
Abstract
miRNAs are endogenous small non-coding RNAs of 20–22 nucleotides that repress gene expression at the posttranscriptional level. There is growing interest in the role of miRNAs in cancer chemoprevention, and several naturally occurring chemopreventive agents have been found to be modulators of miRNA expression both in vitro and in vivo. Moreover, these chemopreventive phytochemicals commonly possess anti-oxidative and/or anti-inflammatory properties, and Nrf2 has been extensively studied as a molecular target in cancer prevention. The crosstalk between miRNAs and the traditional cellular signaling pathways of chemoprevention remain to be fully elucidated. This review summarizes the data regarding the potential interactions between miRNAs and anti-oxidative and anti-inflammatory pathways. Cellular redox homeostasis can affect the biogenesis and processing of miRNAs, which in turn regulate the Nrf2 pathway of detoxifying/anti-oxidative genes. We also discuss the miRNA regulatory mechanisms in relation to inflammation-related cancer signaling pathways.
Similar content being viewed by others


Introduction
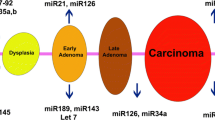
Cancer chemoprevention involves the use of chemical agents that naturally occur in food or are administered as pharmaceuticals to inhibit or reverse the carcinogenic process. Increasing evidence indicates that epigenetic modifications can initiate cancer. Unlike genetic mutations, epigenetic changes are modifications of gene expression that occur in the absence of alterations in DNA sequences. Thus, epigenetic changes have been identified as new targets for chemopreventive strategies because they are considered to be reversible via certain chemicals. In addition to DNA methylation and histone modifications, gene expression is regulated by microRNAs (miRNAs) in what is considered to be a “fine-tuning” mechanism of epigenetic modulation. MiRNAs are endogenous small non-coding RNAs of 20–22 nucleotides that repress gene expression through the degradation of mRNA or the inhibition of protein translation [1, 2]. Diverse miRNAs have been identified to control cell growth, the cell cycle, cell differentiation, tumor suppression, and apoptosis [3–5]. Genome-wide profiling has shown that the miRNA expression patterns of healthy and pathological tissues vary, as do those of different types of cancer. Therefore, increasing efforts have focused on analyzing these miRNA expression pattern alterations to identify novel cancer biomarkers and therapeutic targets. Interestingly, as shown in Table 1, dietary chemopreventive agents from widely different sources have also been evaluated as modulators of miRNA expression in a variety of cancers [6••].
Moreover, a number of naturally occurring chemopreventive compounds possess anti-oxidative and anti-inflammatory properties that are associated with their protective effects against tumorigenesis. Oxidative stress can be a consequence of the increased generation of reactive oxygen/nitrogen species (ROS/RNS) and/or decreased functioning of the anti-oxidative stress defense systems of the body [7]. In immune cells, endogenous ROS/RNS are generated to eliminate invading pathogens [8, 9]. In the context of oxidative stress, the secretion of a large amount of ROS/RNS recruits more activated inflammatory immune cells. When the crosstalk between inflammation and oxidative stress becomes chronic, excessive cellular ROS/RNS is produced [10–12]. This exacerbating loop can result in the oxidation of intracellular proteins, lipids, and nucleic acids, leading to aberrant genetic changes and/or epigenetic alterations such as the dysregulation of oncogene and tumor-suppressor gene expression [13, 14, 12].
Phase II detoxifying/antioxidant enzymes, such as glutathione S-transferase (GST), UDP-glucuronosyltransferase (UGT), heme oxygenase-1 (HO-1), NADP(H):quinone oxidoreductase (NQO), glutamate cysteine ligase (GCL), and gamma glutamylcysteine synthetase (γGCS), among others, produce a cytoprotective environment by reducing the toxicity of reactive intermediates, which augments cellular defense against stress and maintains intracellular redox homeostasis [10–15]. The genes that encode detoxifying/antioxidant enzymes typically share a consensus cis-element in their promoter regions that is known as the antioxidant response element (ARE) or electrophile response element (EpRE). The transcriptional activation of ARE-mediated genes is primarily regulated by nuclear factor erythroid-2 related factor-2 (Nrf2 or NFE2L2), a helix-loop-helix basic leucine zipper transcription factor. Extensive research both in vitro and in vivo has demonstrated the important role of Nrf2 as a molecular target for naturally derived chemopreventive agents.
How miRNAs interact with anti-oxidative and anti-inflammatory pathways in diverse physiological and pathophysiological processes remains to be fully elucidated. Given that the Nrf2/ARE and inflammatory pathways have long been the focus of cancer prevention studies, uncovering the relationship between miRNAs and these signaling pathways could reveal an important regulatory mechanism that blocks carcinogenesis.
MiRNAs and Carcinogenesis
The relationship between aberrant miRNA expression and carcinogenesis may be attributed to the fact that greater than 50 % of the genes that encode these miRNAs are located in the cancer-associated regions of fragile sites; these sites exhibit chromosomal instability via events associated with amplification, translocation or deletion [16]. Based on the properties of their target genes, miRNAs can function as either oncogenic miRNAs or tumor-suppressive miRNAs. Because miRNAs are commonly expressed as polycistronic transcripts, the dysregulation of one member of the cluster often coincides with the dysregulation of other members of the cluster. Therefore, miRNome-wide aberrations are more likely to be involved in carcinogenesis than mutations in a single miRNA gene that targets one oncogene or tumor suppressor.
A study conducted by Calin et al. was the first to establish the existence of altered levels of the miR-15a/miR-16-1 cluster in B-cell chronic lymphocytic leukemia (CLL) [17]. Both miRNAs are located at chromosomal position 13q14.3, which is a region that is often deleted in CLL and prostate cancer [18, 19]. One of the identified targets of miR-16-1 is B-cell lymphoma 2 (BCL2), which is an anti-apoptotic gene associated with cell survival and carcinogenesis. Therefore, the impaired expression of miR-15a/miR-16-1 is inversely correlated with BCL2 expression in CLL; additional genes affected by this cluster include MCL1, ETS1, and JUN, among others [20]. The tumor-suppressive function of this miRNA cluster was further validated by the finding that the overexpression of the miR-15a/16-1 cluster induces apoptosis in the chronic myeloid leukemia MEG-01 cell line by activating the intrinsic apoptosis pathway and inhibiting tumor growth in a nude mouse engraftment model [20, 21]. In addition to miR-15 and miR-16, other miRNAs with a tumor-suppressive role (anti-oncomirs) are frequently downregulated in cancer cells. For example, p53 suppresses the proto-oncogene c-Myc via the induction of miR-146 [22], which is commonly downregulated in lung and breast cancer and is deleted in prostate cancer [23]. However, remarkably, some miRNAs can function as either oncogenic or tumor-suppressive factors depending on the cell type and the pattern of gene expression [24]. Most oncomirs downregulate the expression of tumor suppressors and are overexpressed in cancer cells. For example, the consistent up-regulation of oncogenic miR-21 expression has been reported in various cancers, including those of the breast, colon, lung, stomach, and pancreas, as well as squamous cell cancer [25]. Taken together, these data indicate that profiling differential miRNA patterns in a variety of cancer types may provide potential cancer-specific “miRNA signatures” for diagnostic and therapeutic applications.
Although a number of studies have described the alteration of miRNAs in cancer cells in response to various chemopreventive agents [26], the mechanisms underlying the changes in the expression and the anticancer activities of miRNA induced by these phytochemicals remain to be fully elucidated. Recent investigations into the causes and consequences of miRNA dysfunction indicate that miRNAs are subject to higher levels of control regulating both miRNA biogenesis and function. For example, environmental factors, such as redox homeostasis and inflammatory conditions, play an important role in the modulation of miRNAs.
Redox Homeostasis Regulates miRNA Biogenesis
To date, few reports have indicated that Nrf2 regulates the transcriptional activities of the genes encoding miRNAs through Nrf2/ARE-mediated mechanisms. However, several clues in this rapidly developing field support the hypothesis that miRNA biogenesis can be affected by cellular redox homeostasis, which is maintained by the Nrf2 pathway under a number of physiological conditions. The biogenesis of miRNAs occurs similarly to that of other RNA molecules, which begins with DNA transcription. Immature primary miRNAs (pri-miRNAs) are synthesized via the transcription of the miRNA genes by RNA polymerase II [27]. The pri-miRNAs are then processed by a nuclear microprocessor complex consisting of RNase III (Drosha) and the co-factor DiGeorge critical region 8 (DGCR8) to form precursor miRNAs (pre-miRNAs) that are approximately 70 nt long [28]. After their production in the nucleus, the pre-miRNAs are transported to the cytoplasm by the nuclear export factor exportin 5 (XPO5) where they are bound by the RNase III-type endonuclease, Dicer, which can cleave the hairpins of the pre-miRNAs to form double-stranded RNAs [29]. Finally, the mature miRNAs are assembled into a multi-protein effector RNA-induced silencing complex (RISC) with members of the Argonaute protein family (Ago 1–4), which are the essential core components of the RISC [30]. Recently, increasing evidence has shown that the key molecules in each step of miRNA biogenesis are affected by the cellular redox environment. For instance, the binding of ferric heme enhances the activity of DGCR8 [31, 32], which plays a role in the maturation of all of the canonical miRNAs. Therefore, the Nrf2-regulated antioxidant enzyme HO-1 affects miRNA biogenesis by competing for the heme substrate, thereby decreasing the activity of DGCR8. This finding is supported by the observation that the overexpression of HO-1 inhibits the differentiation of murine myoblasts by downregulating miRNA biogenesis, thus altering gene expression by modulating the global level of functional miRNAs [33]. In addition, the transporter activity of XPO5 can be affected by oxidative stress in a manner similar to that in other nuclear exporters [34], resulting in another molecular hub in which the cellular redox conditions may affect the miRNA transcriptome. The expression of cytoplasmic Dicer has been shown to be downregulated by aging and hydrogen peroxide-induced oxidative stress in rat cerebral microvascular endothelial cells (CMVECs) [35]. Aging-associated vascular dysfunction in rats is characterized by decreased Nrf2-mediated antioxidant signaling and increased oxidative stress [36]. Additionally, Dicer protein levels have also been found to be downregulated in aged human adipocytes, 3T3-F442 preadipocytes, and JAR trophoblast cells exposed to hydrogen peroxide [37, 38]. These findings further support the hypothesis that miRNA biogenesis is affected by redox dysregulation via Dicer. Interestingly, through the in silico screening of transcription factor binding sites and the comparative database analysis of DNA sequences, an ARE consensus sequence was discovered in the promoter region of human and mouse Dicer genes. The existence of this consensus sequence may partially explain why chemopreventive phytochemicals, such as sulforaphane and resveratrol, restore the expression of Dicer in CMVEC cells [35]. In addition to the key molecules mentioned above, two families of enzymes, the DNA methyltransferase (DNMT) and histone deacetylase (HDAC) families, can regulate the expression of genes encoding miRNAs via epigenetic mechanisms [39]. Both DNMT and HDAC activities are affected by oxidative stress [40•], providing other routes by which cellular redox signaling may alter miRNA biogenesis. Furthermore, miRNAs themselves can be modified by ROS, resulting in the alteration of their integrity, stability, binding affinity, and function, thereby contributing to perturbed redox signaling-associated disease mechanisms [41]. Because the knockout of the Nrf2 gene significantly impairs cellular redox homeostasis, it is feasible that miRNA processing is altered by cellular Nrf2 levels, thereby resulting in changes of the expression of the miRNAs that modulate antioxidant responses and/or exacerbate oxidative stress.
Role of miRNAs in Regulating the Nrf2 Pathway
One interesting mode of miRNA action is the ability to self-regulate. As mentioned above, miRNAs can participate in the regulation of their own biogenesis by altering redox homeostasis. miRNAs have been observed to be involved in the posttranscriptional regulation of Nrf2 levels. Using an in silico analysis, Narasimhan et al. demonstrated that the ectopic expression of four miRNA mimics, miR-27a, miR-142-5p, miR-144, and miR-153, can affect the nucleo-cytoplasmic level of Nrf2 protein in a Keap1-independent manner [42•]. This observation together with the decreased abundance of Nrf2 mRNA by these miRNA mimics indicates that Nrf2 is a direct target of these miRNAs. Thus, inefficient activating ability of Nrf2 results in diminished GCLC and GSR expression, leading to a decrease in cellular glutathione (GSH) levels. miR-144 was also found to be highly expressed in reticulocytes from patients with sickle cell disease, which may attenuate the antioxidant defense capacity by altering Nrf2 signaling [43]. In human mammary epithelial cells and the breast cancer MCF-7 cell line, miR-28 affects Nrf2 mRNA stability by facilitating degradation, which may also modulate the rate of tumor cell growth [44]. A systematic analysis of the Nrf2 interactome and regulome by Papp et al. revealed that 85 miRNAs are predicted to bind Nrf2 mRNA to downregulate its translation [45••]. Among these, 63 miRNAs were defined as potential members of negative feedback loops for Nrf2 signaling in which an activating transcription factor could increase the level of expression of miRNAs that downregulate Nrf2. Additionally, positive feedback loops exist whereby an activated transcription factor represses the expression of Nrf2-downregulating miRNAs to enhance Nrf2 translation.
Given that the cellular activity of Nrf2 can be regulated by factors other than miRNAs, such as Kelch-like ECH-associated protein 1 (Keap1), small masculoaponeurotic fibrosarcoma (Maf), BTB and CNC homologue 1 (Bach1), and Parkinson protein 7 (PARK7/DJ-1), the miRNAs that interact with these Nrf2 modulators would also be expected to regulate Nrf2/ARE-mediated signaling in an indirect manner. Keap1 serves as a suppressor of the Nrf2 pathway by binding to Nrf2 and facilitating its ubiquitination and degradation. In the human breast cancer cell line MDA-MB-231, miR-200a can target Keap1 mRNA, leading to its degradation [46]. In the same study, Eades et al. found that the epigenetic silencing of miR-200a may contribute to the dysregulation of Nrf2 activity in breast cancer, whereas restoring miR-200a expression leads to enhanced Nrf2 nuclear translocation and the activation of NAD(P)H-quinone oxidoreductase 1(NQO1) gene transcription, which consequently inhibits the anchorage-independent growth of breast cancer cells. The transcriptional repressor Bach1, which functions in association with small Maf proteins, sequesters ARE-like enhancers within cells and antagonizes Nrf2 binding until it is inactivated by pro-oxidants [47]. Several studies have shown that the miRNAs let-7b, let-7c, miR-98, and miR-196 can downregulate Bach1 expression in human hepatoma Huh-7 cells [48], thereby increasing Nrf2-mediated HO-1 gene expression and attenuating oxidative stress. Notably, pro-inflammatory miR-155 has also been reported to enhance the induction of HO-1 expression via the inhibition of Bach1 translation [49], which indicates that miRNA regulation could be a potential factor in the cross-talk between the inflammatory and oxidative stress pathways. However, several phytochemicals, such as quercetin, allyl-isothiocyanate, and sulforaphane, have been reported to decrease miR-155 levels in lipopolysaccharide-stimulated murine RAW264.7 macrophages; the induction of HO-1 expression was also observed [50, 51]. Because interactions between the inflammatory response and Nrf2 pathways may occur at multiple levels, the precise mechanisms by which miR-155 affects the cellular anti-oxidative system remain to be fully elucidated. In addition to directly targeting Nrf2 mRNA, miR-34a has also been reported to regulate sirtuin1 (Sirt1) [52], a nicotinamide adenine dinucleotide-dependent class III HDAC that serves as a crucial regulator of metabolism and aging [53]. Following the release of Nrf2 from Keap1, the acetylation of Nrf2 by CREB-binding protein (CBP) has been shown to increase the nuclear localization of Nrf2, thus activating Nrf2-dependent transcription through target gene promoters and enhanced ARE binding, whereas the deacetylation of Nrf2 by Sirt1 disengages Nrf2 from the ARE region of the downstream gene promoter, thereby resulting in Nrf2 transcription and its subsequent nuclear export.
Inflammatory Conditions and miRNAs
In addition to inducing the expression of detoxifying and antioxidant enzymes, the Nrf2 pathway appears to mediate a strong anti-inflammatory response [54•]. This finding is supported by the attenuation of the anti-inflammatory effect of sulforaphane in LPS-treated primary peritoneal macrophages from Nrf2 (−/−) mice [55]. Furthermore, Nrf2-KO mice are more susceptible to dextran sulfate (DSS)-induced colitis and to AOM-DSS-induced colorectal cancer [56, 57], which are common models for characterizing chronic inflammation and carcinogenesis. Targeting anti-inflammation is another important approach for cancer prevention. In addition to the presence of oxidative stress, the tumor microenvironment contains various infiltrating immune cells due to inflammatory stimuli [58, 59]. Epidemiological studies suggest that chronic inflammation may contribute to the development of 25 % of all cancer cases [60]. Through cross-talk with cancer cells and tumor stromal cells, tumor-associated macrophages play key roles in cancer-related inflammation by secreting a number of growth factors, angiogenic factors, proteinases, chemokines, and cytokines [61–63]. This inflammatory environment stimulates cell proliferation, cell survival, angiogenesis, cell migration, and tumor metastasis, thereby driving cancer progression.
A variety of miRNAs have been associated with cancer-related inflammation. Of these, miR-21, miR-125b, miR-155, miR-196, and miR-210 are the most extensively studied [64, 65]. As mentioned in the previous section, miR-21 is overexpressed in nearly all carcinoma and malignancy types [66], and its expression can also be induced in human peripheral blood mononuclear cells via LPS treatment [67]. Surprisingly, miR-21 is implicated in both positive and negative feedback loops depending on the type of pro-/anti-inflammatory cytokines regulated by miR-21. Similar to miR-21, the level of miR-125b was drastically changed in mouse macrophage RAW264.7 cells challenged with LPS [68] where it functions in preventing excessive pro-inflammatory responses. miR-125b expression is downregulated in metastatic cutaneous malignant melanomas [69]. The abnormal overexpression of miR-155 is often associated with enhanced cytokine production. Increased miR-155 expression in tumors does not appear to result from any gene mutation, which suggests that it could instead be a result of inflammatory/oncogenic regulation [70, 71]. MiR-210 has been shown to directly target NF-κB transcripts, which serve as a very important negative feedback regulator of the production of pro-inflammatory cytokines [72].
The expression of miR-155, miR-21, miR-125b, miR-196, and miR-210 is controlled by a number of inflammatory signals originating from cells that participate in innate or adaptive inflammatory responses. The activation of toll-like receptors (TLRs) and the production of cytokines are the most prominent mechanisms linking the functions of these miRNAs with inflammatory events [73••, 74, 75]. For instance, the transcription levels of both pri-miR-21 and pri-miR-125b were up-regulated by LPS treatment in human biliary epithelial cells through NF-κB activation [76]. Furthermore, high levels of miR-155 in diffuse large B-cell lymphoma (DLBCL) are related to the endogenous production of TNF, which induces miR-155 expression in these cells, in turn blocking the expression of SHIP1, promoting the conversion of PIP2 to PIP3 by PI3K and thus further augmenting the pro-inflammatory effects of TNF [77]. Additionally, the LPS-induced expression of miR-155 in macrophages can be inhibited by IL-10, a potent anti-inflammatory cytokine that dampens the expression of pro-inflammatory genes [78]. The inhibition of miR-155 expression in the presence of IL-10 is associated with B cell integration cluster gene (BIC) transcription repression via its Ets1 site in a STAT3-dependent manner, and the IL-10-dependent inhibition of miR-155 expression may be a potential mechanism used to halt the immune response after the pathogens have been cleared.
MiRNAs Link Inflammation and Cancer
The activation of NF-κB and the activation of the transcription factors of activator protein-1 (AP-1) and the members of the STAT families through nuclear translocation are essential for a balanced immune response under inflammatory conditions. However, their continuous activation or overexpression has been found in many different malignancies [60–72, 73••, 74–79]. MiR-21 and miR-155 expression have been reported to be under the control of NF-κB, AP-1, STATs, and SMADs; thus, the enhanced levels of these miRNAs (which are at the junctions between inflammatory and oncogenic signaling pathways) could at least partially explain the link between inflammation and cancer at the molecular level.
The aberrant activation of the JAK/STAT pathway has been demonstrated to be one of the most important inflammatory signals in malignant cells [80]. The level of both miR-21 and miR-155 are affected by the members of the STAT families. For example, the NF-κB-dependent recruitment of STAT3 to the miR-21 promoter leads to the up-regulation of miR-2 expression, which may contribute to both the therapeutic resistance and metastasis of breast cancer cells [81]. Stimulating breast cancer cells using the inflammatory cytokines IFN-gamma, interleukin-6 (IL-6) and LPS can significantly up-regulate miR-155 expression [82]. In the same study, Jiang et al. found that the suppressor of cytokine signaling 1 (SOCS1), a negative regulator of JAK/STAT signaling, is an evolutionarily conserved target of miR-155 in breast cancer cells. The overexpression of miR-155 leads to the constitutive activation of STAT3, which promotes the proliferation and transformation of breast cancer cells and breast tumor growth in nude mice [82].
TGF-β is a pleiotropic cytokine that exerts important effects on processes such as fibrosis, angiogenesis, and immunosuppression [83]. Typically, TGF-β controls gene expression through transcription factors known as SMAD proteins. However, TGF-β facilitates metastasis by altering the homeostasis of TGF-β signaling in advanced malignancies. Using a bioinformatics approach, SMAD2 was predicted to be a target of miR-155 [84]. The overexpression of miR-155 altered cellular responses to TGF-β, consequently modulating tissue repair and remodeling that occur in response to macrophages [84]. Additionally, miR-155 directly targets the bone morphogenetic protein (BMP)-responsive transcriptional factor SMAD5 in DLBCLs, resulting in resistance to the growth inhibitory effects of both TGF-β and BMPs via the defective induction of p21 expression and impaired cell cycle arrest [85]. MiR-21 is also a downstream effector of TGF-β in facilitating cellular invasion and metastasis. Colon carcinoma organoids have been reported to undergo the endothelial-mesenchymal transition in response to TGF-β; this process can be further accelerated by TNF-α, whereas the level of miR-21 is prominently elevated by the synergistic activities of TGF-β/TNF-α [86].
The constitutive activation of phosphatidylinositol 3-kinase (PI3K)/AKT signaling is one of the most common molecular signatures of human cancers [87]. Under physiological conditions, the phosphorylation of AKT is antagonized and balanced by phosphatases such as PTEN, SHIP1, and phosphatase protein phosphatase 2A catalytic subunit alpha (PPP2CA). Among these, SHIP1 and PPP2CA have been identified to be targets of miR-155 [88, 77]; therefore, a high miR-155 level decreases the level of SHIP1 and PPP2CA expression, thereby resulting in the prolonged activation of the PI3K/AKT pathway. Additionally, a proteomics investigation of diffuse large B-cell lymphoma (DLBCL) revealed that PIK3R1 (p85a), which functions as a cellular suppressor of the PI3K/AKT pathway, is also targeted by miR-155 [89]. Furthermore, the lack of PTEN expression is found to be associated with an increased level of miR-21 in numerous malignancies. The inhibition of miR-21 expression in cultured human hepatocellular cancer cells increased the expression of the PTEN tumor suppressor and reduced cell proliferation, migration, and invasion [90].
Conclusion
The deregulation of miRNA expression is implicated in carcinogenesis due to its promotion of cell proliferation, survival, invasion, and metastasis as well as its ability to inhibit cell apoptosis and alter the transcription levels of a variety of genes. Naturally occurring chemopreventive compounds have been reported to dramatically affect miRNA expression to produce anti-cancer effects. However, most of the studies of the effects of dietary phytochemicals on miRNA regulation are highly descriptive, and this field remains in its infancy. Because miRNAs regulate mRNA levels in a “fine tuning” manner, they are sensitive to changes in the cellular environment such as those induced by oxidative stress and inflammatory conditions. Given that a number of dietary chemopreventive agents possess anti-oxidative and anti-inflammatory properties, investigations into the biogenesis, metabolism, and function of miRNAs under cellular homeostasis/stress conditions would provide new mechanistic insights into the roles of miRNAs in cancer pathogenesis as well as into approaches using chemopreventive agents. Finally, current studies investigating the expression of a limited number of miRNAs using antagomir or miRNA-mimicking techniques need to be elaborated upon by further studies that profile the genome-wide miRNome.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Garzon R, Pichiorri F, Palumbo T, Visentini M, Aqeilan R, Cimmino A, et al. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene. 2007;26(28):4148–57. doi:10.1038/sj.onc.1210186.
Liu C, Tang DG. MicroRNA regulation of cancer stem cells. Cancer Res. 2011;71(18):5950–4. doi:10.1158/0008-5472.CAN-11-1035.
Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64(11):3753–6. doi:10.1158/0008-5472.CAN-04-0637.
Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–69. doi:10.1038/nrc1840.
Lee YS, Kim HK, Chung S, Kim KS, Dutta A. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J Biol Chem. 2005;280(17):16635–41. doi:10.1074/jbc.M412247200.
Parasramka MA, Ho E, Williams DE, Dashwood RH. MicroRNAs, diet, and cancer: new mechanistic insights on the epigenetic actions of phytochemicals. Mol Carcinog. 2012;51(3):213–30. doi:10.1002/mc.20822. This manuscript reviews several natural dietary factors that exert anti-cancer properties through regulating miRNAs.
Beckman KB, Ames BN. Mitochondrial aging: open questions. Ann N Y Acad Sci. 1998;854:118–27.
Babior BM. Phagocytes and oxidative stress. Am J Med. 2000;109(1):33–44.
Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–82. doi:10.1016/j.cell.2010.02.029.
Kong AN, Mandlekar S, Yu R, Lei W, Fasanmande A. Pharmacodynamics and toxicodynamics of drug action: signaling in cell survival and cell death. Pharm Res. 1999;16(6):790–8.
Ma Q. Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacol Ther. 2010;125(3):376–93. doi:10.1016/j.pharmthera.2009.11.004.
Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31(1):37–49. doi:10.1093/carcin/bgp272.
Chen C, Kong AN. Dietary cancer-chemopreventive compounds: from signaling and gene expression to pharmacological effects. Trends Pharmacol Sci. 2005;26(6):318–26. doi:10.1016/j.tips.2005.04.004.
Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int JCancer J Int du Cancer. 2007;121(11):2373–80. doi:10.1002/ijc.23173.
Kwak MK, Egner PA, Dolan PM, Ramos-Gomez M, Groopman JD, Itoh K, et al. Role of phase 2 enzyme induction in chemoprotection by dithiolethiones. Mutat Res. 2001;480–481:305–15.
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101(9):2999–3004. doi:10.1073/pnas.0307323101.
Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A. 2004;101(32):11755–60. doi:10.1073/pnas.0404432101.
Dong JT, Boyd JC, Frierson Jr HF. Loss of heterozygosity at 13q14 and 13q21 in high grade, high stage prostate cancer. Prostate. 2001;49(3):166–71.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99(24):15524–9. doi:10.1073/pnas.242606799.
Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105(13):5166–71. doi:10.1073/pnas.0800121105.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102(39):13944–9. doi:10.1073/pnas.0506654102.
Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009;106(9):3207–12. doi:10.1073/pnas.0808042106.
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65(16):7065–70. doi:10.1158/0008-5472.CAN-05-1783.
Melo SA, Esteller M. Dysregulation of microRNAs in cancer: playing with fire. FEBS Lett. 2011;585(13):2087–99. doi:10.1016/j.febslet.2010.08.009.
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–61. doi:10.1073/pnas.0510565103.
Yi B, Piazza GA, Su X, Xi Y. MicroRNA and cancer chemoprevention. Cancer Prev Res (Phila). 2013;6(5):401–9. doi:10.1158/1940-6207.CAPR-13-0032.
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23(20):4051–60. doi:10.1038/sj.emboj.7600385.
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the microprocessor complex. Nature. 2004;432(7014):231–5. doi:10.1038/nature03049.
Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11(3):228–34. doi:10.1038/ncb0309-228.
Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404(6775):293–6. doi:10.1038/35005107.
Barr I, Smith AT, Chen Y, Senturia R, Burstyn JN, Guo F. Ferric, not ferrous, heme activates RNA-binding protein DGCR8 for primary microRNA processing. Proc Natl Acad Sci U S A. 2012;109(6):1919–24. doi:10.1073/pnas.1114514109.
Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Heme is involved in microRNA processing. Nat Struct Mol Biol. 2007;14(1):23–9. doi:10.1038/nsmb1182.
Kozakowska M, Ciesla M, Stefanska A, Skrzypek K, Was H, Jazwa A, et al. Heme oxygenase-1 inhibits myoblast differentiation by targeting myomirs. Antioxid Redox Signal. 2012;16(2):113–27. doi:10.1089/ars.2011.3964.
Crampton N, Kodiha M, Shrivastava S, Umar R, Stochaj U. Oxidative stress inhibits nuclear protein export by multiple mechanisms that target FG nucleoporins and Crm1. Mol Biol Cell. 2009;20(24):5106–16. doi:10.1091/mbc.E09-05-0397.
Ungvari Z, Tucsek Z, Sosnowska D, Toth P, Gautam T, Podlutsky A, et al. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A: Biol Med Sci. 2013;68(8):877–91. doi:10.1093/gerona/gls242.
Ungvari Z, Bailey-Downs L, Gautam T, Jimenez R, Losonczy G, Zhang C, et al. Adaptive induction of NF-E2-related factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am J Physiol Heart Circ Physiol. 2011;300(4):H1133–40. doi:10.1152/ajpheart.00402.2010.
Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol Immunol. 2009;46(6):1222–8. doi:10.1016/j.molimm.2008.11.012.
Mori MA, Raghavan P, Thomou T, Boucher J, Robida-Stubbs S, Macotela Y, et al. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab. 2012;16(3):336–47. doi:10.1016/j.cmet.2012.07.017.
Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J. 2011;278(10):1598–609. doi:10.1111/j.1742-4658.2011.08089.x.
Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal. 2011;15(2):551–89. doi:10.1089/ars.2010.3492. This manuscript reviews epigenetic modifications in the context of redox basis.
Poulsen HE, Specht E, Broedbaek K, Henriksen T, Ellervik C, Mandrup-Poulsen T, et al. RNA modifications by oxidation: a novel disease mechanism? Free Radic Biol Med. 2012;52(8):1353–61. doi:10.1016/j.freeradbiomed.2012.01.009.
Narasimhan M, Patel D, Vedpathak D, Rathinam M, Henderson G, Mahimainathan L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS One. 2012;7(12):e51111. This manuscript describes novel miRNAs targeting Nrf2.
Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116(20):4338–48. doi:10.1182/blood-2009-04-214817.
Yang M, Yao Y, Eades G, Zhang Y, Zhou Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res Treat. 2011;129(3):983–91. doi:10.1007/s10549-011-1604-1.
Papp D, Lenti K, Modos D, Fazekas D, Dul Z, Turei D, et al. The NRF2-related interactome and regulome contain multifunctional proteins and fine-tuned autoregulatory loops. FEBS Lett. 2012;586(13):1795–802. doi:10.1016/j.febslet.2012.05.016. This manuscript extends Nrf2-related interactome to miRNA level.
Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem. 2011;286(47):40725–33. doi:10.1074/jbc.M111.275495.
Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007;35(21):7074–86. doi:10.1093/nar/gkm638.
Hou W, Tian Q, Steuerwald NM, Schrum LW, Bonkovsky HL. The let-7 microRNA enhances heme oxygenase-1 by suppressing Bach1 and attenuates oxidant injury in human hepatocytes. Biochim Biophys Acta. 2012;1819(11–12):1113–22. doi:10.1016/j.bbagrm.2012.06.001.
Pulkkinen KH, Yla-Herttuala S, Levonen AL. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic Biol Med. 2011;51(11):2124–31. doi:10.1016/j.freeradbiomed.2011.09.014.
Wagner AE, Boesch-Saadatmandi C, Dose J, Schultheiss G, Rimbach G. Anti-inflammatory potential of allyl-isothiocyanate–role of Nrf2, NF-(kappa) B and microRNA-155. J Cell Mol Med. 2012;16(4):836–43. doi:10.1111/j.1582-4934.2011.01367.x.
Boesch-Saadatmandi C, Loboda A, Wagner AE, Stachurska A, Jozkowicz A, Dulak J, et al. Effect of quercetin and its metabolites isorhamnetin and quercetin-3-glucuronide on inflammatory gene expression: role of miR-155. J Nutr Biochem. 2011;22(3):293–9. doi:10.1016/j.jnutbio.2010.02.008.
Li N, Muthusamy S, Liang R, Sarojini H, Wang E. Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech Ageing Dev. 2011;132(3):75–85. doi:10.1016/j.mad.2010.12.004.
Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13(4):225–38. doi:10.1038/nrm3293.
Hu R, Saw CL, Yu R, Kong AN. Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: antioxidant coupled with antiinflammatory. Antioxid Redox Signal. 2010;13(11):1679–98. doi:10.1089/ars.2010.3276. This manuscript reviews the important role of Nrf2 signaling pathway for cancer chemoprevention.
Lin W, Wu RT, Wu T, Khor TO, Wang H, Kong AN. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem Pharmacol. 2008;76(8):967–73. doi:10.1016/j.bcp.2008.07.036.
Khor TO, Huang MT, Kwon KH, Chan JY, Reddy BS, Kong AN. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006;66(24):11580–4. doi:10.1158/0008-5472.CAN-06-3562.
Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res (Phila). 2008;1(3):187–91. doi:10.1158/1940-6207.CAPR-08-0028.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45. doi:10.1016/S0140-6736(00)04046-0.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7. doi:10.1038/nature01322.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44. doi:10.1038/nature07205.
Ono M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008;99(8):1501–6. doi:10.1111/j.1349-7006.2008.00853.x.
Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8. doi:10.1038/nrc1256.
Yoshimura A. Signal transduction of inflammatory cytokines and tumor development. Cancer Sci. 2006;97(6):439–47. doi:10.1111/j.1349-7006.2006.00197.x.
Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28(5):264–84. doi:10.1080/08830180903093796.
Tili E, Michaille JJ. Resveratrol, microRNAs, inflammation, and cancer. J Nucleic Acids. 2011;2011:102431. doi:10.4061/2011/102431.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–66. doi:10.1038/nrc1997.
Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O'Leary JJ, Ruan Q, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 2010;11(2):141–7. doi:10.1038/ni.1828.
Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179(8):5082–9.
Glud M, Rossing M, Hother C, Holst L, Hastrup N, Nielsen FC, et al. Downregulation of miR-125b in metastatic cutaneous malignant melanoma. Melanoma Res. 2010;20(6):479–84. doi:10.1097/CMR.0b013e32833e32a1.
Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, et al. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;286(2):1436–44. doi:10.1074/jbc.M110.145870.
Piccinini AM, Midwood KS. Endogenous control of immunity against infection: tenascin-C regulates TLR4-mediated inflammation via microRNA-155. Cell Rep. 2012;2(4):914–26. doi:10.1016/j.celrep.2012.09.005.
Qi J, Qiao Y, Wang P, Li S, Zhao W, Gao C. microRNA-210 negatively regulates LPS-induced production of proinflammatory cytokines by targeting NF-kappaB1 in murine macrophages. FEBS Lett. 2012;586(8):1201–7. doi:10.1016/j.febslet.2012.03.011.
O’Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of toll-like receptor signalling. Nat Rev Immunol. 2011;11(3):163–75. doi:10.1038/nri2957. This manuscript reviews the role of miRNAs in Toll-like receptor signaling, which is a major signaling of inflammation.
O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104(5):1604–9. doi:10.1073/pnas.0610731104.
Nahid MA, Satoh M, Chan EK. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol Immunol. 2011;8(5):388–403. doi:10.1038/cmi.2011.26.
Zhou R, Hu G, Gong AY, Chen XM. Binding of NF-kappaB p65 subunit to the promoter elements is involved in LPS-induced transactivation of miRNA genes in human biliary epithelial cells. Nucleic Acids Res. 2010;38(10):3222–32. doi:10.1093/nar/gkq056.
Pedersen IM, Otero D, Kao E, Miletic AV, Hother C, Ralfkiaer E, et al. Onco-miR-155 targets SHIP1 to promote TNFalpha-dependent growth of B cell lymphomas. EMBO Mol Med. 2009;1(5):288–95. doi:10.1002/emmm.200900028.
McCoy CE, Sheedy FJ, Qualls JE, Doyle SL, Quinn SR, Murray PJ, et al. IL-10 inhibits miR-155 induction by toll-like receptors. J Biol Chem. 2010;285(27):20492–8. doi:10.1074/jbc.M110.102111.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. doi:10.1016/j.cell.2010.01.025.
Li WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008;18(11):545–51. doi:10.1016/j.tcb.2008.08.008.
Niu J, Shi Y, Tan G, Yang CH, Fan M, Pfeffer LM, et al. DNA damage induces NF-kappaB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J Biol Chem. 2012;287(26):21783–95. doi:10.1074/jbc.M112.355495.
Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H, et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70(8):3119–27. doi:10.1158/0008-5472.CAN-09-4250.
Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19(8):1745–54. doi:10.1093/emboj/19.8.1745.
Louafi F, Martinez-Nunez RT, Sanchez-Elsner T. MicroRNA-155 targets SMAD2 and modulates the response of macrophages to transforming growth factor-{beta}. J Biol Chem. 2010;285(53):41328–36. doi:10.1074/jbc.M110.146852.
Rai D, Kim SW, McKeller MR, Dahia PL, Aguiar RC. Targeting of SMAD5 links microRNA-155 to the TGF-beta pathway and lymphomagenesis. Proc Natl Acad Sci U S A. 2010;107(7):3111–6. doi:10.1073/pnas.0910667107.
Cottonham CL, Kaneko S, Xu L. miR-21 and miR-31 converge on TIAM1 to regulate migration and invasion of colon carcinoma cells. J Biol Chem. 2010;285(46):35293–302. doi:10.1074/jbc.M110.160069.
Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi:10.1146/annurev.biochem.68.1.965.
Bakirtzi K, Hatziapostolou M, Karagiannides I, Polytarchou C, Jaeger S, Iliopoulos D, et al. Neurotensin signaling activates microRNAs-21 and −155 and Akt, promotes tumor growth in mice, and is increased in human colon tumors. Gastroenterology. 2011;141(5):1749–61. doi:10.1053/j.gastro.2011.07.038. e1.
Huang X, Shen Y, Liu M, Bi C, Jiang C, Iqbal J, et al. Quantitative proteomics reveals that miR-155 regulates the PI3K-AKT pathway in diffuse large B-cell lymphoma. Am J Pathol. 2012;181(1):26–33. doi:10.1016/j.ajpath.2012.03.013.
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2):647–58. doi:10.1053/j.gastro.2007.05.022.
Hu S, Dong TS, Dalal SR, Wu F, Bissonnette M, Kwon JH, et al. The microbe-derived short chain fatty acid butyrate targets miRNA-dependent p21 gene expression in human colon cancer. PLoS One. 2011;6(1):e16221. doi:10.1371/journal.pone.0016221.
Yang J, Cao Y, Sun J, Zhang Y. Curcumin reduces the expression of Bcl-2 by upregulating miR-15a and miR-16 in MCF-7 cells. Med Oncol. 2010;27(4):1114–8. doi:10.1007/s12032-009-9344-3.
Zhang J, Zhang T, Ti X, Shi J, Wu C, Ren X, et al. Curcumin promotes apoptosis in A549/DDP multidrug-resistant human lung adenocarcinoma cells through an miRNA signaling pathway. Biochem Biophys Res Commun. 2010;399(1):1–6. doi:10.1016/j.bbrc.2010.07.013.
Sun M, Estrov Z, Ji Y, Coombes KR, Harris DH, Kurzrock R. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol Cancer Ther. 2008;7(3):464–73. doi:10.1158/1535-7163.MCT-07-2272.
Ali S, Ahmad A, Banerjee S, Padhye S, Dominiak K, Schaffert JM, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70(9):3606–17. doi:10.1158/0008-5472.CAN-09-4598.
Tsang WP, Kwok TT. Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. J Nutr Biochem. 2010;21(2):140–6. doi:10.1016/j.jnutbio.2008.12.003.
Siddiqui IA, Asim M, Hafeez BB, Adhami VM, Tarapore RS, Mukhtar H. Green tea polyphenol EGCG blunts androgen receptor function in prostate cancer. FASEB J : Off Pub Fed Am Soc Exp Biol. 2011;25(4):1198–207. doi:10.1096/fj.10-167924.
Wen XY, Wu SY, Li ZQ, Liu ZQ, Zhang JJ, Wang GF, et al. Ellagitannin (BJA3121), an anti-proliferative natural polyphenol compound, can regulate the expression of MiRNAs in HepG2 cancer cells. Phytother Res : PTR. 2009;23(6):778–84. doi:10.1002/ptr.2616.
Chen Y, Zaman MS, Deng G, Majid S, Saini S, Liu J, et al. MicroRNAs 221/222 and genistein-mediated regulation of ARHI tumor suppressor gene in prostate cancer. Cancer Prev Res (Phila). 2011;4(1):76–86. doi:10.1158/1940-6207.CAPR-10-0167.
Majid S, Dar AA, Saini S, Chen Y, Shahryari V, Liu J, et al. Regulation of minichromosome maintenance gene family by microRNA-1296 and genistein in prostate cancer. Cancer Res. 2010;70(7):2809–18. doi:10.1158/0008-5472.CAN-09-4176.
Sun Q, Cong R, Yan H, Gu H, Zeng Y, Liu N, et al. Genistein inhibits growth of human uveal melanoma cells and affects microRNA-27a and target gene expression. Oncol Rep. 2009;22(3):563–7.
Li Y, Vandenboom TG, Wang Z, Kong D, Ali S, Philip PA, et al. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res. 2010;70(4):1486–95. doi:10.1158/0008-5472.CAN-09-2792.
Melkamu T, Zhang X, Tan J, Zeng Y, Kassie F. Alteration of microRNA expression in vinyl carbamate-induced mouse lung tumors and modulation by the chemopreventive agent indole-3-carbinol. Carcinogenesis. 2010;31(2):252–8. doi:10.1093/carcin/bgp208.
Izzotti A, Calin GA, Steele VE, Cartiglia C, Longobardi M, Croce CM, et al. Chemoprevention of cigarette smoke-induced alterations of microRNA expression in rat lungs. Cancer Prev Res. 2010;3(1):62–72. doi:10.1158/1940-6207.CAPR-09-0202.
Li Y, VandenBoom 2nd TG, Kong D, Wang Z, Ali S, Philip PA, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69(16):6704–12. doi:10.1158/0008-5472.CAN-09-1298.
Izzotti A, Larghero P, Cartiglia C, Longobardi M, Pfeffer U, Steele VE, et al. Modulation of microRNA expression by budesonide, phenethyl isothiocyanate and cigarette smoke in mouse liver and lung. Carcinogenesis. 2010;31(5):894–901. doi:10.1093/carcin/bgq037.
Davidson LA, Wang N, Shah MS, Lupton JR, Ivanov I, Chapkin RS. n-3 Polyunsaturated fatty acids modulate carcinogen-directed non-coding microRNA signatures in rat colon. Carcinogenesis. 2009;30(12):2077–84. doi:10.1093/carcin/bgp245.
Boesch-Saadatmandi C, Wagner AE, Wolffram S, Rimbach G. Effect of quercetin on inflammatory gene expression in mice liver in vivo - role of redox factor 1, miRNA-122 and miRNA-125b. Pharmacol Res : Off J Ital Pharmacol Soc. 2012;65(5):523–30. doi:10.1016/j.phrs.2012.02.007.
Tili E, Michaille JJ, Alder H, Volinia S, Delmas D, Latruffe N, et al. Resveratrol modulates the levels of microRNAs targeting genes encoding tumor-suppressors and effectors of TGFbeta signaling pathway in SW480 cells. Biochem Pharmacol. 2010;80(12):2057–65. doi:10.1016/j.bcp.2010.07.003.
Tili E, Michaille JJ, Adair B, Alder H, Limagne E, Taccioli C, et al. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis. 2010;31(9):1561–6. doi:10.1093/carcin/bgq143.
Gaedicke S, Zhang X, Schmelzer C, Lou Y, Doering F, Frank J, et al. Vitamin E dependent microRNA regulation in rat liver. FEBS Lett. 2008;582(23–24):3542–6. doi:10.1016/j.febslet.2008.09.032.
Acknowledgments
This work was supported in part by institutional funds and by R01-CA118947, R01-CA152826, from the National Cancer Institute (NCI), R01AT007065 from the National Center for Complementary and Alternative Medicines (NCCAM) and the Office of Dietary Supplements (ODS). The authors thank all the members in Dr. Ah-Ng Tony Kong’s lab for their helpful discussion of this work.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Chengyue Zhang, Limin Shu, and Ah-Ng Tony Kong declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on miRNA and Cancer Prevention and Therapeutic Agents
Rights and permissions
About this article

Cite this article
Zhang, C., Shu, L. & Kong, AN.T. MicroRNAs: new Players in Cancer Prevention Targeting Nrf2, Oxidative Stress and Inflammatory Pathways. Curr Pharmacol Rep 1, 21–30 (2015). https://doi.org/10.1007/s40495-014-0013-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-014-0013-7