
Abstract
Background
Frailty is a state of increased vulnerability to poor resolution of homeostasis after a stressor event, which increases the risk of adverse outcomes including falls, disability and death. The underlying pathophysiological pathways of frailty are not known but the hypothalamic–pituitary–adrenal axis and heightened chronic systemic inflammation appear to be major contributors.
Methods
We used the English Longitudinal Study of Ageing dataset of 3160 individuals over the age of 50 and assessed their frailty status according to the Fried-criteria. We selected single nucleotide polymorphisms in genes involved in the steroid hormone or inflammatory pathways and performed linear association analysis using age and sex as covariates. To support the biological plausibility of any genetic associations, we selected biomarker levels for further analyses to act as potential endophenotypes of our chosen genetic loci.
Results
The strongest association with frailty was observed in the Tumor Necrosis Factor (TNF) (rs1800629, P = 0.001198, β = 0.0894) and the Protein Tyrosine Phosphatase, Receptor type, J (PTPRJ) (rs1566729, P = 0.001372, β = 0.09397) genes. Rs1800629 was significantly associated with decreased levels of high-density lipoprotein (HDL) (P = 0.00949) and cholesterol levels (P = 0.00315), whereas rs1566729 was associated with increased levels of HDL (P = 0.01943). After correcting for multiple testing none of the associations remained significant.
Conclusions
We provide potential evidence for the involvement of a multifunctional proinflammatory cytokine gene (TNF) in the frailty phenotype. The implication of this gene is further supported by association with the endophenotype biomarker results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Frailty refers a state of increased vulnerability to poor resolution of homeostasis after a stressor event, which increases the risk of adverse outcomes, including falls, delirium and disability [1]. The mechanistic pathophysiological pathways of frailty are not known, but there is evidence for the involvement of steroid hormones and the immune system [2, 3]. During ageing, physiological decline occurs in steroid hormone levels, as observed in males where free testosterone declines at the rate of 1 % per year [4]. Similarly, in females cycling estradiol production is replaced by very low, constant, estradiol levels after the age of about 50 [5]. In both sexes, dehydroepiandrosterone (DHEA) and its sulphated derivate (DHEAS) levels gradually decline in later life due to the decreased activity of adrenal glands [6]. These hormones are known to effect changes in proinflammatory cytokine levels, for example, testosterone was shown to suppress interleukin-6 (IL-6) mRNA levels in osteoblastic cell line [7] and reduce the expression and secretion of tumor necrosis factor (TNF) in macrophages [8]. Estradiol in human bone-derived cells inhibits TNF-induced IL-6 production [9]. Finally, DHEA concentration has been shown to inhibit IL-6 secretion from peripheral blood mononuclear cells [10]. The increased levels of proinflammatory cytokines (IL-1, IL-6 and TNF) and acute-phase proteins, such as C-reactive protein (CRP), results in a chronic, low-grade systemic inflammatory state of the ageing immune system, termed “inflamm-ageing”. This chronic inflammation leads to long-term tissue damage and is detrimental for longevity [11]. Increased levels of CRP and IL-6 have been associated with increased mortality in the elderly [12, 13]. Studies have shown a large body of evidence suggestive of a heightened inflammatory state in frail older individuals, as marked by increase of these inflammatory markers compared with levels observed in robust older controls [14–16].
There have been only a few studies on the genetic determinants of frailty and those that have been conducted have somewhat conflicting results. A candidate gene study found that two single nucleotide polymorphs (SNP) of the IL-6 gene were weakly associated with frailty but not with serum IL-6 levels [17], whilst another showed that a SNP in the CRP gene (rs1205) is associated with frailty scores, albeit counter intuitively with presence of frailty related to lower levels of CRP [18]. The largest study so far (in terms of number of SNPs investigated) has found weak associations between frailty and genetic variants of genes involved in pathways related to apoptosis and metabolism of amino acids in a sample of elderly women [19]. However, these results did not survive correction for multiple testing.
In this study, we conducted a candidate gene association study using a standardized frailty phenotype [20] in 3160 community dwelling individuals over the age of 50 in the English Longitudinal Study of Ageing (ELSA) cohort. We selected genes involved in the steroid hormone biosynthesis and inflammation pathways, as evidence in the literature indicates their possible involvement in frailty pathophysiology (for example, DHEAS [21], IL-6, TNF and C-reactive protein [14]). We also selected genetic variants of key genes of the steroid hormone and inflammatory pathways, such as HSD11B1, SULT1E1, SULT2A1, IL1B and IFNG. We hypothesized that SNPs within these genes, especially those ones which have been shown to affect the expression of their genes such as rs1800795 for IL6 [22] and rs1800629 for TNF [23] or have shown associations with metabolite levels such as rs2547231 and rs182420 for SULT2A1 [24], will show significant association to frailty phenotype.
To support the biological plausibility of any genetic association with the frailty phenotype, we also conducted association analyses with selected biomarkers that act as potential endophenotypes for our chosen genetic loci: high-sensitivity C-reactive protein (hsCRP) as inflammatory marker, high-density lipoprotein (HDL), total cholesterol and DHEAS as markers for steroid metabolism.
Materials and methods
Participants
The analyses are performed on a sample of 3160 participants drawn from Wave 2 (2004) and Wave 4 (2008) of the English Longitudinal Study of Ageing (ELSA). Detailed description of ELSA can be found elsewhere [25], (http://www.ifs.org.uk/ELSA). Briefly ELSA is prospective cohort study representative of older men and women living in England who originally participated in the Health Survey for England in 1998, 1999 or 2001 (http://www.natcen.ac.uk/series/health-survey-for-england). The participants have been participated biannually in a computer-assisted personal interview (Core dataset) and every 4 years for a separate nurse assessment (Nurse dataset), carried out shortly after the interview. During the nurse assessment participants gave blood for genetic and biomarker analysis.
Genetic and biomarker data
Genotype data of 620 Single Nucleotide Polymorphisms (SNP) for 3160 participants were obtained from a publicly available ELSA DNA Repository (EDNAR).
Genotyping was performed by Illumina (San Diego, CA) as part of a 1536 Goldengate custom SNP panel using high-throughput BeadArrayTM technology.
For the comparative endophenotype analysis, biomarker data were drawn from both the Wave 2 (2004) and Wave 4 (2008) Nurse dataset. We used the following analytes from W2: blood total cholesterol (mmol/l), HDL (mmol/l), and hsCRP (mg/l). HsCRP >10 mg/l results were excluded from the analysis as they indicate ongoing acute-phase response [26]. From W4, we used the DHEAS level (μmol/l). Blood samples were analysed at the Royal Victoria Infirmary laboratory in Newcastle upon Tyne, UK (detailed description of blood analyses can be found in [27]).
Phenotypic measures
Frailty status assessment was based on the 5-item criteria by Fried and colleagues [20], derived for both Wave 2 and Wave 4. This is a widely used measure which determines the condition based on specific criteria, such as unintentional weight loss, exhaustion, low physical activity, slowness and weakness. As with previous studies using Frailty Phenotype (FP) criteria, we conducted a study specific operationalization in ELSA, as follows. Weight loss (item 1) was defined as present if changes in measured weight loss between waves were equal or greater than 8 % baseline values. Exhaustion (item 2) was recorded present if a positive answer to both of the questions from the Center for Epidemiological Studies depression questionnaire (CES-D) [28]: whether respondent felt everything they did during the past week was an effort and whether respondent could not get going much of the time during the past week.
Low physical activity (item 3) used self-report questions on work activity and recreational physical activity. In short, if subject reported they did not work and performed only mild sport activity not more than once a week, they were considered positive for this item.
Respondent is considered positive for slowness (item 4) if the average of the two timed 8 feet (=240 cm) walk test falls into the slowest 20 % of population, adjusting for sex and height.
Respondent was assessed as frail for weakness (item 5) if the average of the 3 dominant hand grip strength (GS) measures by handheld dynamometer falls into lowest 20 % of the population stratified by sex and body mass index (BMI). The latter 2 items were assessed in the whole ELSA wave 2 and wave 4 cohorts with available data then the cut-off points were applied for the genotyped sub-cohort. (Detailed description of variables used for frailty assessment can be found in the online Supplementary material Appendix 1.)
For analysis purposes, we used these variables both as a summed count of the number of frailty items present (0–5) and as an ordinal frailty status measure (frail if they possessed 3–5 items, pre-frail if 1–2 items and robust if none). Biomarker data were treated as continuous variables and were normally distributed. However, hsCRP and DHEAS data were skewed; therefore a square root transformation was performed for genetic association analysis purposes to normalize the distribution.
Statistical analysis
We used the Plink software for genetic association analyses [29]. Associations between genotypes and number of frailty items present (0–5) and between genotypes and biomarker levels, and were tested with linear regression using sex and age as covariates. Results of the most significant associations were re-tested by ordered logistic regression using frailty status as dependent variable within the Stata12 software (Stata Corporation, http://www.stata.com/). Stata12 was also used for demographic, phenotypic and biomarker analysis. Linkage disequilibrium blocks in PTPRJ gene were generated and visualized with HaploView software [30]. Genetic power calculation was performed by Quanto software (http://biostats.usc.edu/Quanto.html).
Results
Demographic, phenotypic and biomarker distributions
Tables 1 and 2 show the demographic and frailty phenotype results for the participants,, respectively. There were more females than males in the sample with no significant differences between the mean ages. There is evidence of a relationship between sex and the FP but this did not achieve statistical significance at the 5 % level in Wave 2 (P = 0.068) and was not significant in Wave 4 (P = 0.106) (Fischer’s exact test).
Table 3 shows the mean biomarker levels for the entire population and for the two sexes. All the biomarker levels were significantly different between sexes on the P < 0.05 level.
To explore the predictive value of these biomarkers, we conducted a Spearman’s correlation test. The analysis revealed that all biomarkers were significantly correlated with the number of frailty items at least on P < 0.005 level (HDL, Spearman’s rho = −0.054) or below the P < 0.0001 level (cholesterol, hsCRP and DHEAS, Sperman’s rho: −0.118, 0.098 and −0.143, respectively). Table 4 shows the mean biomarker levels, according to frailty categories (robust, pre-frail and frail).
Genetic association results
Of the available 620 SNPs, 15 were out of Hardy–Weinberg equilibrium (P < 0.01), and a further 15 had a minor allele frequency (MAF) below 5 %, so these were excluded from the genetic association analysis, resulting in 590 SNPs used.
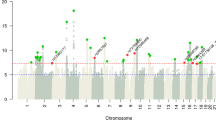
Table 5 shows the most significant results of the genetic association analysis (for results for each of the 590 SNPs see online Supplementary material Appendix 2). The SNP which was most significantly associated with the number of frailty items (0–5) phenotype is rs1800629 (β = 0.0894, P = 0.001198) in the promoter region of Tumour Necrosis Factor gene (TNF) on Chromosome 6. Four other SNPs (rs1566729, rs1566728, rs2047812 and rs611646) were associated below the P = 0.01 level (β values 0.09397, 0.09043, 0.08481 and −0.06407, respectively). The first three of these are located within the Protein Tyrosine Phosphatase, Receptor type, J (PTPRJ) gene on chromosome 11. Two of these SNPs, rs1566729 and rs1566728, are in very high linkage disequilibrium (LD) with each other (r 2 = 0.99) and rs2047812 with both of them (r 2 = 0.85) thus they probably indicate the same signal (Fig. 1). Finally, the fourth SNP, rs611646 which appeared to reduce the number of frailty items (β = −0.06407, P = 0.003904), is an intron variant within the Ataxia Telangiectasia Mutated (ATM) gene on chromosome 11.

Linkage disequilibrium (LD) blocks within the PTPRJ gene. Rs2047812, rs1566729 and rs1566728 are in strong correlation with each other (R 2 ≥ 0.85), indicating that the observed associations are possibly originated from the same signal
None of these results survives the Bonferroni-correction (P < 8.474E–05 for 590 tests).
Retesting the nominally significant results (P < 0.05) of the genetic association analysis with ordered logistic regression using frailty status (robust, pre-frail and frail) phenotype confirmed the previous results. The most significantly associated variant remained rs1800629 of TNF (coefficient = 0.2346, P = 0.001). Of the PTPRJ genetic variants, the same three SNPs were significantly associated with this phenotype below the P = 0.01 threshold (rs2047812, coefficient = 0.2095, P = 0.004; rs1566729, coefficient = 0.2088, P = 0.006 and rs1566728, coefficient = 0.1995, P = 0.008). Finally, another SNP reached the P = 0.01 significance level, rs4646316 (coefficient = 0.1695, P = 0.01) an intronic variant within the Catechol-O-methyltransferase (COMT) gene on chromosome 22.
To provide support for biological plausibility for genetic findings, we selected steroid hormone biosynthesis and inflammatory pathway-related biomarkers as endophenotypes and examined if any of the SNPs were significantly associated with their levels.
TNF SNP rs1800629 was significantly associated with decreased cholesterol level (β = −0.11, P = 0.003151) and decreased HDL level (β = −0.03022, P = 0.009489). Of the 3 PTPRJ SNPs each was significantly associated with increased HDL levels (β values 0.02898–0.03593, P values 0.002294–0.01943), and at least on a suggestive level with increased square root levels of DHEAS (β values 0.03404–0.03505, P values 0.0527–0.07158). Finally, rs611646 or rs4646316 did not have an effect on any of the biomarker levels.
Finally, we included HDL, cholesterol and hsCRP levels separately in a multivariate model to test if adding these biomarker levels attenuates the association between SNPs and frailty status. Including hsCRP in the model reduced the coefficient to 0.1776 for rs1566728 and to 0.1898 for rs2047812, whereas cholesterol and HDL in the model affected the coefficient for rs611646 (−0.118 and −0.116, respectively) (Table 6).
Discussion
In this study, we investigated the association between genes involved in steroid hormone metabolism and inflammatory pathways and the frailty phenotype. Our results show evidence of the involvement of TNF, PPRJ, ATM and COMT genes.
Our most significant genetic finding is with the TNF locus on chromosome 6. The TNF gene encodes a proinflammatory cytokine, mainly secreted by macrophages. This cytokine is involved in the regulation of a wide spectrum of biological processes including cell proliferation, differentiation, apoptosis, lipid metabolism, and coagulation and has been implicated in a variety of diseases, such as autoimmune diseases, insulin resistance, and cancer [31]. The most significant SNP, rs1800629 is located in the promoter region of the TNF gene causing a G > A transition. Functional studies suggest that the rare A allele is a much more powerful transcriptional activator than the common G allele [23]. In our study, the rare A allele was significantly associated with possession of increasing numbers of frailty items and becoming frail. This agrees with the observations of elevated TNF levels in frail individuals [15]. This SNP was significantly associated with decreased HDL and cholesterol levels as well in our study. Relationships between TNF and blood lipid levels have been investigated before. During septic shock increased TNF levels were found to be accompanied by a significant decline in total serum cholesterol [32], whilst TNF inhibitor treatment was associated with increased levels of HDL and total cholesterol in rheumatoid arthritis patients [33]. Studies of elderly participants found association between lower blood lipid levels and survival. For example, a 2-year follow-up study of very old and frail individuals has found that HDL cholesterol levels were significantly lower among those who died compared with survivors [34], whilst in another 3-year follow-up study subjects with low total cholesterol levels were at a higher risk of dying [35]. Finally, in a genome-wide association (GWA) study of over 100,000 individuals of European ancestry rs1800629 A allele was associated with lower cholesterol (P = 6.81E−09) and triglyceride (P = 7.90E−09) levels [36]. Despite these findings, it is possible that the frailty increasing and cholesterol and HDL-level decreasing effects of TNF are manifested through different pathways.
The role of the immune system in FP was not corroborated by other genes, such as CRP, IL6 or IL6R in our study. It may indicate that the effect of TNF manifests though other pathways rather than the inflammatory, such as apoptosis.
Our second most significant genetic association hit was with SNP rs1566729 which lies within the PTPRJ gene. PTPRJ is a member of the protein tyrosine phosphatase (PTP) family. PTPs are known to be signalling molecules that regulate a variety of cellular processes, including cell growth, differentiation, mitotic cycle and cancer-associated signalling processes. PTPRJ has been implicated in breast, colon, lung and thyroid cancers [37], and it is expressed by all resting leukocytes and upregulated following in vitro stimulation, thus it appears to be a T cell activating molecule [38]. A recent study suggests a possible connection between TNF and PTPRJ. In a mouse melanoma model treatment with TNF yielded the downregulation of PTPRJ [39]. The 3 PTPRJ SNPs were investigated by Teslovich and colleagues [36] who reported an increase in HDL levels (P values range between 1.77E−08: rs1566729 to 2.71E−08: rs1566728), as we did, but this contradicts the literature showing a negative association with frailty that we have just described [34]. A further contradiction is that these SNPs appear to be associated with increased DHEAS levels, which is thought to be protective against frailty [40]. A possible interpretation of this counter-intuitive finding may be that an effect of PTPRJ antagonises the HDL, DHEAS level-increasing effects and increases frailty.
The third gene below the 0.01 significance threshold is ATM. ATM protein is a high-molecular-weight PI3K-family kinase. It plays many important cytoplasmic roles where it phosphorylates hundreds of protein substrates that activate and coordinate cell-signalling pathways involved in cell-cycle checkpoints, nuclear localization, gene transcription and expression, the response to oxidative stress, apoptosis and nonsense-mediated decay, among others [41]. With this wide-range of functions in the cell, this gene and its protein product are plausible candidates for frailty. Our top SNP within this gene, rs611646 has been associated with breast cancer in interaction with BRCA1 in a study of Chinese Han subjects [42] indicating its possible functionality.
The ordered logistic regression showed the possible role of a fourth gene, COMT. The protein product of this gene catalyses the transfer of a methyl group to catecholamines, including neurotransmitters dopamine, epinephrine and norepinephrine, but it also acts on oestrogen, a member of the steroid biosynthesis pathway [43]. Rs4646316 within this gene has been shown to be associated with schizophrenia [44] but not with any biomarker level in our study, making this result difficult to interpret.
Including the biomarkers into the model only slightly attenuated the relationship between frailty status and the SNPs in PTPRJ and ATM genes. It may indicate that the relationship between these variables is not a direct mechanistic one or that biomarker levels and SNP variants are independently associated to frailty.
The measured biomarkers were all significant predictors for the number of frailty items possessed, but their individual effects were small. This is in agreement with the genetic findings which indicate the involvement of several pathways, each with limited effects.
This study has a number of limitations. First, only a proportion of the ELSA sample were genotyped and the prevalence of frailty in this group was slightly lower than in the full sample, thus it may not be representative for the whole United Kingdom population of older adults. Second, the frailty phenotype is constructed using study specific items from the extensive ELSA public archive, although as in many published studies in we used an approach based on the method published by Fried and colleagues [20]. Finally, the analysed biomarkers, genes and genotypic variants were selected from a publicly available dataset rather than specifically for the purpose of this study. However, this did include SNPs and biomarkers relevant to the pathways we set out to investigate.
The strength of our study is the adequate sample size. The effective sample size required to test whether a gene main effect is associated with a continuous trait for 99 % power, α = 0.05 (two-sided test), assuming outcomes that are modelled using linear regression (mean 0.52, SD 0.83, genetic effect = 0.01, MAF = 0.05) is 1828, which is given in our study. Despite this, it would advantageous to replicate our findings. Another advantage is the use of biomarker levels as endophenotypes, which offers additional biological plausibility for our findings.
In conclusion, our study provides evidence for the involvement of the TNF in the pathophysiology of frailty in line with previous observations in the literature. We also report new observations of the involvement of a protein tyrosine phosphatase (PTPRJ), Ataxia Telangiectasia Mutated (ATM) and Catechol-O-methyltransferase (COMT) in the frailty phenotype. Although we use biomarker endophenotypes to support plausibility of these genetic associations, replication in other cohorts is encouraged. Nevertheless, these findings provide opportunities to better understand the biological processes underpinning frailty, which may over time lead to possible interventions.
References
Clegg A, Young J, Iliffe S et al (2013) Frailty in elderly people. Lancet 381:752–762
Carcaillon L, Blanco C, Alonso-Bouzón C et al (2012) Sex differences in the association between serum levels of testosterone and frailty in an elderly population: the Toledo Study for Healthy Aging. PLoS One 7:e32401
Baylis D, Bartlett DB, Syddall HE et al (2013). Immune-endocrine biomarkers as predictors of frailty and mortality: a 10-year longitudinal study in community-dwelling older people. Age (Dordrecht, Netherlands) 35:963–971
Morley JE, Kaiser FE, Perry HM 3rd et al (1997) Longitudinal changes in testosterone, luteinizing hormone, and follicle-stimulating hormone in healthy older men. Metabolism 46:410–413
Lamberts SW, van den Beld AW, van der Lely AJ (1997) The endocrinology of aging. Science 278:419–424
Ferrari E, Cravello L, Muzzoni B et al (2001) Age-related changes of the hypothalamic-pituitary-adrenal axis: pathophysiological correlates. Eur J Endocrinol 144:319–329
Hofbauer LC, Ten RM, Khosla S (1999) The anti-androgen hydroxyflutamide and androgens inhibit interleukin-6 production by an androgen-responsive human osteoblastic cell line. J Bone Miner Res 14:1330–1337
Corcoran MP, Meydani M, Lichtenstein AH et al (2010) Sex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal women. J Endocrinol 206:217–224
Girasole G, Jilka RL, Passeri G et al (1992) 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J Clin Investig 89:883–891
Straub RH, Konecna L, Hrach S et al (1998) Serum dehydroepiandrosterone (DHEA) and DHEA sulfate are negatively correlated with serum interleukin-6 (IL-6), and DHEA inhibits IL-6 secretion from mononuclear cells in man in vitro: possible link between endocrinosenescence and immunosenescence. J Clin Endocrinol Metab 83:2012–2017
De Martinis M, Franceschi C, Monti D et al (2006) Inflammation markers predicting frailty and mortality in the elderly. Exp Mol Pathol 80:219–227
Harris TB, Ferrucci L, Tracy RP et al (1999) Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med 106:506–512
Reuben DB, Cheh AI, Harris TB et al (2002) Peripheral blood markers of inflammation predict mortality and functional decline in high-functioning community-dwelling older persons. J Am Geriatr Soc 50:638–644
Collerton J, Martin-Ruiz C, Davies K et al (2012) Frailty and the role of inflammation, immunosenescence and cellular ageing in the very old: cross-sectional findings from the Newcastle 85+ Study. Mech Ageing Dev 133:456–466
Hubbard RE, O’Mahony MS, Savva GM et al (2009) Inflammation and frailty measures in older people. J Cell Mol Med 13:3103–3109
Leng S, Chaves P, Koenig K et al (2002) Serum interleukin-6 and hemoglobin as physiological correlates in the geriatric syndrome of frailty: a pilot study. J Am Geriatr Soc 50:1268–1271
Walston J, Arking DE, Fallin D et al (2005) IL-6 gene variation is not associated with increased serum levels of IL-6, muscle, weakness, or frailty in older women. Exp Gerontol 40:344–352
Almeida OP, Norman PE, van Bockxmeer FM et al (2012) CRP 1846G>A polymorphism increases risk of frailty. Maturitas 71:261–266
Ho YY, Matteini AM, Beamer B et al (2011) Exploring biologically relevant pathways in frailty. J Gerontol Ser A Biol Sci Med Sci 66:975–979
Fried LP, Tangen CM, Walston J et al (2001) Frailty in older adults: evidence for a phenotype. J Gerontol Ser A Biol Sci Med Sci 56:M146–M156
Tajar A, O’Connell MD, Mitnitski AB et al (2011) Frailty in relation to variations in hormone levels of the hypothalamic-pituitary-testicular axis in older men: results from the European male aging study. J Am Geriatr Soc 59:814–821
Fishman D, Faulds G, Jeffery R et al (1998) The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Investig 102:1369–1376
Wilson AG, Symons JA, McDowell TL et al (1997) Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc Natl Acad Sci USA 94:3195–3199
Shin SY, Fauman EB, Petersen AK et al (2014) An atlas of genetic influences on human blood metabolites. Nat Genet 46:543–550
Steptoe A, Breeze E, Banks J et al (2013) Cohort profile: the English longitudinal study of ageing. Int J Epidemiol 42:1640–1648
Pepys MB, Hirschfield GM (2003) C-reactive protein: a critical update. J Clin Investig 111:1805–1812
Sproston K, Mindell J (ed) (2004) Health Survey for England. Methodology and documentation, vol 2, pp 34–41. http://www.hscic.gov.uk/catalogue/PUB01170/hea-surv-ethn-min-eng-2004-rep-v2.pdf
Radloff LS (1977) The CES-D scale: a self-report depression scale for research in the general population. Appl Psychol Meas 1:385–401
Purcell S, Neale B, Todd-Brown K et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Barrett JC, Fry B, Maller J et al (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3:745–756
Fraunberger P, Pilz G, Cremer P et al (1998) Association of serum tumor necrosis factor levels with decrease of cholesterol during septic shock. Shock 10:359–363
Daïen CI, Duny Y, Barnetche T et al (2012) Effect of TNF inhibitors on lipid profile in rheumatoid arthritis: a systematic review with meta-analysis. Ann Rheum Dis 71:862–868
Landi F, Russo A, Pahor M et al (2008) Serum high-density lipoprotein cholesterol levels and mortality in frail, community-living elderly. Gerontology 54:71–78
Brescianini S, Maggi S, Farchi G et al (2003) Low total cholesterol and increased risk of dying: are low levels clinical warning signs in the elderly? Results from the Italian longitudinal study on aging. J Am Geriatr Soc 51:991–996
Teslovich TM, Musunuru K, Smith AV et al (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466:707–713
Ostman A, Hellberg C, Böhmer FD (2006) Protein-tyrosine phosphatases and cancer. Nat Rev Cancer 6:307–320
Tangye SG, Phillips JH, Lanier LL et al (1998) CD148: a receptor-type protein tyrosine phosphatase involved in the regulation of human T cell activation. J Immunol 161:3249–3255
de Lima VC, de Carvalho AF, Morato-Marques M et al (2013) TNF-alpha and melphalan modulate a specific group of early expressed genes in a murine melanoma model. Cytokine 62:217–225
Leng SX, Cappola AR, Andersen RE et al (2004) Serum levels of insulin-like growth factor-I (IGF-I) and dehydroepiandrosterone sulfate (DHEA-S), and their relationships with serum interleukin-6, in the geriatric syndrome of frailty. Aging Clin Exp Res 16:153–157
Ambrose M, Gatti RA (2013) Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood 121:4036–4045
Wang K, Ye Y, Xu Z et al (2010) Interaction between BRCA1/BRCA2 and ATM/ATR associate with breast cancer susceptibility in a Chinese Han population. Cancer Genet Cytogenet 200:40–46
Gaikwad NW, Yang L, Pruthi S et al (2009) Urine biomarkers of risk in the molecular etiology of breast cancer. Breast Cancer Basic Clin Res 3:1–8
Voisey J, Swagell CD, Hughes IP et al (2012) HapMap tag-SNP analysis confirms a role for COMT in schizophrenia risk and reveals a novel association. Eur Psychiatry 27:372–376
Acknowledgments
The authors are very grateful for all participants in ELSA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors confirm that there are no conflicts of interest. The authors agree to allow the Journal to review the data if requested and they have full control of the primary data.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Ethical approval was granted by the London Multi-Centre Research Ethics Committee.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Funding
This work was supported by the Medical Research Council (G1001375), United Kingdom. The funder had no further role in the conduct of the research.
Electronic supplementary material
Below is the link to the electronic supplementary material.
40520_2015_419_MOESM1_ESM.docx
Appendix 1 Variables used in the English Longitudinal Study of Ageing to operationalize the Frailty Phenotype (DOCX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mekli, K., Nazroo, J.Y., Marshall, A.D. et al. Proinflammatory genotype is associated with the frailty phenotype in the English Longitudinal Study of Ageing. Aging Clin Exp Res 28, 413–421 (2016). https://doi.org/10.1007/s40520-015-0419-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40520-015-0419-z