging process of an organism results from a combination of stochastic events including both genetic and environmental factors (e.g., diet, smoking, obesity and stress), which, at the molecular level, cause changes in gene expression underlying the decline of physiological function. For example, aging of the brain is accompanied by changes of expression of genes encoding proteins involved in inflammatory and stress responses and neuropeptide metabolism, while the elderly heart has an altered transcription profile that contributes to impairment of heart function [2].
Furthermore, aging-related diseases are consequences of an impairment of gene expression. For example, cancer is caused by alteration of gene expression that leads to cellular acquisition of neoplastic characteristics (e.g., proliferation that is not under the control of the organism, loss of differentiation and ability to metastasize to distal tissues). These changes can be triggered by genetic mutations or stochastic events that cause changes in the gene expression programs [3]. In general, the subjection of DNA, RNA, and proteins to chemical alterations throughout the lifespan impairs both their structure and function. The consequences of nucleic acid and protein damage during aging have been linked to functional deterioration of cells and organs, inevitably leading to disease [4]. While heart failure is accompanied by two pathological processes, cardiac hypertrophy and cardiac fibrosis, the underlying source results from alterations of gene expression. Cardiac hypertrophy is accompanied by an increase of expression of fetal cardiac genes (e.g., Nppa, Nppb, Myh7 and Skeletal Alpha-Actin) and repression of adult genes (e.g., Myh6), while cardiac fibrosis is a result of an increase of expression of genes encoding proteins of the extracellular matrix (e.g., collagens), processes for which the specific underlying mechanisms still remain unknown [5].
Several studies over the last decade have strongly implicated epigenetic mechanisms in the (dys)regulation of the gene expression changes regulating several aging-related diseases such as cancer and heart failure, and in promoting the alteration of gene expression responsible for the aging process of different tissues. Therefore, alteration of the epigenetic mechanisms occurring during aging renders cells more prone to the transcriptional changes responsible for aging-related diseases. The impairment of gene expression, and the multifactorial nature of epigenetic changes throughout the aging process and disease onset render their mechanisms of action enigmatic: it is, thus, of utmost importance to elucidate these mechanisms to understand the origins of age-related disease. In this short review, we will discuss the possible role of epigenetics in regulating the onset of two aging-related diseases, cancer and cardiovascular disease.
A brief overview of epigenetics
Epigenetics refers to all the mechanisms regulating gene expression independent of the DNA sequence, which can be grouped into four main processes: ATP-dependent chromatin-remodeling complexes, DNA and histone modifications, and non-coding RNAs. These processes regulate gene expression by modulating the structure of chromatin or by regulating the binding of transcriptional machinery to DNA. One important feature of these mechanisms is that they can be modulated by a myriad of factors including physiological and pathological stimuli, as well as by environmental factors such as diet, stress, physical activity, working habits, smoking and alcohol consumption (Fig. 1) [6,7,8].

Overview of the epigenetic mechanisms that mediate the effects of physiological and pathological stimuli and environmental factors on gene expression
ATP-dependent chromatin-remodeling complexes are multi-protein complexes that regulate gene expression by modifying the nucleosome organization of DNA using energy derived from ATP hydrolysis. Members of these families function as transcriptional activators by promoting the formation of an open and accessible chromatin structure, allowing the recruitment of several proteins involved in transcription. For example, SWI/SNF complexes promote the formation of this structure through a mechanism involving sliding nucleosomes, evicting the H2A/H2B dimers or removing the histone octamers from DNA. Other chromatin-remodeling factors cause gene silencing by organizing the nucleosomes on DNA in a manner that results in the chromatin structure taking a highly compacted form, and thus preventing accessibility to transcription factors [9].
DNA modifications are covalent modifications of DNA bases: the most commonly studied modifications are methylation and hydroxymethylation of cytosine bases. DNA methylation occurs predominantly on the cytosine within CG-dinucleotide-rich genomic regions. These genomic regions are called CpG islands and are found in the majority of promoters in both the human and mouse genomes. The methylation of cytosine promotes transcriptional repression by acting as docking sites of methyl-CpG binding domain (MBD) proteins, a family of proteins that promotes the formation of silent chromatin. In mammals, the DNA methylation pattern is established and maintained by three DNA methyltransferase enzymes (DNMTs): DNMT3A and DNMT3B are essential for de novo DNA methylation during development [10], while DNMT1 is required for maintaining methylation patterns during cell division. Moreover, DNA hydroxymethylation is the product of hydroxylation of 5-mC (5-methylcytosine) catalyzed by ten–eleven translocation (TET) enzymes. High levels of 5-hmC (5-hydroxymethylcytosine) in promoter and enhancer regions are linked with high levels of transcription [11].
Histone modifications are covalent post-translational modifications, which include acetylation, methylation, phosphorylation, ubiquitylation and sumoylation. Among these, the best studied are acetylation and methylation. Acetylation occurs on lysine residues present in the tails of histones, allowing transcription factor accessibility as a result of neutralization of the positive charge of histone tails [7]. The level of acetylation of chromatin depends on the activity of two classes of enzymes, histone acetyltransferases, which catalyze the transfer of an acetyl group from acetyl-coenzyme A to lysine residues present in the tails of histones, and histone deacetylases which remove this acetyl group [7]. Histone methylation is another important epigenetic marker, whose effects on transcription depend on the specific position and degree of lysine and arginine methylation on histone tails. Briefly, histone methylation associated with transcription activation occurs as di- or tri-methylation of histone H3 at lysines 4, 36 and 79 (H3K4, H3K36 and H3K79, respectively) and mono-methylated H3K9 and H4K20; whereas, transcriptional repression is characteristic of tri-methylation of H3K9, H3K27, H4K20 and di-methylation of H3K9, which are involved in gene silencing through formation of facultative and constitutive heterochromatic regions [6, 7]. Just like histone acetylation, methylation is a dynamic process resulting from the activity of two classes of enzymes: histone methyltransferases, which catalyze the transfer of a methyl group from S-adenosyl-methionine to lysine or arginine residues of histones, and histone demethylases, which catalyze the demethylation of histone tails.
The last class of epigenetic mechanisms involves noncoding RNAs (ncRNAs), which include a variety of RNAs that are not translated into proteins. Non-coding RNAs are classified based on their length: short and long. The class of short ncRNAs includes molecules of RNA shorter than 200 nucleotide such as PIWI-interacting RNAs, small interfering RNAs (siRNAs), and microRNAs (miRNAs). In contrast, long ncRNAs (lncRNAs) includes molecules of RNA longer than 200 nucleotides. NcRNAs regulate the expression of proteins at the transcriptional and translational levels. The lack of species conservation of lncRNA renders its study more difficult, although their temporal and spatial expression could be key to understanding the regulation of chromatin structure, recruitment of transcriptional machinery and gene expression. Conversely, microRNAs inhibit the expression of genes by binding to the 3′-UTR (untranslated region) of their target mRNAs, resulting in a degradation of the target mRNA and a subsequent inhibition protein translation [12].
Over the last few decades, the involvement of epigenetic mechanisms has been implicated in several aging-related diseases. In this review, we will focus our attention on two key pathologies, cancer and cardiovascular disease.
Ageing and epigenetics in pathologies
Aging, cancer and epigenetics
Cancer is a disease that results from accumulation of genetic and epigenetic alterations, and for several cancers, the most important risk factor is age [13]. Perturbations to the genome as a result of changes to cellular environment, inflammation, decrease immune function and accumulation of DNA damage, result in malignant transformation and carcinogenesis [14].
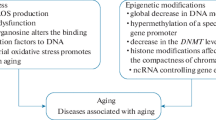
In an effort to elucidate the role of epigenetic regulation in cancer, several groups over the last few decades have focused their attention on the canonical epigenetic mechanisms that are perturbed during cancer. Dysregulation of DNA methylation is one of the most common epigenetic perturbations in cancer. Loss of DNA methylation at specific regulatory and repetitive elements such as Alu elements (Alu) and long interspersed element-1 (LINE1) has been associated with an increase in genomic instability, making chromosomal arrangements that lead to tumor formation more likely [15, 16]. Conversely, it has been shown that in certain types of tumors, the hypermethylation of CpG islands in the promoter regions of tumor-suppressor genes has been associated with development of cancer (Fig. 2) [17].

Summary of DNA methylation changes in carcinogenesis and aging
Moreover, aberrant activity of the enzymes catalyzing histone modifications contributes to carcinogenesis [18]. For example, EZH2 (enhancer of zeste homologue 2), the catalytic subunit of PRC2, which mediates the deposition of the H3K27me3 repressive mark, has been implicated in several cancer types, whereby its expression is increased in prostate cancer, breast cancer, lymphomas and glioblastomas [19,20,21]. Conversely, the histone lysine demethylase JMJD2C, which catalyzes the demethylation of H3K9, has been associated with breast and esophageal cancers, de-repressing genes involved in these pathologies [22, 23]. Moreover, the dynamic process of histone acetylation, which is generally associated with transcriptional activation, is involved in a variety of cancer types; the balance of the activity of histone acetylases (HATs) and histone deacetylases (HDACs) is necessary in the maintenance of cellular homeostasis by regulating chromatin conformation and transcription states [24]. In fact, aberrant regulation of several HATs has been found to be responsible for gene expression changes underlying carcinogenesis. For example, the Gcn5 HAT has been implicated in breast cancer due to dysregulation of Wnt signaling [25]. Furthermore, the altered expression of the MOZ gene and its paralog MORF, which encode for two histone acetyltransferases that act as co-transcriptional activators, contributes to the development of myeloid leukemia [26]. Finally, p300 and CBP HATs have been identified as key tumor-suppressor proteins, and their dysregulation was described in many cancer types [27]. Conversely, HDACs are also involved in several cancers, whereby increase in their expression potentiates tumorigenesis in breast, prostate and colorectal tissues as a result of hypoacetylation of several gene loci [24]. Interestingly, deacetylation of non-histone proteins such as p53 and YY1 transcriptional repressors and the STAT3 transcriptional activator has been implicated in carcinogenesis in many cell types [28].
Studies regarding the role of epigenetics in aging have been greatly focused on the role of DNA methylation in this physiological process. Global levels and genomic distribution of 5-methylcytosine change across the genome and at specific loci during aging. Interestingly, DNA methylation has been evaluated as a biomarker for determining the age of cells and tissues, and loci-specific methylation patterns can be very telling as to the age of various tissues. Exposure to ROS, which increases DNA damage, inflammation and the activity and function of DNMTs, has been shown to be correlated with the deposition of 5mC and 5hmc and an increase in mutation frequency [29]. Furthermore, studies have shown that damage occurring to DNMT genes during aging causes a significant decrease in global methylation levels in repetitive elements such as LTRs (long terminal repeat), SINEs (short interspersed nuclear element) and LINE-1 (long interspersed element-1) (Fig. 2) [30,31,32,33,34]. Studies have also shown that specific loci containing hypomethylated enhancers are linked to genes whose expression is regulated during aging, and the presence of hypermethylation was shown to be enriched at CpG islands, which is associated with carcinogenesis onset [35].
Dysregulation of genes during aging is also linked to an alteration of histone methylation: in murine models, changes in H3K4me3 (activating) and H3K27me3 (repressing) levels have been directly linked to lifespan. Therefore, the DNA and histone modification changes occurring during aging could contribute to the definition of an epigenome more inclined to acquire epigenetic changes responsible for tumor onset.
Finally, several non-coding RNAs have been described in regulating cell mechanisms associated with aging, including proliferation, differentiation, apoptosis and senescence, which in turn, have been implicated in potentiation of carcinogenesis [36]. Some examples of long non-coding RNAs that have been studied in this context are MALAT1 (metastasis-related lung adenocarcinoma transcript 1), SALNR and HOTAIR (HOX transcript antisense RNA), which are necessary in regulating cellular processes such as promotion of tumor cell proliferation, invasion, metastasis, drug resistance, and angiogenesis, thus playing a key role cancer progression as a result of aging [37].
Aging and cardiovascular diseases and epigenetics
Cardiovascular disease (CVD), leading to heart failure, and subsequently death, is the major cause of morbidity and mortality worldwide [38]. There are several risk factors linked to the development of CVD, including hypertension, diabetes and obesity [39]. However, one of the main risk factors of CVD is age, with its prevalence, including atherosclerosis, stroke and myocardial infarction, increasing in the elderly [40]. Aging patients present with several functional changes to the heart, including diastolic and systolic dysfunction, arrhythmias, and atrial fibrillation, to name a few [41]. As with all aging pathophysiologies, the high prevalence of CVD in the aged population has been linked to inflammation, oxidative stress, production of ROS, apoptosis, myocardial deterioration and degeneration [42]. Furthermore, the inflammatory response results in cardiac remodeling, with significant changes to the extracellular matrix, and the presence of proinflammatory and inflammatory markers (IL-6, TNFα, CRP) [42]. Cardiac remodeling subsequently leads to the development of cardiac hypertrophy and fibrosis, an effect that is pronounced in aged hearts, leading to impairment of cardiac function [43]. Development of fibrosis has been considered to be the starting point of several pathophysiologies, which is initiated by an aberrant inflammatory response, and results in structural and functional deterioration of many organs [44].
Of great importance to the proper functioning of the heart is the mitochondria, which is necessary for the metabolic activity of the heart and the production of ATP [45]. Cardiac dysfunction in the elderly has been correlated with mitochondrial dysfunction as a result of oxidative stress and production of ROS [41], factors which play a detrimental role to the capacity of mitochondrial respiration [46]. Studies have shown that the development of atherosclerosis in the aged population has been linked to lipid oxidation as a result of mitochondrial dysfunction [47]. Oxidative stress also contributes to impaired calcium signaling, which is required for maintenance of the sarcoplasmic reticulum and muscle contraction [48].
Epigenetic dysregulation has been associated with several cardiovascular pathologies and cardiovascular aging: some of the key epigenetic mechanisms include DNA methylation and hydroxymethylation, chromatin remodeling, aberrant changes in histone modifications, and dysregulation of non-coding RNAs [49, 50]. DNA methylation, which has been shown to be impacted by several environmental factors, is a key player in the genetic regulation of genes necessary for cardiac homeostasis, regulating various cell processes required for proper cardiac function. DNA methylation patterns are maintained by DNMT1, whereas de novo DNA methylation is mediated by DNMT3A and DNMT3B [50]. Furthermore, DNA methylation can be reversed through oxidation of 5-methylcytosine to 5-hydroxymethylcytosine (5-hmC) by the TET (ten–eleven translocation) proteins [50]. Global changes in DNA methylation have been shown to change during aging, and have been correlated with the onset of several cardiovascular pathologies [51].
Chronic inflammation, a key factor of initiation of cardiomyocyte stress and initiation of pathological onset, has been associated with unbalanced lipid levels occurring during atherosclerosis [49]. Studies in patients with dyslipidemia have revealed a subset of genes including CPT1A, with differential methylation profiles in control versus affected individuals. CPT1A is necessary for the maintenance of healthy lipid profiles by regulating mitochondrial function, and aberrant changes in its methylation status have been associated with lipoprotein and triglycerides in the blood [52]. Hypermethylation of the promoter region of ATP-binding cassette A1 (ABCA1), which is required for cholesterol transfer from blood to high-density lipoprotein particles, was found in patients with familial hypercholesterolemia [53]. Furthermore, epigenetic changes in several genes including RELA, NOS3, KLF4 and APOE have been linked to progression of atherosclerosis [54]. An important mechanism in the progression of atherosclerosis is smooth muscle cell (SMC) proliferation; under quiescent conditions, SMCs maintain a differentiated phenotype, aiding in vascular tone. Under pathological conditions of stress or inflammation, SMCs become more proliferative and migratory, contributing to the formation of atherosclerotic lesions [55]. This dysregulation has been associated with changes in methylation of several genes, including the Estrogen Receptor gene (ESR-a) [56], collagen type XV alpha 1 (COL15A1) [57] and transforming growth factor, beta receptor III (TGFBR3) [58].
As with the onset of cardiac hypertrophy, in which cardiac remodeling results in a gene expression shift mimicking that of the fetal stage [5], the same changes in DNA methylation patterns in aging have been observed [50]. Furthermore, changes in 5-hmC patterns in hypertrophic hearts also resemble the neonatal stage: these changes in methylation and hydroxymethylation have been associated with the regulation of key cardiac genes such as MYH7, MYOCD, SRF and KLF4 [50], although such changes during cardiac aging still remain elusive. Although the study of histone modifications and chromatin remodeling has been a topic of great interest in the study of cardiovascular disease, and there have been great advances in understanding the specific mechanisms regulating cardiac homeostasis, many aspects in the context of aging are still unclear. The histone methyltransferase G9a/Ehmt2 has been implicated in the maintenance of cardiac homeostasis by interacting with EZH2, and MEF2C (myocyte-specific enhancer factor 2C) to maintain the heterochromatin needed for repressing fetal gene expression in the adult heart [5]. Conversely, after onset of pathological hypertrophy, expression of miR-217 inhibits G9a, resulting in a reduction of H3K9me2, thus de-repressing the fetal gene program and resulting in pathological remodeling of cardiomyocytes [59]. Moreover, the study of histone deacetylases (HDACs) in the regulation of cardiac hypertrophy has been a topic of great interest over the last few decades, and several studies have shown that this class of enzymes is necessary in the maintenance of cardiomyocyte homeostasis [60, 61]. Finally, the chromatin remodeling complex, BAF (SWI/SNF), has been shown to be critical to cardiomyocyte function, and its dysregulation has been implicated in loss of cardiovascular function and onset of pathology by regulating several cardiac-specific genes including MYH6. Furthermore, studies have revealed that in injured adult mouse hearts, there is an alteration of binding of BAF to genomic DNA, thus altering gene expression profiles (Fig. 3) [62].

The classes of epigenetic mechanism that define the transcriptional changes of cardiac hypertrophy
Non-coding RNAs have been of great interest in the regulation of cardiomyocyte homeostasis and onset of cardiovascular disease. Over the last few years, many studies have outlined the importance of several ncRNAs that are altered in cardiac and age-related cardiac pathologies. For example, microRNAs miR-21, miR-22, miR-34a and the miR17-92 cluster are significantly altered in cardiac aging, regulating proteins important in fibroblast–myofibroblast transition [63], cardiac fibroblast senescence [64], cardiac contraction [65] and collagen synthesis [66], respectively.
Finally, of great importance, and of great interest over the last few years, is whether RNA methylation could be subject to epigenetic modifications. Although several chemical modifications can be found on RNA, RNA methylation (N6-methyladenosine, m6A) is the most abundant modification in eukaryotic messenger RNAs. This chemical modification has been found to be present in intronic regions of messenger RNA, transfer RNA, ribosomal RNA, and noncoding RNAs [67]. RNA methylation is a dynamic process and is mediated by writers, readers and erasers, just like DNA methylation, and the role of these proteins has been gaining much interest in several biological contexts and pathophysiologies [67]: the role of this chemical modification of RNA in pathogenesis and aging, however, remains largely unknown. Recent studies have revealed that RNA methylation is necessary for the maintenance of cardiac homeostasis via a mechanism driven by METTL3, a protein responsible for RNA methylation. This study revealed that in vivo overexpression of METTL3 increased m6A and resulted in hypertrophic growth of the heart, and furthermore, that cardiomyocyte-specific knockout of METTL3 resulted in a decrease of cardiac function after pressure overload and during aging [68]. Further, elucidation of the role of METTL3 and the role of RNA methylation in aging is necessary to understand the mechanisms by which perturbations in these modifications may play a role in aging and the onset of pathogenesis. Conversely, FTO (fat mass and obesity-associated protein), an m6A eraser, has been shown to decrease in mammalian failing hearts thus reducing the contractile function of cardiomyocytes [69]. Although its role in aging has not yet been elucidated, the understanding of the balance between RNA methylation writers and erasers will be necessary in uncovering the mechanisms perturbed during aging-induced pathologies.
Taken together, these (and other) studies have unveiled several epigenetic mechanisms regulating cardiac homeostasis and revealed how dysregulation of these pathways can lead to onset of cardiovascular pathologies. Many of epigenetic mechanisms involved in CVD are also implicated in aging of several tissues. Despite this, the role of epigenetics in cardiovascular aging and in the onset of cardiovascular disease in the elderly still remains to be elucidated.
Conclusions and future directions
Over the last decade, our understanding and knowledge of the epigenetic mechanisms involved in cellular homeostasis and disease have greatly increased. Mechanisms involving histone post-translational modifications, chromatin remodeling and non-coding RNA have revealed multi-layered mechanisms regulating the homeostatic state of several cell type (cardiomyocytes, fibroblasts, immune cells, etc.); mechanisms which when dysregulated result in the onset of cancer and cardiac pathologies. Although epigenomic dysregulation in CVD alone is still not fully understood, the role of epigenetics in aging and age-related diseases remains even more elusive. Much progress is still needed to understand changes in the epigenetic landscape of the aged cardiomyocyte (both physiological and pathological), the pathways of which are critical for the maintenance of cardiomyocyte homeostasis, and which are the key factors (environmental or other) that influence chromatin architecture and govern gene expression changes that promote disease.
Furthermore, despite decades of study of carcinogenesis, the mechanisms (both genetic and epigenetic) governing the aberrant gene expression changes, cellular identity and responsiveness to cellular and environmental cues, ultimately resulting in cancer, still remain unknown. Studies have revealed that targeting epigenetic mechanisms could play a profound and possibly ubiquitous role in abolishing tumorigenesis. As with other age-related pathologies, there are several unanswered questions with respect to correlating mechanisms of carcinogenesis and aging.
Moreover, recent work has shown, using RNA sequencing of whole blood in healthy aged subjects, that there is a small subset of genes that is characterized by changes in expression and splicing and a reduction in genetic regulation, late in life. Quantifying allele-specific gene expression, alternative splicing and genetic regulation, this study showed that there is a strong correlation between genetic effects and the increase in allelic imbalance with age, which is furthermore correlated with increasing environmental variance [70]. Recent studies have also emphasized the importance of the role of maintenance of heterochromatin, and the dysregulation of chromatin structure during the aging process. Indeed, the heterochromatin protein HP1 has been suggested to be required for longevity, and its dysregulation has been implicated in progeroid syndromes [71]. Using genome-wide approaches in aged animal models will be key to understanding changes in the epigenetic landscape of aged cells and tissues to better correlate pathology-specific gene expression changes. Exploiting new technologies, such as assay for transposase accessible chromatin sequencing (ATAC-seq), in human aging models, to elucidate changes in chromatin accessibility in cardiac and tissues during and post-disease onset, will be critical in understanding changes at the epi-transcriptome level [72]. Correlation of these changes in chromatin accessibility with changes in gene expression profiles can be further linked using RNA-sequencing, Hi-C analysis, and ChIP-sequencing of aged cardiac tissue to specifically map changes in genomic and epigenetic domains.
Since epigenetic changes have been shown to be reversible, manipulation of epigenomic pathways and epigenetic enzymes using CRISPR/Cas9 technology or targeted pharmacological inhibition could prove to be crucial in reversing epigenetic aberrations that are a hallmark of aging. Currently, there are several epigenetic drugs that are either commercially available, or under clinical phase trials [73]. These drugs target an array of epigenetic enzymes including histone modifying enzymes and DNA methyltransferases [74, 75]. Exploiting these drugs for pathologies beyond cancer will be critical for the understanding mechanisms and onset of several age-related diseases. The main challenge will be to test these drugs in aged animal models, and elucidate their function and role in a tissue-specific manner, avoiding off-target effects.
Finally, single-cell transcriptome analysis will be crucial to uncovering epigenomic changes within various cellular and sub-cellular populations to further shed light on the correlation of (epi)transcriptomic changes in aged and diseased tissue. Single-cell RNA sequencing of a murine cardiac pressure-overload HF model revealed that induction of HF leads to an intricate immune activation, whereby a myriad of cells, including neutrophils, B cells, NK cells and mast cells are activated [76]. Using this approach, and correlation of aberrant or acute immune responses with disease onset during aging can further shed light onto the underlying mechanisms of the failing heart. To this end, a recent study in muscle stem cells revealed that there are context-dependent alterations of DNA methylation with age. Using single-cell transcriptomic analysis, this study showed that there is a degradation of functional transcriptional networks correlated with an increase in heterogeneity between the epigenome and the transcriptome during aging [77]. Using these approaches in a context- and tissue-specific manner can reveal the subtle, yet critical changes underlying pathophysiologies associated with aging.
References
Shioi T, Inuzuka Y (2012) Aging as a substrate of heart failure. J Cardiol 60:423–428
Kennedy BK, Berger SL, Brunet A et al (2014) Geroscience: linking aging to chronic disease. Cell 159:709–713
Flavahan WA, Gaskell E, Bernstein BE (2017) Epigenetic plasticity and the hallmarks of cancer. Science 357:eaal2380
Ou HL, Schumacher B (2018) DNA damage responses and p53 in the aging process. Blood 131:488–495
Papait R, Serio S, Pagiatakis C et al (2017) Histone methyltransferase G9a is required for cardiomyocyte homeostasis and hypertrophy. Circulation 136:1233–1246
Papait R, Cattaneo P, Kunderfranco P et al (2013) Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci U S A 110:20164–20169
Papait R, Corrado N, Rusconi F et al (2015) It’s time for An epigenomics roadmap of heart failure. Curr Genomics 16:237–244
Papait R, Kunderfranco P, Stirparo GG et al (2013) Long noncoding RNA: a new player of heart failure? J Cardiovasc Transl Res 6:876–883
Sun X, Hota SK, Zhou YQ et al (2018) Cardiac-enriched BAF chromatin-remodeling complex subunit Baf60c regulates gene expression programs essential for heart development and function. Biol Open 7:bio029512
Okano M, Bell DW, Haber DA et al (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257
Greco CM, Kunderfranco P, Rubino M et al (2016) DNA hydroxymethylation controls cardiomyocyte gene expression in development and hypertrophy. Nat Commun 7:12418
Salamon I, Jotti GS, Condorelli G (2018) The long noncoding RNA landscape in cardiovascular disease: a brief update. Curr Opin Cardiol 33:282–289
Cao Q, Guo Z, Yan Y et al (2019) Exosomal long noncoding RNAs in aging and age-related diseases. IUBMB Life 71:1846–1856
Naylor RM, Baker DJ, van Deursen JM (2013) Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin Pharmacol Ther 93:105–116
Sartor MA, Dolinoy DC, Jones TR et al (2011) Genome-wide methylation and expression differences in HPV(+) and HPV(–) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics 6:777–787
Zheng Y, Joyce BT, Liu L et al (2017) Prediction of genome-wide DNA methylation in repetitive elements. Nucleic acids Res 45:8697–8711
Di Croce L, Raker VA, Corsaro M et al (2002) Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295:1079–1082
Wang GG, Allis CD, Chi P (2007) Chromatin remodeling and cancer, Part I: covalent histone modifications. Trends Mol Med 13:363–372
Varambally S, Dhanasekaran SM, Zhou M et al (2002) The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419:624–629
Kleer CG, Cao Q, Varambally S et al (2003) EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A 100:11606–11611
Suva ML, Riggi N, Janiszewska M et al (2009) EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res 69:9211–9218
Liu G, Bollig-Fischer A, Kreike B et al (2009) Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene 28:4491–4500
Yang ZQ, Imoto I, Fukuda Y et al (2000) Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res 60:4735–4739
Virani S, Colacino JA, Kim JH et al (2012) Cancer epigenetics: a brief review. ILAR J 53:359–369
Chen J, Luo Q, Yuan Y et al (2010) Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Mol Cell Biol 30:5621–5635
Yang XJ, Ullah M (2007) MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene 26:5408–5419
Tillinghast GW, Partee J, Albert P et al (2003) Analysis of genetic stability at the EP300 and CREBBP loci in a panel of cancer cell lines. Genes Chromosomes Cancer 37:121–131
Glozak MA, Sengupta N, Zhang X et al (2005) Acetylation and deacetylation of non-histone proteins. Gene 363:15–23
Morris BJ, Willcox BJ, Donlon TA (2019) Genetic and epigenetic regulation of human aging and longevity. Biochim Biophys Acta Mol Basis Dis 1865:1718–1744
Nakano K, Whitaker JW, Boyle DL et al (2013) DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis 72:110–117
Bollati V, Schwartz J, Wright R et al (2009) Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev 130:234–239
Ben-Avraham D, Muzumdar RH, Atzmon G (2012) Epigenetic genome-wide association methylation in aging and longevity. Epigenomics 4:503–509
Day K, Waite LL, Thalacker-Mercer A et al (2013) Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol 14:R102
Jones MJ, Goodman SJ, Kobor MS (2015) DNA methylation and healthy human aging. Aging Cell 14:924–932
Cole JJ, Robertson NA, Rather MI et al (2017) Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol 18:58
Gomez-Verjan JC, Vazquez-Martinez ER, Rivero-Segura NA et al (2018) The RNA world of human ageing. Hum Genet 137:865–879
Sun Z, Yang S, Zhou Q et al (2018) Emerging role of exosome-derived long non-coding RNAs in tumor microenvironment. Mol Cancer 17:82
Gensous N, Bacalini MG, Pirazzini C et al (2017) The epigenetic landscape of age-related diseases: the geroscience perspective. Biogerontology 18:549–559
Rodgers JL, Jones J, Bolleddu SI et al (2019) Cardiovascular risks associated with gender and aging. J Cardiovasc Dev Dis 6:19
Yazdanyar A, Newman AB (2009) The burden of cardiovascular disease in the elderly: morbidity, mortality, and costs. Clin Geriatr Med 25:563–577
Steenman M, Lande G (2017) Cardiac aging and heart disease in humans. Biophys Rev 9:131–137
Curtis AB, Karki R, Hattoum A et al (2018) Arrhythmias in patients ≥ 80 years of age: pathophysiology, management, and outcomes. J Am Coll Cardiol 71:2041–2057
Martos R, Baugh J, Ledwidge M et al (2007) Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 115:888–895
Yang Z, Jiang S, Shang J et al (2019) LncRNA: shedding light on mechanisms and opportunities in fibrosis and aging. Ageing Res Rev 52:17–31
Martin-Fernandez B, Gredilla R (2016) Mitochondria and oxidative stress in heart aging. Age (Dordr) 38:225–238
Nakou ES, Parthenakis FI, Kallergis EM et al (2016) Healthy aging and myocardium: a complicated process with various effects in cardiac structure and physiology. Int J Cardiol 209:167–175
Carew TE (1989) Role of biologically modified low-density lipoprotein in atherosclerosis. Am J Cardiol 64:18G–22G
Xie W, Santulli G, Reiken SR et al (2015) Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep 5:11427
Gentilini D, Garagnani P, Pisoni S et al (2015) Stochastic epigenetic mutations (DNA methylation) increase exponentially in human aging and correlate with X chromosome inactivation skewing in females. Aging (Albany NY) 7:568–578
Zhang W, Song M, Qu J et al (2018) Epigenetic modifications in cardiovascular aging and diseases. Circ Res 123:773–786
Lind L, Ingelsson E, Sundstrom J et al (2018) Methylation-based estimated biological age and cardiovascular disease. Eur J Clin Invest 48:e12872
Irvin MR, Zhi D, Joehanes R et al (2014) Epigenome-wide association study of fasting blood lipids in the genetics of lipid-lowering drugs and diet network study. Circulation 130:565–572
Guay SP, Brisson D, Munger J et al (2012) ABCA1 gene promoter DNA methylation is associated with HDL particle profile and coronary artery disease in familial hypercholesterolemia. Epigenetics 7:464–472
Han P, Hang CT, Yang J et al (2011) Chromatin remodeling in cardiovascular development and physiology. Circ Res 108:378–396
Pagiatakis C, Sun D, Tobin SW et al (2017) TGFbeta-TAZ/SRF signalling regulates vascular smooth muscle cell differentiation. FEBS J 284:1644–1656
Post WS, Goldschmidt-Clermont PJ, Wilhide CC et al (1999) Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res 43:985–991
Connelly JJ, Cherepanova OA, Doss JF et al (2013) Epigenetic regulation of COL15A1 in smooth muscle cell replicative aging and atherosclerosis. Hum Mol Genet 22:5107–5120
Liang H, Zhang C, Ban T et al (2012) A novel reciprocal loop between microRNA-21 and TGFbetaRIII is involved in cardiac fibrosis. Int J Biochem Cell Biol 44:2152–2160
Thienpont B, Aronsen JM, Robinson EL et al (2017) The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J Clin Invest 127:335–348
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42
Montgomery RL, Davis CA, Potthoff MJ et al (2007) Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev 21:1790–1802
Vieira JM, Howard S, Villa Del Campo C et al (2017) BRG1-SWI/SNF-dependent regulation of the Wt1 transcriptional landscape mediates epicardial activity during heart development and disease. Nat Commun 8:16034
Zhou XL, Xu H, Liu ZB et al (2018) miR-21 promotes cardiac fibroblast-to-myofibroblast transformation and myocardial fibrosis by targeting Jagged1. J Cell Mol Med 22:3816–3824
Jazbutyte V, Fiedler J, Kneitz S et al (2013) MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr) 35:747–762
Boon RA, Iekushi K, Lechner S et al (2013) MicroRNA-34a regulates cardiac ageing and function. Nature 495:107–110
van Almen GC, Verhesen W, van Leeuwen RE et al (2011) MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell 10:769–779
Pagiatakis C, Condorelli G (2019) The RNA methylome blackboard. Circulation 139:546–548
Dorn LE, Lasman L, Chen J et al (2019) The N(6)-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation 139:533–545
Mathiyalagan P, Adamiak M, Mayourian J et al (2019) FTO-dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation 139:518–532
Balliu B, Durrant M, Goede O et al (2019) Genetic regulation of gene expression and splicing during a 10-year period of human aging. Genome Biol 20:230
Lee SK, Wang W (2019) Roles of topoisomerases in heterochromatin, aging, and diseases. Genes (Basel) 10:884
Daugherty AC, Yeo RW, Buenrostro JD et al (2017) Chromatin accessibility dynamics reveal novel functional enhancers in C. elegans. Genome Res 27:2096–2107
Heerboth S, Lapinska K, Snyder N et al (2014) Use of epigenetic drugs in disease: an overview. Genet Epigenet 6:9–19
Altucci L, Rots MG (2016) Epigenetic drugs: from chemistry via biology to medicine and back. Clin Epigenetics 8:56
Brunet A, Berger SL (2014) Epigenetics of aging and aging-related disease. J Gerontol A Biol Sci Med Sci 69:S17–S20
Martini E, Kunderfranco P, Peano C et al (2019) Single cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.119.041694
Hernando-Herraez I, Evano B, Stubbs T et al (2019) Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat Commun 10:4361
Acknowledgements
This work was supported by the National Research Council of Italy (CNR), Research Project “Aging: molecular and technological innovations for improving the health of the elderly population” (Prot. MIUR 2867 25.11.2011) and by the CARIPLO Foundation (Grant Number #2015-573) to RP.
Funding
This study was funded by grants of CARIPLO Foundation (# 2015-573) and the Flagship. Project Invecchiamento of the National Research Council of Italy (Prot. MIUR 2867 25.11.2011).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest (financial or non-financial).
Statement of human and animal rights
The authors declare that the research did not involve human participants and animals.
Informed consent
No informed consent is required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pagiatakis, C., Musolino, E., Gornati, R. et al. Epigenetics of aging and disease: a brief overview. Aging Clin Exp Res 33, 737–745 (2021). https://doi.org/10.1007/s40520-019-01430-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40520-019-01430-0