
Abstract
Multiple myeloma (MM) is a clonal plasma cell disorder defined by bone marrow infiltration and osteolytic bone lesions and is the second most common hematologic malignancy after non-Hodgkin lymphoma. The landscape of MM treatment was transformed at the dawn of the twenty-first century by the introduction of novel agents including proteasome inhibitors (bortezomib) and immunomodulatory drugs (thalidomide, lenalidomide), which have prolonged the survival of MM patients. The recently revised International Myeloma Working Group diagnostic criteria for MM added validated biomarkers (clonal bone marrow plasma cell ≥60%, involved:uninvolved serum free light chain ratio ≥100, or >1 focal lesion on magnetic resonance imaging) to identify near inevitable progression to symptomatic MM requiring therapy. In addition, the definition of myeloma-defining CRAB features (hypercalcemia, renal failure, anemia, and bone lesions) has been refined based on advances in imaging and laboratory techniques since the 2003 IMWG consensus. Despite expanded treatment options, MM remains an incurable disease. Drug resistance and clonal evolution remain problematic, and novel therapeutic agents are needed. New approaches to myeloma treatment include anti-CD38 antibodies, next generation proteasome inhibitors, epigenetic modulation with histone deacetylase inhibitors, and targeting the tumor microenvironment. In this article, the diagnosis, staging, and prognostic stratification of newly diagnosed MM will be reviewed. Clinical data pertaining to the emerging targeted agents will be discussed, and a suggested framework for integration of these new therapeutic options will be provided.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma (MM) is the second most common hematologic malignancy and is characterized by the presence of a monoclonal protein detectable in the blood and/or urine, clonal plasma cell involvement of the bone marrow, and lytic bone lesions, anemia, renal insufficiency, and hypercalcemia. In 2014, an estimated 26,850 new cases of MM were diagnosed and were responsible for 11,240 deaths [1]. Monoclonal gammopathy of undetermined significance (MGUS) precedes MM in virtually all cases [2]. The immunomodulatory drugs (IMiDs) thalidomide and lenalidomide and the proteasome inhibitors bortezomib and carfilzomib have led to substantial improvement in patient outcomes. Median overall survival (OS) in patients diagnosed after the year 2000 has more than doubled compared to patients diagnosed prior to the era of novel agents [3]. In this review, diagnosis and risk stratification, recommendations for initial and subsequent therapy, and a discussion regarding emerging therapies for MM will be provided. This review is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Diagnosis
A monoclonal protein may be discovered in the serum or urine following diagnostic testing for a variety of conditions including the evaluation of anemia, renal impairment, hypercalcemia, musculoskeletal pain, or neurologic conditions. MGUS and smoldering MM must be differentiated from MM necessitating treatment. Pathologic examination of the bone marrow with aspirate and biopsy are essential for making the diagnosis of MM, which is defined by the presence of ≥10% clonal bone marrow plasma cells (BMPCs) with one or more myeloma-defining features [4]. The revised International Myeloma Working Group diagnostic criteria for MM incorporated the validated biomarkers of BMPCs ≥60%, involved:uninvolved serum free light chain (FLC) ratio of ≥100, or >1 focal lesion on magnetic resonance imaging (MRI) spine as myeloma-defining events, and patients with these features are no longer considered smoldering MM [4]. Due to the greater than 80 % risk of these patients developing symptomatic disease within 2 years without therapy, treatment may now be offered prior to the onset of CRAB features (hypercalcemia, renal failure, anemia, and bone lesions). CRAB criteria have also been updated recognizing the advances in the detection of bone and extramedullary lesions by computed tomography (CT) and positron emission tomography (PET)/CT as well as better estimation of renal impairment by measurement of creatinine clearance in addition to serum creatinine.
A diagnostic panel of serum protein electrophoresis (SPEP), serum immunofixation (IFE), and the serum FLC assay should be obtained when MM is suspected. The serum FLC assay quantifies kappa and lambda light chains circulating unbound from the heavy chain and detects a clonal process by the alteration of the kappa/lambda ratio. In a study involving 1877 samples, sensitivity for detection of monoclonal proteins in MM was 87.6% for SPEP, 94.4% for serum IFE, and 98.6% for serum FLC, with a combined 100% diagnostic sensitivity [5]. If a patient is diagnosed with a plasma cell disorder by initial screening, then a 24-h urine protein electrophoresis (UPEP) and immunofixation should be obtained. With the use of the serum FLC along with SPEP, IFE, and UPEP, only approximately 2% of patients are determined to have non-secretory disease [6].
Multiparameter flow cytometry has become a valuable addition to the morphologic assessment of the bone marrow and may provide additional prognostic information. Immunophenotyping allows precise separation of aberrant plasma cells (typically CD19, CD27, and CD45-negative, CD56-positive) from normal plasma cells (CD19 and CD45-positive, CD56-negative) [7, 8]. Paiva et al. [9] reported that newly diagnosed MM patients with ≤15% aberrant BMPCs had a progression-free survival (PFS) of 43 vs. 36 months (P = 0.003) and OS of 97 vs. 54 months (P < 0.001). Quantification of circulating clonal plasma cells by flow cytometry is also a useful prognostic marker, and the presence of >400 circulating plasma cells shortened time to next treatment and median OS (14 vs. 26 months; 32 months vs. not reached; P < 0.001) [10]. Plasma cell proliferation may also be precisely evaluated by flow cytometry and has replaced the slide-based plasma cell labeling index (PCLI) [11]. Aspirate samples from the first pull should be used for plasma cell quantification to minimize sampling error.
The whole-body skeletal survey by conventional radiography (WBXR) has been utilized for decades in the evaluation for osteolytic lesions; however, more advanced imaging technologies including whole-body CT, MRI, and PET/CT are being increasingly utilized to detect bone and extramedullary disease. In 2009, the IMWG recommended the continued use of WBXR despite its limitations in detecting spine and pelvic bone involvement. Spine and pelvic or whole-body MRIs are suggested in symptomatic patients if WBXR is negative as well as those with compression fractures to exclude spinal cord compression [12]. In patients with smoldering MM or solitary plasmacytoma, 18F-FDG PET/CT or axial MRI should be performed to evaluate for the presence of >1 focal lesion as results may influence management in one-third of patients [13, 14]. To compare the performance of axial MRI and WBXR, 611 myeloma patients underwent both studies. In 267 patients with normal WBXR, 52% had focal lesions on MRI, whereas 20% of the 160 patients with normal MRI results had abnormal WBXR. Twenty-one percent of patients had no focal lesions by either technique, whereas 51% had lesions on both MRI and WBXR. Importantly, MRI was superior in detection of lesions in the spine (78% vs. 16%; P < 0.001), pelvis (64% vs. 28%; P < 0.001), and sternum (24% vs. 3%; P < 0.001).
Risk Stratification and Molecular Classification of MM
Despite therapeutic advances, outcomes remain heterogeneous with an approximate 10% mortality rate within the first year following diagnosis, while one-third of patients diagnosed earlier than age 65 years live beyond a decade [15]. The Durie Salmon Staging system and International Staging System (ISS) were developed to classify patients into risk groups and provide prognostic information using readily available clinical information. The ISS was developed from data on patient outcomes from 1981 to 2002 in over 10,000 patients and consists of three stages: stage I (β-2-microglobulin <3.5 mg/dl and albumin ≥3.5 g/dl; median OS 62 months), stage II (neither stage I or II; median OS 44 months), and stage III (β-2-microglobulin ≥5.5 g/dl; median OS 29 months) [16]. The ISS has also been validated in patients treated with novel agents, and 5-year survival in stages I, II, and III patients was significantly different between groups at 66 %, 45 %, and 18 %, respectively (P < 0.001) [17].
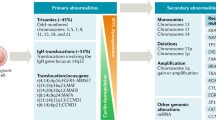
The development of interphase fluorescent in situ hybridization (FISH) to detect recurrent cytogenetic abnormalities has been a critical factor in identifying variability intrinsic to the plasma cell clone [18]. Translocations involving the immunoglobulin heavy-chain locus (IgH; 14q32) and hyperdiploidy of odd-numbered chromosomes are commonly detected by FISH [19, 20]. Monosomy of chromosome 13 or 17, deletions involving chromosomes 1, 13, or 17, and amplification of chromosome 1q are also frequent findings. Recurrent chromosomal translocations have both prognostic and therapeutic implications and have led to the development of risk-adapted treatment strategies [21]. Institutionally, we have developed a FISH-based stratification model known as mSMART (Mayo Stratification of Myeloma and Risk-Adapted Therapy) that aims to individualize therapy based on tumor biology and patient-related factors. The mSMART classification stratifies patients into three risk groups: high risk [deletion 17p, t(14;16), t(14;20) or high risk gene expression profile]; intermediate risk [t(4;14), 1q amplification, metaphase deletion 13, or high plasma cell proliferative rate S-phase]; or standard risk [hyperdiploid, t(11;14), or t(6;14)] (Table 1). Traditionally, median OS for high, intermediate, and standard risk disease was less than 3 years, 4–5 years, and 8–10 years, respectively [21, 22]. In an effort to improve outcomes for high-risk patients, combination induction therapy with both an IMiD and proteasome inhibitor (bortezomib, lenalidomide, dexamethasone; VRd) followed by bortezomib maintenance is recommended. In standard risk trisomy-only patients, an oral-based induction with lenalidomide/dexamethasone (Rd) until progression is suggested, with consideration of delayed autologous stem cell transplantation (ASCT) in eligible patients. For intermediate or standard risk patients, weekly VCd (bortezomib, cyclophosphamide, dexamethasone; CyBorD) is recommended, followed by ASCT and varying post-transplant consolidation and maintenance strategies based on risk group.
Beyond detection of primary genetic alterations by FISH, gene expression profiling (GEP), single nucleotide polymorphism (SNP) arrays, and next generation sequencing have confirmed significant clonal heterogeneity with few recurrent driver mutations [23]. Whole-exome sequencing of three myeloma patient cohorts demonstrated subclonal KRAS, NRAS, and BRAF mutations in one-third of patients and significant heterogeneous subclonal structure [24]. At present, clear biologic subgroups and predictive markers of therapeutic response have yet to be discovered.
Treatment of Newly Diagnosed MM
Critical factors to be considered during the initial evaluation of newly diagnosed MM are the performance status, age, medical comorbidities, and preferences of the patient in addition to the intrinsic tumor biology. ASCT should be considered for all eligible patients younger than age 70 years with good performance status and absence of significant comorbidities. Multiple randomized studies performed prior to the development of IMiDs and proteasome inhibitors demonstrated a survival advantage with ASCT compared to nonintensive therapy [25, 26]. Induction therapy utilizing novel agents results in higher response rates post-induction and post-transplantation when compared with VAD (vincristine, doxorubicin, dexamethasone) [27]. VAD achieved at least partial response (PR) in 50% of patients, complete response (CR) in <10%, and ≥VGPR in 15% [28]. Thalidomide-dexamethasone (TD) improved the post-induction objective response rate (ORR; ≥PR) to 75% of patients, however, achieved CR in only 10% and ≥VGPR in less than 20% of patients. TD has now been replaced by more effective and better tolerated lenalidomide-based regimens. Lenalidomide and bortezomib are now routinely incorporated into pre-ASCT induction regimens and do not interfere with adequate stem cell collection. More recently, Palumbo et al. [29] compared ASCT to melphalan, prednisone, and lenalidomide (MPR) consolidation therapy following four cycles of induction therapy with lenalidomide-dexamethasone (Rd). Both PFS and OS were significantly better in the ASCT group [median PFS 43.0 vs. 22.4 months, hazard ratio (HR) 0.44; P < 0.001 and 4-year OS 82% vs. 65%, HR 0.55; P = 0.02]. The rising number of effective and well-tolerated treatment options in recent years has led to a debate over early versus delayed ASCT, and two large intergroup studies have been initiated to examine this issue. In a review of 290 newly diagnosed MM patients treated at the Mayo Clinic with IMiD-based induction, there was no difference in time to progression or 4-year OS between the early or delayed ASCT groups [30].
In the USA, ASCT with high-dose melphalan may be performed safely in very fit patients >70 years old with advances in supportive care; therefore, age alone should not be the primary limiting factor in patient selection. Patients above age 65 years with good performance status who are not felt to be suitable for melphalan 200 mg/m2 may still benefit from reduced intensity autologous transplantation with melphalan 100 mg/m2. The IFM 99-06 trial-randomized patients aged 65–75 years to treatment with melphalan and prednisone (MP), melphalan and prednisone plus thalidomide (MPT), or VAD induction followed by ASCT with reduced intensity melphalan 100 mg/m2 (MEL100) [31]. Median OS was superior in the MPT group at 51.6 vs. 33.2 months for MP and 38.3 months for MEL100; however, the VAD induction arm may have contributed to inferior outcomes in the transplant arm. Reduced intensity ASCT was again evaluated using a more active induction regimen of bortezomib plus liposomal doxorubicin and dexamethasone (PAD) in patients aged 65–75 years followed by tandem MEL100 and lenalidomide plus prednisone consolidation. The median PFS was 48 months and 5-year OS was 63 %, demonstrating bortezomib-based induction followed by reduced ASCT may be an option in select elderly patients [32].
Significant variability exists in the choice of primary therapy for both transplant and non-transplant candidates, and head-to-head comparisons of the commonly used triplet regimens of bortezomib, lenalidomide, dexamethasone (VRd) and bortezomib, cyclophosphamide, dexamethasone (VCd or CyBorD) are lacking. Table 2 lists commonly used frontline regimens, their respective response rates, and available survival data for transplant-eligible and transplant-ineligible patients. Data pertaining to Rd and the triplet induction regimens of VTD, VCD, VRD, and KRD (carfilzomib, lenalidomide, dexamethasone) are further described below.
A decision regarding the frontline management of elderly patients not eligible for transplantation must balance adequate disease control while avoiding excess treatment-related toxicities. The VISTA trial, a phase III comparison of VMP to MP in elderly newly diagnosed MM patients, demonstrated a significant improvement in time to next treatment (31 vs. 21 months) and median OS (56 vs. 43 months) with the addition of bortezomib [33, 34]. Subsequent modifications to the VMP regimen have reduced treatment-related toxicities by moving to once weekly dosing and subcutaneous rather than intravenous administration of bortezomib. Bortezomib, thalidomide, and prednisone (VTP) were compared to VMP in an effort to reduce toxicities; however, higher rates of treatment discontinuation and serious adverse events occurred in the VTP group without improvement in efficacy [35]. The UPFRONT phase III trial compared bortezomib/dexamethasone (VD), bortezomib/dexamethasone plus thalidomide (VTD), and VMP in transplant-ineligible patients treated in the US community practice setting, and the triplet combinations of VTD and VMP did not offer a significant progression-free or OS benefit [36]. In fit elderly patients, the VCD and VRD regimens have been adopted based on phase II studies and are often substituted for VMP and VTD, respectively. In less fit elderly patients, less intensive therapy with doublet combinations (VD or Rd) and dose reductions are recommended. Continuous lenalidomide/dexamethasone (Rd) until progression was demonstrated to be superior in PFS and OS compared to fixed duration Rd for 18 cycles and MPT for 12 cycles and may be considered a new standard of care for newly diagnosed transplant-ineligible patients [37]. The routine use of maintenance therapy following fixed duration first-line treatment has not demonstrated an OS benefit consistently and is not recommended in standard practice outside of a clinical trial.
Lenalidomide/Dexamethasone (Rd)
Lenalidomide is an analog of thalidomide with more potent anti-MM activity and ability to stimulate T cell proliferation, interleukin (IL)-2, and interferon-γ without the problematic somnolence, constipation, and neuropathy that limited thalidomide [38]. The mechanism of IMiDs has been elucidated following the identification of the cereblon protein (CRBN) as the target of thalidomide [39]. IMiDs bind to CRBN and cause degradation of transcription factors Ikaros and Aiolos, resulting in downregulation of IRF4 and Myc and cytotoxicity to myeloma cells [40, 41]. Significant clinical activity was demonstrated in a phase II study of lenalidomide plus high-dose dexamethasone. An ORR of 91% was achieved with ≥VGPR of 32% and CR rate of 6% [42]. Subsequently, lenalidomide plus weekly dexamethasone (low dose) was compared to lenalidomide plus high-dose dexamethasone [43]. At the 1-year interim analysis, OS was 96% in the low-dose arm compared to 87% in the high-dose dexamethasone arm, and the trial was stopped early. Grade 3 or higher toxicities were more frequent with high-dose dexamethasone (DVT or PE in 26%, 16% grade 3 infection, 15% fatigue, 11% non-neuropathic weakness, 11% hyperglycemia). Lenalidomide with low-dose dexamethasone (Rd) has since become a standard initial therapy. Thromboprophylaxis with aspirin, warfarin, or low-molecular-weight heparin is essential for all patients treated with IMiDs to reduce the near 25% incidence of thrombotic complications in patients without prophylaxis [44].
Fixed duration therapy has been a mainstay for decades; however, this paradigm is also changing. The FIRST trial randomized transplant-ineligible patients to either lenalidomide/dexamethasone for 18 cycles, until disease progression, or MPT for 18 cycles [37]. Median PFS was superior with continuous Rd at 25.5 months (HR 0.72; P < 0.001) versus fixed duration Rd (PFS 20.7 months) or MPT (PFS 21.2 months). The safety profile of Rd was also more favorable than MPT with fewer grade ≥3 hematologic toxicities, significantly less neuropathy, and fewer second primary cancers.
Bortezomib, Thalidomide, and Dexamethasone (VTD)
Significantly higher response rates during induction therapy for untreated MM were achieved with the introduction of bortezomib/dexamethasone (VD) [45] and thalidomide/dexamethasone (TD) [28, 46] compared to VAD. The triplet combination of bortezomib, thalidomide, and dexamethasone (VTD) was the first regimen to utilize both an IMiD and a proteasome inhibitor. A GIMEMA phase III study of 474 patients compared VTD (bortezomib 1.3 mg/m2 IV on days 1, 4, 8, 11; thalidomide 200 mg daily; dexamethasone 320 mg per 21 day cycle) to TD [47]. Post-induction rates of VGPR or better were significantly higher with VTD compared to TD (62% vs. 28%; P < 0.0001), and PFS at 3 years was also improved (68% vs. 56%). The improved depth of response was offset by a higher incidence of grade ≥3 peripheral neuropathy in the VTD arm compared to TD (10% vs. 2%; P = 0.0004), and there was no difference in OS at 3 years (86% vs. 84%). The PETHEMA/GEM phase III study compared VTD to TD and alternating cycles of VBMCP/VBAD and demonstrated similar findings as patients treated with VTD achieved higher response rates compared to TD and VBMCP/VBAD (≥VGPR in 60% vs. 29% vs. 36%); however, VTD resulted in significantly higher rates of peripheral neuropathy [48]. In the VTD arm, treatment-emergent grade ≥3 peripheral neuropathy developed in 14% compared to 5% with TD, and 46% of patients in the VTD arm developed grade 2 peripheral neuropathy. Subsequently, Moreau et al. [49] compared reduced-dose bortezomib and thalidomide plus dexamethasone (vtD) to VD and demonstrated improved efficacy with use of vtD (≥VGPR 49% vs. 36%; P = 0.05) with lower rates of peripheral neuropathy (grade 1–4 53% vs. 70%; P = 0.01).
Cyclophosphamide, Bortezomib, and Dexamethasone (VCD or CyBorD)
Preclinical data suggest synergistic anti-MM activity with concurrent proteasome inhibition and chemotherapeutic agents such as doxorubicin and alkylating agents to induce DNA damage [50]. In a phase II trial of 33 patients with untreated MM, bortezomib 1.3 mg/m2 intravenously on days 1, 4, 8, and 11, cyclophosphamide 300 mg/m2 orally once weekly, and high-dose dexamethasone were given each 28 day cycle. After four cycles, patients who achieved an ORR of 88% and 61% had a VGPR or better. A second cohort of 30 patients was treated with a modified schedule using the same cyclophosphamide dose, weekly bortezomib 1.5 mg/m2, and weekly dexamethasone 40 mg [51]. The modified CyBorD regimen had fewer grade 3/4 adverse events (37%/3% vs. 48%/12%) and fewer required dose reductions of bortezomib and dexamethasone. Subcutaneous bortezomib administration has also replaced intravenous infusion because of the reduced incidence of peripheral neuropathy with this route of administration [52].
In the EVOLUTION study, the efficacy and tolerability of combining bortezomib, cyclophosphamide, lenalidomide, and dexamethasone (VDCR) was studied in comparison to VDR (bortezomib, dexamethasone, lenalidomide) and VDC in two separate dosing schemes [53]. VDCR demonstrated similar activity to triplet combinations with a VGPR or better rate of 58% and CR rate of 25%; however, grade 3 hematologic toxicity was significantly higher. VDR and VCD were recommended for further comparative testing.
The VCd combination does not interfere with stem cell harvest and is an effective pretransplant induction regimen. A retrospective comparison of VCD and VTD as pretransplant induction therapy reported VTD may achieve higher rates of ≥VGPR and CR and be better tolerated compared to VCD; however, questions about the methodology of the study have been raised [54, 55]. The ongoing IFM 2013-04 randomized study is comparing VTD versus VCD for four cycles prior to ASCT in order to compare post-induction response rates and safety of the two commonly used regimens.
Bortezomib, Lenalidomide, and Dexamethasone (VRD)
Bortezomib, lenalidomide, and dexamethasone (VRD) were combined with the aim of achieving the higher rates of response observed with VTD while improving upon the safety profile. In a phase I/II study, the regimen of bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11; lenalidomide 25 mg on days 1–14; and dexamethasone 20 mg days 1, 2, 4, 5, 8, 9, 11, and 12 per 21 day cycle was selected for the phase II component [56]. In this cohort of 35 patients, 37% had a CR, 74% had ≥VGPR, and all patients had an objective response. The most common adverse events were peripheral neuropathy in 80% (2% grade 3), fatigue (64%), constipation (61%), myalgia (44%), rash (36%), diarrhea (35%), nausea (32%), neuropathic pain (32%), and insomnia (30%).
The combination of an IMiD and a proteasome inhibitor leads a greater depth of response compared to historic regimens, but at a higher rate of toxicity and cost. Prospective data on whether achieving the surrogate endpoints of CR or ≥VGPR result in improved OS are lacking; however, retrospective studies have attempted to answer this question. In transplant-ineligible patients treated with MP, MPT, VMP, or VMPT, PFS at 3 years was 67% in those who achieved a CR versus 27% in those without a CR [57]. Three-year OS was 91% in the group that obtained a CR versus 67% in those with a VGPR or PR.
Carfilzomib, Lenalidomide, and Dexamethasone (KRd)
Newer frontline regimens substituting carfilzomib for bortezomib have also been studied. Carfilzomib is a second-generation proteasome inhibitor with distinct chemical properties from bortezomib with highly chymotrypsin-specific irreversible proteasome inhibition [58]. Twice-weekly carfilzomib (36 mg/m2 based on dose escalation) was combined with lenalidomide 25 mg days 1–21 plus weekly dexamethasone at 40 mg in a phase I/II study of newly diagnosed MM patients [59]. At the 36 mg/m2 dose level, 39% achieved stringent CR, 55% near CR, 72% VGPR or better, and 97% of patients had an objective response. The incidence of peripheral neuropathy compared to VRD was lower at 23% and was limited to grade 1 (17%) and grade 2 (6%). Treatment-related non-hematologic adverse events were generally mild to moderate and included hyperglycemia in 72% (23% grade 3/4), edema (47%), hypophosphatemia (45%), fatigue (38%), muscle cramping (32%), rash (28%), elevated liver function tests (28%), diarrhea (26%), dyspnea (15%), DVT (11%), pulmonary embolism (6%), and nausea (13%). Grade 3/4 thrombocytopenia, anemia, and neutropenia developed in approximately 20% of patients. The ECOG E1A11 ENDURANCE study is currently enrolling patients to compare the efficacy and safety of KRd to VRD and will also analyze the role of limited versus indefinite lenalidomide maintenance following induction therapy.
Treatment of Relapsed and Relapsed/Refractory MM
Despite highly active combination regimens with novel agents, high-dose melphalan with stem cell transplant and an increasing use of consolidation and maintenance therapy, MM remains an incurable disease in the vast majority of patients, and subsequent lines of therapy are required to address disease relapse. The treatment of patients with dual-refractory disease to lenalidomide and bortezomib remains challenging, as median survival was 9 months in this population prior to the introduction of carfilzomib and pomalidomide [60]. Given the abundance of available treatment options for relapsed disease, patient factors (performance status, renal function, neuropathy), tumor biology (risk status, pace of relapse, presence of extramedullary disease or plasma cell leukemia), and treatment-related factors (duration of response to prior therapies, exposure to major classes of therapies including IMiDs, proteasome inhibitors, and alkylating agents) need to be considered before selecting a salvage therapy [61]. Close observation without treatment is reasonable in the case of indolent biochemical progression, whereas patients with rapid relapse and evidence of end-organ dysfunction benefit from combination therapy utilizing multiple novel agents. Combination therapy utilizing multiple mechanisms of action to overcome drug resistance and clonal heterogeneity is being increasingly utilized. The duration of response to each subsequent line of therapy decreases consistently [62]; therefore, retreatment with a prior regimen may be considered if remission off therapy has lasted more than 6 months. This approach is supported by data from the VISTA [63] and RETRIEVE [64] studies, which demonstrated ORR in 40–60% of patients upon retreatment with bortezomib or lenalidomide. Transplant-eligible patients who delayed ASCT initially are recommended to undergo ASCT upon first relapse. Relapse following initial treatment with an IMiD based regimen (Rd or VRd) may be treated with VCd, KRd, or pomalidomide/dexamethasone. Several promising combination regimens using next generation novel agents are undergoing clinical testing, and available outcome data using these regimens are listed in Table 3.
Next Generation Proteasome Inhibitors
The proteasome is a key structure within the nucleus and cytoplasm of eukaryotic cells that degrades ubiquinated proteins and is involved in cell homeostasis [65]. The ubiquitin-proteasome pathway is a rational therapeutic target as proteasome inhibition results in accumulation of misfolded proteins, endoplasmic reticulum stress, and induction of apoptosis [66]. Bortezomib was the first compound to attain regulatory approval and was initially used as a single-agent in relapsed/refractory disease but now is a fundamental component of several combination therapies. Bortezomib is a dipeptide boronic acid and reversible inhibitor of the 20S proteasome primarily at the chymotrypsin-like and caspase-like active sites [67]. Following the success of bortezomib, several other compounds with unique chemical characteristics have entered clinical testing in an attempt to overcome bortezomib resistance as well as to improve the safety profile of proteasome inhibition.
Carfilzomib has several unique chemical properties compared to bortezomib. Carfilzomib is a peptide epoxyketyone that is an irreversible, highly selective inhibitor of chymotrypsin-like activity with fewer off-target effects on non-proteasomal targets and significantly less neurotoxicity compared to bortezomib [58]. In the PX-171-004 phase II study, relapsed/refractory bortezomib-naive patients received either carfilzomib 20 mg/m2 for 12 cycles or 20 mg/m2 in cycle 1 then 27 mg/m2 in cycles 2–12 [68]. The ORR was higher in the 20/27-mg/m2 group compared to the 20-mg/m2 group (52.2% vs. 42.4%), and the incidence of treatment-emergent peripheral neuropathy was low (17.1% overall) with no treatment discontinuations due to neuropathy. Patients with heavily pretreated MM including bortezomib-treatment were enrolled in the PX-171-003-A1 phase II study of single-agent carfilzomib and were treated with 20 mg/m2 twice weekly 3 weeks out of 4 during cycle 1 then 27 mg/m2 thereafter. In this study, the ORR was 23.7% with a median PFS of 3.7 months and median duration of response of 7.8 months [69]. Based on the encouraging single-agent activity and favorable toxicity profile, carfilzomib has been incorporated into several combination regimens. Carfilzomib, lenalidomide, and dexamethasone (KRd) were compared to Rd in the ASPIRE study and demonstrated a significant improvement in the PFS (26.3 vs. 17.6 months) and CR rate (31.8 % vs. 9.3 %). KRd compares favorably in both efficacy and tolerability to VTD (median PFS 19.5 months; 29 % rate of grade ≥3 peripheral neuropathy) [70]. In a phase II study of relapsed/refractory MM patients treated with VRd, median PFS was 9.5 months with a CR rate of 11 %. The incidence of treatment-related peripheral neuropathy was higher at 53 % with VRd compared to 17 % in the KRd ASPIRE study.
Ixazomib (MLN9708) is a second-generation, orally bioavailable 20S proteasome inhibitor with improved tissue penetration and antitumor activity [71]. Ixazomib has significant single-agent activity even with prior bortezomib and carfilzomib exposure, and an ORR of 27 % was seen in patients treated at the maximum tolerated dose [72]. The phase III TOURMALINE-MM1 study compared ixazomib 4 mg PO on days 1, 8, and 15 plus lenalidomide and dexamethasone to Rd in patients with relapsed/refractory MM. The primary endpoint of improvement in PFS was met at a prespecified interim analysis, but the data have not yet been disclosed. The phase I/II trial of ixazomib, lenalidomide, and dexamethasone in untreated MM demonstrated an ORR of 92 % with ≥VGPR in 53 % of patients [73]. Primary treatment-related toxicities were generally mild and most commonly were fatigue, rash, nausea, vomiting, diarrhea, and thrombocytopenia. Ixazomib is the first oral proteasome inhibitor to enter phase III testing but additional third generation proteasome inhibitors including marizomib and oprozomib have also entered clinical testing.
Pomalidomide
Pomalidomide is the most potent IMiD and demonstrates significant activity even in lenalidomide-refractory patients. Similar to lenalidomide and thalidomide, pomalidomide binds to CRBN, and a potential mechanism of resistance is downregulation of the target. CRBN expression may be a potential predictive biomarker for the selection of patients most likely to respond to pomalidomide, as pretreatment CRBN levels correspond to the response to pomalidomide [74]. Pomalidomide has broad immunomodulatory effects including T-cell stimulation, inhibition of T regulatory cells, NK cell activation, enhancement of antibody-dependent cell-mediated cytotoxicity (ADCC), and osteoclast inhibition [75].
Thirty-eight relapsed/refractory patients were initially treated at a continuous dose of 2 mg daily with weekly low-dose dexamethasone, 63% achieved an objective response, and a median PFS of 11.6 months was reported. Grade 3/4 myelotoxicity was the primary treatment-related toxicity [76]. The randomized IFM 2009-02 [77] and MM-003 [78] studies both demonstrated the efficacy of pomalidomide 4 mg on days 1–21 plus dexamethasone 40 mg weekly, although the median PFS of 4–5 months was less than the initial reports. Pomalidomide was granted accelerated approval in 2013 for patients who have received at least two prior therapies with disease progression on or within 60 days of the previous therapy. The side effect profile is similar to other IMiDs with a low rate of sensory neuropathy (10 % all grades) however grade 3/4 neutropenia was observed in 47 % of patients. The safety as well as optimal dosing of pomalidomide in combination with carfilzomib and dexamethasone in patients with relapsed/refractory MM is a current area of investigation (NCT01665794).
Panobinostat
A number of histone deacetylase inhibitors (HDACi) have exhibited synergistic cytotoxicity with bortezomib and may overcome resistance to proteasome inhibition through HDACi-induced blockage of the aggresome pathway, which is a complementary pathway of intracellular protein degradation [79]. Single-agent vorinostat and romidepsin achieve very few objective responses; however, phase I/II studies in combination with bortezomib demonstrated ORR of 20–40 % [80]. The VANTAGE-008 study was a randomized, phase III study of bortezomib with or without vorinostat, which demonstrated no clinically meaningful improvement in median PFS (7.6 vs. 6.8 months; P = 0.01) but an increased rate of nausea, vomiting, and fatigue in the vorinostat group [81]. San-Miguel et al. [100] compared bortezomib, dexamethasone with or without panobinostat in a randomized phase III trial of 768 relapsed/refractory MM patients (PANORAMA-1). A 4-month improvement in median PFS was seen in the panobinostat group (12 vs. 8.1 months; HR 0.63; P < 0.0001) as well as an increased rate of CR/nCR (28% vs. 16%; P = 0.0001), although the ORR was the same (61% vs. 55%; P = 0.09). The rate of grade 3/4 treatment-related adverse events was higher in the panobinostat arm, including thrombocytopenia (67% vs. 31%), diarrhea (26% vs. 8%), and fatigue (24% vs. 12%). Panobinostat was approved in 2015 in combination with bortezomib and dexamethasone and is the first HDACi approved for the treatment of MM. HDAC6 selectivity may enhance the potency and reduce off-target effects compared to non-selective pan-HDAC inhibitors (vorinostat, panobinostat) [82]. Ricolinostat (ACY-1215), a selective HDAC6 inhibitor, is now in clinical testing in several combinations with other novel agents including pomalidomide (NCT01997840) and bortezomib (NCT01323751).
Monoclonal Antibodies
The next major breakthrough in the therapy of MM is likely to be the effective incorporation of monoclonal antibodies. Multiple novel targets have been identified, and the three agents in phase III testing include elotuzumab (antiCS-1/SLAM7) and the anti-CD38 antibodies daratumumab and SAR650984. Elotuzumab was tested in combination with bortezomib in a phase I dose-escalation study and achieved an ORR of 48% and median PFS of 9.5 months with no dose-limiting toxicities [83]. Elotuzumab in combination with lenalidomide and dexamethasone demonstrated a significant increase in activity (ORR 82%, ≥VGPR 29%), suspected to be due to a synergistic induction of ADCC-mediated apoptosis [84]. In the recent phase III study of Rd with or without elotuzumab (ELOQUENT-2), median PFS was 19.4 vs. 14.9 months in the control group (HR 0.70; P < 0.001), and ORR improved to 79% in the elotuzumab arm versus 66% with Rd (P < 0.001) [85].
Daratumumab demonstrated single-agent dose-dependent efficacy in a phase I dose-escalation study with the most common adverse event of mild infusion-related events, especially during the initial treatment [86]. ORR was 46% in patients treated at the 16 mg/kg dose level, and no dose-related increase in adverse effects was observed. In preclinical studies, lenalidomide enhances the anti-MM activity of daratumumab [87]. Daratumumab 16 mg/kg plus lenalidomide and dexamethasone in an expansion cohort achieved an ORR of 86.7%, and in patients treated with ≥6 cycles, VGPR or better was seen in 64.7% with CR in 11.8% [88]. Most infusion-related reactions (86%) occurred with the first infusion, and 18/19 patients were able to continue the subsequent infusion.
SAR650984 plus lenalidomide and dexamethasone was evaluated in a phase Ib trial including heavily pretreated patients with a median of seven prior therapies (including pomalidomide and carfilzomib), and an ORR of 63% was achieved with a median PFS of 6.2 months. No dose-limiting toxicities were observed, and the most common treatment-related adverse effects were fatigue (42%), nausea (39%), upper respiratory tract infection (39%), diarrhea (36%), and infusion-related reactions in 39% (predominantly cycle 1). The dose of 10 mg/kg every 2 weeks was selected for an expansion cohort [89].
Siltuximab is a chimeric monoclonal antibody against IL-6 and has been evaluated as an adjunct to novel agents in the treatment of MM. In a phase II randomized study of VMP or VMP plus siltuximab followed by siltuximab maintenance in newly diagnosed MM patients, median PFS and 1-year OS were identical in both arms (17 months and 88%, respectively), while more hematologic events and infections occurred in the siltuximab group [90]. In the relapsed setting, the addition of siltuximab to bortezomib did not significantly improve the response rate or PFS [91].
Future Directions
Chimeric antigen T-cell therapy (CAR-T) with CTL019, an anti-CD19 CAR transduced via lentiviral vector, is a novel immunotherapeutic approach currently in pilot studies in advanced myeloma and holds promise as a salvage therapy in MM. Further efforts to stimulate immunity against MM cells has led to the development of a dendritic cell-myeloma fusion cell vaccine, which has been combined with anti-PD-1 antibodies to stimulate antitumor immunity in the post-ASCT setting and is currently under investigation (NCT01067287) [92]. Peptide vaccines targeting NY-ESO-1 and MAGE-A3 are also in development. Cell cycle inhibitors including dinaciclib, an oral cyclin-dependent kinase inhibitor, and filanesib (ARRY-520), a kinesin spindle protein inhibitor that arrests mitosis, both have demonstrated anti-MM activity in combination with bortezomib and dexamethasone and are being evaluated in relapsed disease. Targeting MYC dysregulation is being explored via CPI-0610, a selective inhibitor of bromodoman and extra-terminal protein (BET), which is currently in phase I testing (NCT02157636). Multiple other small-molecule inhibitors of deubiquitylating enzymes (DUBs), the PI3 K/Akt/mTOR pathway, aurora kinase (MLN8237), and Hedgehog pathway signaling are undergoing investigation.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29.
Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412–7.
Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–20.
Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–48.
Katzmann JA, Dispenzieri A, Kyle RA, et al. Elimination of the need for urine studies in the screening algorithm for monoclonal gammopathies by using serum immunofixation and free light chain assays. Mayo Clin Proc Mayo Clin. 2006;81(12):1575–8.
Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215–24.
Paiva B, Almeida J, Perez-Andres M, et al. Utility of flow cytometry immunophenotyping in multiple myeloma and other clonal plasma cell-related disorders. Cytom B Clin Cytom. 2010;78(4):239–52.
Kumar S, Kimlinger T, Morice W. Immunophenotyping in multiple myeloma and related plasma cell disorders. Best Pract Res Clin Haematol. 2010;23(3):433–51.
Paiva B, Vidriales MB, Perez JJ, et al. Multiparameter flow cytometry quantification of bone marrow plasma cells at diagnosis provides more prognostic information than morphological assessment in myeloma patients. Haematologica. 2009;94(11):1599–602.
Gonsalves WI, Rajkumar SV, Gupta V, et al. Quantification of clonal circulating plasma cells in newly diagnosed multiple myeloma: implications for redefining high-risk myeloma. Leukemia. 2014;28(10):2060–5.
Paiva B, Vidriales MB, Montalban MA, et al. Multiparameter flow cytometry evaluation of plasma cell DNA content and proliferation in 595 transplant-eligible patients with myeloma included in the Spanish GEM2000 and GEM2005 < 65y trials. Am J Pathol. 2012;181(5):1870–8.
Dimopoulos M, Terpos E, Comenzo RL, et al. International myeloma working group consensus statement and guidelines regarding the current role of imaging techniques in the diagnosis and monitoring of multiple Myeloma. Leukemia. 2009;23(9):1545–56.
Dimopoulos MA, Hillengass J, Usmani S, et al. Role of magnetic resonance imaging in the management of patients with multiple myeloma: a consensus statement. J Clin Oncol. 2015;33(6):657–64.
Kim PJ, Hicks RJ, Wirth A, et al. Impact of 18F-fluorodeoxyglucose positron emission tomography before and after definitive radiation therapy in patients with apparently solitary plasmacytoma. Int J Radiat Oncol Biol Phys. 2009;74(3):740–6.
Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28(5):1122–8.
Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23(15):3412–20.
Kastritis E, Zervas K, Symeonidis A, et al. Improved survival of patients with multiple myeloma after the introduction of novel agents and the applicability of the International Staging System (ISS): an analysis of the Greek Myeloma Study Group (GMSG). Leukemia. 2009;23(6):1152–7.
Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23(12):2210–21.
Avet-Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109(8):3489–95.
Kumar S, Fonseca R, Ketterling RP, et al. Trisomies in multiple myeloma: impact on survival in patients with high-risk cytogenetics. Blood. 2012;119(9):2100–5.
Mikhael JR, Dingli D, Roy V, et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus guidelines 2013. Mayo Clin Proc. 2013;88(4):360–76.
Rajkumar SV. Treatment of multiple myeloma. Nat Rev Clin Oncol. 2011;8(8):479–91.
Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91–101.
Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997.
Attal M, Harousseau JL, Stoppa AM, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med. 1996;335(2):91–7.
Child JA, Morgan GJ, Davies FE, et al. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. N Engl J Med. 2003;348(19):1875–83.
Sonneveld P, Schmidt-Wolf IG, van der Holt B, et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/GMMG-HD4 trial. J Clin Oncol. 2012;30(24):2946–55.
Cavo M, Zamagni E, Tosi P, et al. Superiority of thalidomide and dexamethasone over vincristine-doxorubicindexamethasone (VAD) as primary therapy in preparation for autologous transplantation for multiple myeloma. Blood. 2005;106(1):35–9.
Palumbo A, Cavallo F, Gay F, et al. Autologous transplantation and maintenance therapy in multiple myeloma. N Engl J Med. 2014;371(10):895–905.
Kumar SK, Lacy MQ, Dispenzieri A, et al. Early versus delayed autologous transplantation after immunomodulatory agents-based induction therapy in patients with newly diagnosed multiple myeloma. Cancer. 2012;118(6):1585–92.
Facon T, Mary JY, Hulin C, et al. Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99-06): a randomised trial. Lancet. 2007;370(9594):1209–18.
Gay F, Magarotto V, Crippa C, et al. Bortezomib induction, reduced-intensity transplantation, and lenalidomide consolidation-maintenance for myeloma: updated results. Blood. 2013;122(8):1376–83.
Harousseau JL, Palumbo A, Richardson PG, et al. Superior outcomes associated with complete response in newly diagnosed multiple myeloma patients treated with nonintensive therapy: analysis of the phase 3 VISTA study of bortezomib plus melphalan–prednisone versus melphalan–prednisone. Blood. 2010;116(19):3743–50.
San Miguel JF, Schlag R, Khuageva NK, et al. Persistent overall survival benefit and no increased risk of second malignancies with bortezomib–melphalan–prednisone versus melphalan–prednisone in patients with previously untreated multiple myeloma. J Clin Oncol. 2013;31(4):448–55.
Mateos MV, Oriol A, Martinez-Lopez J, et al. Bortezomib, melphalan, and prednisone versus bortezomib, thalidomide, and prednisone as induction therapy followed by maintenance treatment with bortezomib and thalidomide versus bortezomib and prednisone in elderly patients with untreated multiple myeloma: a randomised trial. Lancet Oncol. 2010;11(10):934–41.
Niesvizky R, Flinn IW, Rifkin R, et al. Community-based phase IIIB trial of three UPFRONT bortezomib-based myeloma regimens. J Clin Oncol. 2015. doi:10.1200/JCO.2014.58.7618.
Benboubker L, Dimopoulos MA, Dispenzieri A, et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371(10):906–17.
Richardson PG, Schlossman RL, Weller E, et al. Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood. 2002;100(9):3063–7.
Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–50.
Zhu YX, Braggio E, Shi CX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118(18):4771–9.
Lu G, Middleton RE, Sun H, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–9.
Rajkumar SV, Hayman SR, Lacy MQ, et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood. 2005;106(13):4050–3.
Rajkumar SV, Jacobus S, Callander NS, et al. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open-label randomised controlled trial. Lancet Oncol. 2010;11(1):29–37.
Larocca A, Cavallo F, Bringhen S, et al. Aspirin or enoxaparin thromboprophylaxis for patients with newly diagnosed multiple myeloma treated with lenalidomide. Blood. 2012;119(4):933–9 (quiz 1093).
Harousseau JL, Attal M, Avet-Loiseau H, et al. Bortezomib plus dexamethasone is superior to vincristine plus doxorubicin plus dexamethasone as induction treatment prior to autologous stem-cell transplantation in newly diagnosed multiple myeloma: results of the IFM 2005-01 phase III trial. J Clin Oncol. 2010;28(30):4621–9.
Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2006;24(3):431–6.
Cavo M, Tacchetti P, Patriarca F, et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet. 2010;376(9758):2075–85.
Rosinol L, Oriol A, Teruel AI, et al. Superiority of bortezomib, thalidomide, and dexamethasone (VTD) as induction pretransplantation therapy in multiple myeloma: a randomized phase 3 PETHEMA/GEM study. Blood. 2012;120(8):1589–96.
Moreau P, Avet-Loiseau H, Facon T, et al. Bortezomib plus dexamethasone versus reduced-dose bortezomib, thalidomide plus dexamethasone as induction treatment before autologous stem cell transplantation in newly diagnosed multiple myeloma. Blood. 2011;118(22):5752–8.
Mitsiades N, Mitsiades CS, Richardson PG, et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101(6):2377–80.
Reeder CB, Reece DE, Kukreti V, et al. Once- versus twice-weekly bortezomib induction therapy with CyBorD in newly diagnosed multiple myeloma. Blood. 2010;115(16):3416–7.
Moreau P, Pylypenko H, Grosicki S, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011;12(5):431–40.
Kumar S, Flinn I, Richardson PG, et al. Randomized, multicenter, phase 2 study (EVOLUTION) of combinations of bortezomib, dexamethasone, cyclophosphamide, and lenalidomide in previously untreated multiple myeloma. Blood. 2012;119(19):4375–82.
Leiba M, Kedmi M, Duek A, et al. Bortezomib–cyclophosphamide–dexamethasone (VCD) versus bortezomib–thalidomide–dexamethasone (VTD) -based regimens as induction therapies in newly diagnosed transplant eligible patients with multiple myeloma: a meta-analysis. Br J Haematol. 2014;166(5):702–10.
Moreau P, Mary JY, Attal M. Bortezomib–thalidomide–dexamethasone versus bortezomib–cyclophosphamide–dexamethasone as induction therapy prior to autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2015;168(4):605–6.
Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010;116(5):679–86.
Gay F, Larocca A, Wijermans P, et al. Complete response correlates with long-term progression-free and overall survival in elderly myeloma treated with novel agents: analysis of 1175 patients. Blood. 2011;117(11):3025–31.
Kuhn DJ, Chen Q, Voorhees PM, et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007;110(9):3281–90.
Jakubowiak AJ, Dytfeld D, Griffith KA, et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood. 2012;120(9):1801–9.
Kumar SK, Lee JH, Lahuerta JJ, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia. 2012;26(1):149–57.
Mikhael JR. A practical approach to relapsed multiple myeloma. Hematol Am Soc Hematol Educ Progr. 2014;2014(1):262–7.
Kumar SK, Therneau TM, Gertz MA, et al. Clinical course of patients with relapsed multiple myeloma. Mayo Clin Proc. 2004;79(7):867–74.
Mateos MV, Richardson PG, Schlag R, et al. Bortezomib plus melphalan and prednisone compared with melphalan and prednisone in previously untreated multiple myeloma: updated follow-up and impact of subsequent therapy in the phase III VISTA trial. J Clin Oncol. 2010;28(13):2259–66.
Petrucci MT, Giraldo P, Corradini P, et al. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. Br J Haematol. 2013;160(5):649–59.
Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79(1):13–21.
Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–16.
Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4(5):349–60.
Vij R, Wang M, Kaufman JL, et al. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood. 2012;119(24):5661–70.
Siegel DS, Martin T, Wang M, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120(14):2817–25.
Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372(2):142–52.
Kupperman E, Lee EC, Cao Y, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70(5):1970–80.
Kumar SK, Bensinger WI, Zimmerman TM, et al. Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood. 2014;124(7):1047–55.
Kumar SK, Berdeja JG, Niesvizky R, et al. A phase 1/2 study of weekly MLN9708, an investigational oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma. ASH Annu Meet Abstr. 2012;120:332.
Schuster SR, Kortuem KM, Zhu YX, Braggio E, Shi C-X, Bruins L. Cereblon expression predicts response, progression free and overall survival after pomalidomide and dexamethasone therapy in multiple myeloma. ASH Annu Meet Abstr. 2012;120:194.
Chanan-Khan AA, Swaika A, Paulus A, et al. Pomalidomide: the new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J. 2013;3:e143.
Lacy MQ, Hayman SR, Gertz MA, et al. Pomalidomide (CC4047) plus low-dose dexamethasone as therapy for relapsed multiple myeloma. J Clin Oncol. 2009;27(30):5008–14.
Leleu X, Attal M, Arnulf B, et al. Pomalidomide plus low-dose dexamethasone is active and well tolerated in bortezomib and lenalidomide-refractory multiple myeloma: intergroupe Francophone du Myelome 2009-02. Blood. 2013;121(11):1968–75.
San Miguel J, Weisel K, Moreau P, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(11):1055–66.
Hideshima T, Bradner JE, Chauhan D, Anderson KC. Intracellular protein degradation and its therapeutic implications. Clin Cancer Res. 2005;11(24 Pt 1):8530–3.
Siegel DS, Richardson P, Dimopoulos M, et al. Vorinostat in combination with lenalidomide and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood Cancer J. 2014;4:e182.
Dimopoulos M, Siegel DS, Lonial S, et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol. 2013;14(11):1129–40.
Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119(11):2579–89.
Jakubowiak AJ, Benson DM, Bensinger W, et al. Phase I trial of anti-CS1 monoclonal antibody elotuzumab in combination with bortezomib in the treatment of relapsed/refractory multiple myeloma. J Clin Oncol. 2012;30(16):1960–5.
Lonial S, Vij R, Harousseau JL, et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. J Clin Oncol. 2012;30(16):1953–9.
Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015.
Lokhorst H, Laubach J, Nahi H, et al. Dose-dependent efficacy of daratumumab (DARA) as monotherapy in patients with relapsed or refractory multiple myeloma (RR MM). J Clin Oncol. 2014;35(5):8513.
van der Veer MS, de Weers M, van Kessel B, et al. Towards effective immunotherapy of myeloma: enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica. 2011;96(2):284–90.
Plesner T, Arkenau HT, Lokhorst H, et al. Safety and efficacy of daratumumab with lenalidomide and dexamethasone in relapsed or relapsed, refractory multiple myeloma. ASH Annu Meet Abstr. 2014;124:84.
Martin TG III, Baz R, Benson DM Jr, et al. A phase Ib dose escalation trial of SAR650984 (Anti-CD-38 mAb) in combination with lenalidomide and dexamethasone in relapsed/refractory multiple myeloma. ASH Annu Meet Abstr. 2014;124:83.
San-Miguel J, Blade J, Shpilberg O, et al. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti-IL-6) in multiple myeloma. Blood. 2014;123(26):4136–42.
Orlowski RZ, Gercheva L, Williams C, et al. A phase 2, randomized, double-blind, placebo-controlled study of siltuximab (anti-IL-6 mAb) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. Am J Hematol. 2015;90(1):42–9.
Rosenblatt J, Vasir B, Uhl L, et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood. 2011;117(2):393–402.
Reeder CB, Reece DE, Kukreti V, et al. Cyclophosphamide, bortezomib and dexamethasone induction for newly diagnosed multiple myeloma: high response rates in a phase II clinical trial. Leukemia. 2009;23(7):1337–41.
Jakubowiak AJ, Kendall T, Al-Zoubi A, et al. Phase II trial of combination therapy with bortezomib, pegylated liposomal doxorubicin, and dexamethasone in patients with newly diagnosed myeloma. J Clin Oncol. 2009;27(30):5015–22.
Palumbo A, Hajek R, Delforge M, et al. Continuous lenalidomide treatment for newly diagnosed multiple myeloma. N Engl J Med. 2012;366(19):1759–69.
San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906–17.
Palumbo A, Bringhen S, Rossi D, et al. Bortezomib–melphalan–prednisone–thalidomide followed by maintenance with bortezomib–thalidomide compared with bortezomib–melphalan–prednisone for initial treatment of multiple myeloma: a randomized controlled trial. J Clin Oncol. 2010;28(34):5101–9.
Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–98.
Dimopoulos MA, Orlowski RZ, Facon T, et al. Retrospective matched-pairs analysis of bortezomib plus dexamethasone versus bortezomib monotherapy in relapsed multiple myeloma. Haematologica. 2015;100(1):100–6.
San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15(11):1195–206.
Ludwig H, Kasparu H, Leitgeb C, et al. Bendamustine-bortezomib-dexamethasone is an active and well-tolerated regimen in patients with relapsed or refractory multiple myeloma. Blood. 2014;123(7):985–91.
Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357(21):2133–42.
Reece DE, Masih-Khan E, Atenafu EG, et al. Phase I-II trial of oral cyclophosphamide, prednisone and lenalidomide for the treatment of patients with relapsed and refractory multiple myeloma. Br J Haematol. 2015;168(1):46–54.
Lazaryan A, Hussein MA, Reu FJ, et al. Mature results of MM-011: a phase I/II trial of liposomal doxorubicin, vincristine, dexamethasone, and lenalidomide combination therapy followed by lenalidomide maintenance for relapsed/refractory multiple myeloma. Am J Hematol. 2014;89(4):349–54.
Vij R, Siegel DS, Jagannath S, et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol. 2012;158(6):739–48.
Papadopoulos KP, Siegel DS, Vesole DH, et al. Phase I study of 30-minute infusion of carfilzomib as single agent or in combination with low-dose dexamethasone in patients with relapsed and/or refractory multiple myeloma. J Clin Oncol. 2015;33(7):732–9.
Berenson J, Hilger JD, Klein L, et al. Pomalidomide, dexamethasone and pegylated liposomal doxorubicin for patients with relapsed/refractory multiple myeloma: results from a phase 1/2 trial. ASH Annu Meet Abstr. 2013;122(21):3218.
Garderet L, Iacobelli S, Moreau P, et al. Superiority of the triple combination of bortezomib–thalidomide–dexamethasone over the dual combination of thalidomide-dexamethasone in patients with multiple myeloma progressing or relapsing after autologous transplantation: the MMVAR/IFM 2005-04 randomized phase III trial from the chronic leukemia working party of the European group for blood and marrow transplantation. J Clin Oncol. 2012;30(20):2475–82.
Richardson PG, Xie W, Jagannath S, et al. A phase 2 trial of lenalidomide, bortezomib, and dexamethasone in patients with relapsed and relapsed/refractory myeloma. Blood. 2014;123(10):1461–9.
Mikhael J, Roy V, Richardson PG, et al. A phase I/II trial of pomalidomide, bortezomib and dexamethasone in patients with relpased or refractory multiple myeloma. ASH Annu Meet Abstr. 2013;122(21):1940.
Rosenbaum CA, Kukreti V, Zonder J, et al. Phase 1b/2 study of carfilzomib, pomalidomide, and dexamethasone (KPd) in patients (Pts) with lenalidomide-exposed and/or -refractory but proteasome inhibitor (pi)-naive or -sensitive multiple myeloma: a multiple myeloma research consortium multi-center study. ASH Annu Meet Abstr. 2014;124(21):2109.
Acknowledgments
No funding or sponsorship was received for publication of this article. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Conflict of Interest
JTL declares no conflicts of interest. SKK has received research funding from Millennium, Celgene, The Takeda Oncology Co., Onyx, Novartis, Sanofi, and Janssen Oncology. SKK has acted as a consultant with fees paid to the institution from Celgene, Millennium, Onyx, The Takeda Oncology Co., Sanofi, and Janssen Oncology.
Compliance with Ethics Guidelines
This review is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Larsen, J.T., Kumar, S. Evolving Paradigms in the Management of Multiple Myeloma: Novel Agents and Targeted Therapies. Rare Cancers Ther 3, 47–68 (2015). https://doi.org/10.1007/s40487-015-0009-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40487-015-0009-4