
Abstract
Autism is a complex disease with many genetic factors contributing to the variable phenotype presentation. Genome-wide association studies (GWAS) using large cohorts and dense datasets have revealed a myriad of genes implicated in autism. Several of these genes belong to common biological pathways and gene networks that converge on potential therapeutic targets. In this regard, CACNA, GRM, CNTN, ubiquitin, and SLIT gene families have shown promising variants in association with autism and related neurodevelopmental disorders. Both common and rare variants reside on these genes discovered primarily by single nucleotide polymorphism (SNP) arrays and whole exome sequencing. The ability to examine large cohorts through unbiased screening and statistical testing of allele frequencies between cases, families, and controls through GWAS will continue to elucidate and clarify the puzzle of autism and inform clinical care.
Similar content being viewed by others


Introduction
Autism is characterized by impaired social interaction and communication, and by restricted/repetitive behavior typically presenting in early development. Autism affects neural development and information processing in the brain by altering how nerve cells and synapses connect and organize. Per the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-V), the autism spectrum encompasses previous diagnoses of autistic disorder, Asperger syndrome, pervasive developmental disorder not otherwise specified (PDD-NOS), and childhood disintegrative disorder. Autism has a strong genetic component based on very high heritability in families, although the genetics of autism are complex. It is unclear whether autism spectrum disorders (ASDs) are explained more by rare mutations, or by rare combinations of common genetic variants. Debate remains about the genetic architecture of autism, specifically the extent to which common variant–common disease and rare variant–common disease models describe the disorder [1].
Epidemiology
Autism is approximately four times more common in males than in females [2–4]. Additionally, there is a twofold increase in risk if an older sibling is affected by autism [5]. The prevalence increased from 0.05 % of individuals throughout the 1980s to 2 % of school-age children in the USA [6], in part due to a change in the practices of diagnosis and ascertainment. Evidence that autism concordance in monozygotic twins approaches 92 %, in contrast to 10 % in dizygotic twins, underlines a strong genetic component [7–9].
Treatments
Antidepressants, stimulants, and antipsychotics are commonly prescribed, but no known medication relieves autism's core symptoms of social and communication impairments. However, aripiprazole and risperidone are effective for treating irritability in children with autistic disorders [10]. With new genetic insights offered by genome-wide association studies (GWAS) of autism, we can envision new medication development [11].
Families and the educational system are currently the main resources for treatment. Intensive, sustained special education programs and behavior therapy early in life can help children acquire self-care, social, and job skills, and often improve function and decrease symptom severity and maladaptive behaviors [12]. Available approaches include applied behavior analysis (ABA), developmental models, structured teaching, speech and language therapy, social skills therapy, and occupational therapy [13].
Genomic Studies
Large-scale collections of samples have been genotyped, including the Autism Genetic Resource Exchange (AGRE), Simons Simplex Collection (SSC), Autism Genome Project (AGP), Autism Case–control (ACC), and Autism Center of Excellence (ACE).
Evidence from different cases supports the understanding that chromosomal mutation contributes to autism susceptibility [14]. Linkage studies and cytogenetic analysis have led to identification of several novel candidate genes, including neurexins (NRXNs) and neuroligins (NLGNs). Significant genomic linkages have been reported on 2q31, 3q, 7q22, and 7q34 [15, 16].
Several regions of interest, including 1p13.2, 1q31.1, 5p13, 8q24, 13q, 15q, 16p, 17q, 19p, and Xq have been implicated by multiple studies. The majority of these regions, except 1p13.2 and 2q32, have been reported with chromosomal mutations [16].
Several genome-wide studies have been conducted to identify the genetic variants associated with risk for autism. Copy number variations (CNVs), including several large recurrent deletions or duplications have been found. The best established autism-associated CNVs include 7q11.23, 15q11–13, 16p11.2, and 22q11.2 loci, and NRXN1, CNTN4, NLGNs, and SHANK3 genes [17–19]. CNVs have proven a critical source of genetic burden contributing to the autism phenotype [18–22, 23••, 24, 25, 26••, 27–29].
Recently conducted exome sequencing studies suggest that hundreds of de novo mutations have some role in the development of autism, and clear evidence implicates a few specific genes (CHD8, KATANAL2, SCN2A, NTNG1) [30–33]. Together, these structural variants or de novo mutations, many of which are highly penetrant or protein-altering but individually rare, account for a limited proportion of the genetic risk for autism. In contrast, only a modest number of common variants have been reported at CDH9-CDH10, SEMA5A, and MACROD2 loci through GWAS.
Autism is marked with phenotypic heterogeneity and is etiologically multifactorial. The hypothesis that multiple common variants collectively, or through interaction with environmental factors, account for a certain proportion of risk for autism motivates the rationale for GWAS.
Major GWAS
Major GWAS of autism have been conducted (Tables 1 and 2).
The first successful GWAS was by Wang et al. [34] and surveyed 780 families (3,101 subjects) with affected children, a second cohort of 1,204 affected subjects, and 6,491 control subjects of European ancestry. Six single nucleotide polymorphisms (SNPs) between cadherin 10 (CDH10) and cadherin 9 (CDH9), two genes encoding neuronal cell-adhesion molecules, revealed strong association signals, with the most significant SNP being rs4307059 (P = 3.4 × 10−8, odds ratio 1.19). Top hits replicated in two independent cohorts, with combined P values ranging from 7.4 × 10−8 to 2.1 × 10−10.
Later, Kerin et al. [35] used a tiling array within 100-kb linkage disequilibrium (LD) of the GWAS-identified peak and examined relevant expressed sequence tags (ESTs) and RNA. They identified only one functional element, a single non-coding RNA that was found to correspond to the moesin pseudogene 1 (MSNP1). The 3.9 kb RNA had 94 % sequence identity with the mature messenger RNA (mRNA) protein-coding gene MSN, located on the X chromosome. Interestingly, the (noncoding) RNA at 5p14.1 was encoded by the opposite (antisense) strand of MSNP1, and was named moesin pseudogene 1 antisense (MSNP1AS).
Second, Ma et al. [36] used a discovery dataset of 438 autistic Caucasian families and the Illumina Human 1 M beadchip. A total of 96 SNPs demonstrated strong association with autism risk (P < 0.0001). Validation of the top 96 SNPs was performed using an independent dataset of 487 autism families of European ancestry and genotyped on the 550 K Illumina BeadChip. The same region on chromosome 5p14.1 as reported by Wang et al. showed significance in both the discovery and the validation datasets. Joint analysis of all SNPs in this region identified eight SNPs having improved P-values (3.24 × 10−4 to 3.40 × 10−6) than in either dataset alone.
Third, Weiss et al. [37] analyzed 1,031 multiplex autism families, including 1,553 affected children, and identified regions of suggestive and significant linkage on chromosomes 6q27 and 20p13, respectively. Initial analysis did not yield genome-wide significant associations. Genotyping of top hits in additional families revealed an SNP on chromosome 5p15 (between SEMA5A and TAS2R1) that was significantly associated with autism (P = 2 × 10−7).
Fourth, Anney et al. [38] found that rs4141463, located within MACROD2, surpassed the genome-wide association significance threshold of P < 5 × 10−8. When a smaller replication sample was analyzed, the risk allele at rs4141463 was again over-transmitted. However, the effect size was much smaller and, combined, barely met the P < 5 × 10−8 threshold. Exploratory analyses of phenotypic subtypes yielded no significant associations after correction for multiple testing. Best signals were within KIAA0564, PLD5, POU6F2, ST8SIA2, and TAF1C.
Fifth, Cho et al. [39] analyzed 42 Korean ASD patients genotyped with Affymetrix SNP Array 5.0 and detected candidate SNPs in chromosome 11, rs11212733 (P = 9.76 × 10−6) and rs7125479 (P = 1.48 × 10−4), as a marker of language delay in ASD using the transmission disequilibrium test and multifactor dimensionality reduction test.
Sixth, Anney et al. [40] found no single SNP showing significant association with ASD or selected phenotypes at a genome-wide level. The SNP that achieved the smallest P-value from secondary analyses was rs1718101, which falls in CNTNAP2, a gene previously implicated in susceptibility for ASD. rs1718101 also shows modest association with age of word/phrase acquisition in ASD subjects.
Seventh, Connolly et al. [41•] examined 2,165 participants (mean age 8.95 years) and examined associations between genomic loci and individual assessment items from the Autism Diagnostic Interview-Revised, Autism Diagnostic Observation Schedule, and Social Responsiveness Scale. Significant associations with a number of loci were identified, including KCND2 (P = 3.05 × 10−8, overly serious facial expressions), NOS2A (P = 8.12 × 10−7, loss of motor skills), and NELL1 (P = 2.91 × 10−7, faints, fits, or blackouts).
Eighth, Smoller et al. [42••] represents the Psychiatric Genomics Consortium, which analyzed multiple major psychiatric disorders including ASD, attention deficit–hyperactivity disorder, bipolar disorder, major depressive disorder, and schizophrenia. SNP data for the five disorders were analyzed in 33,332 cases and 27,888 controls of European ancestry. SNPs at four loci surpassed the cutoff for genome-wide significance (P < 5 × 10−8) in the primary analysis: regions on chromosomes 3p21 and 10q24, and SNPs within two L-type voltage-gated calcium channel subunits, CACNA1C and CACNB2.
In an earlier CNV association of schizophrenia, CACNA1B (P = 8.68 × 10−4) and DOC2A (P = 5.69 × 10−6), both calcium-signaling genes responsible for neuronal excitation, were deleted in 16 cases and duplicated in ten cases, respectively [43].
Most recently, Xia et al. [44••] utilized two Chinese cohorts as discovery (n = 2,150) and three data sets of European ancestry populations for replication. Meta-analysis identified three SNPs, rs936938 (P = 4.49 × 10−8), non-synonymous rs6537835 (P = 3.26 × 10−8) and rs1877455 (P = 8.70 × 10−8); and related haplotypes, AMPD1-NRAS-CSDE1, TRIM33, and TRIM33-BCAS2, all associated with autism. All were mapped to a linkage region (1p13.2) previously reported as associated with autism. TRIM33 is an E3 ubiquitin ligase, further supporting the role of ubiquitin mutations in autism.
Scope of Autism GWAS and Future
Effect size for singular common variants is small. Although high odds ratios can be detected by GWAS, few greater than 1.5 have been found. Therefore, widespread epistasis likely exists between multiple common variants with small effect sizes. So-called unaffected relatives may have a sub-threshold genetic burden of variants; thus, we need better endophenotypes and intermediate phenotypes classification. Epistatic interactions are important biologically but computationally intractable with current methods.
Postsynaptic components of excitatory synapses and L-type calcium channels have been major biological categories emerging from neuropsychiatric GWAS. Specifically, NRXN, CNTN, GRM, ubiquitin, and SLIT gene members show multiple gene family members with variants associated with autism and related neurodevelopmental disorders.
Larger population samples are required for dissecting the genetics of psychiatric disease compared with other common diseases, most likely due to the complex genetic architecture of psychiatric disorders. Genetic risk is the culmination of many different genes, within individuals and across families, and both rare and common variants working in concert, making psychiatric disorders polygenic.
Progress is being achieved in the GWAS of autism due to improved understanding of the human genome sequence and the tremendous variation among individuals. Functional variants abound in the genome and provide the mode for polygenic inheritance. Technical innovation of genomic assays is also accelerating progress. The large-scale sequencing effort of the 1000 Genomes Project informs new content for SNP microarrays, which can be affordably genotyped across large populations to gain necessary statistical power to meet the multiple testing correction bar of 5 × 10−8. The large-scale worldwide collaborations are vital to enable sharing of data to allow for meta-analysis and mega-analysis.
Many common variants identified by GWAS have modest effects. Specifically, they act as a genetic risk factor within a genetic background of other variants, increasing risk or protection against autism, and acting in a range of magnitudes on the protein sequence or the expression levels[45]. Klei et al. [46] used simulations to examine the additive model of autism susceptibility, whereby autism is underscored by a large number of variants that cumulatively increase ASD risk, but individually have very small effects. Their model suggests that, in multiplex families, approximately 60 % of susceptibility is attributable to additive effects, whereas, in simplex families, the proportion is closer to 40 %.
We can learn some lessons from syndromic forms of autism, but the large-effect singular variants may radically disrupt brain development and function, such that the psychiatric phenotype is obfuscated. Risk variants typically represent more mild tissue-specific alterations in expression rather than knockouts in severe syndromes.
Ubiquitin
TRIM33, an E3 ubiquitin ligase, is the latest significant GWAS hit in autism. Mutations in ubiquitin E3 ligases are reported as associated with other neuropsychiatric disorders, such as Angelman syndrome, which overlaps phenotypically with autism (UBE3A);[47] Charcot–Marie–Tooth disease (LRSAM1);[48] juvenile-onset Parkinson’s disease (PARK2);[49] and X-linked mental retardation (CUL4B)[50, 51]. Also, CNVs not found in controls affecting ubiquitin pathways, including UBE3A, PARK2, RFWD2, and FBXO40 [18], are enriched in autism patients. The SNP in TRIM33, rs6537825, which displays the strongest association with autism, is nonsynonymous, causes an isoleucine-to-threonine substitution, and has a significant cis-acting regulatory effect on TRIM33 expression in the brain. Functional polymorphisms may slightly alter the function of the ubiquitin-proteasome system, decreasing its efficiency. TRIM33 was differentially expressed in human brains between autism patients and normal subjects, and was reported as one of the top loci associated with differential expressions across the whole genome in an independent sample of post-mortem human brains [52]. TRIM32/ASTN2[53] was found disrupted in males with autism and associated conditions.
Each human genome contains its own unique combinations of alleles that give risk to a unique finely interwoven phenotype. There is a limit of isogenic backgrounds and controlled environments to gain clear specific molecular and cellular phenotypes from certain alleles. The technology of precision genome editing within cell lines or model organism also needs development in order to assess precise variant effects.
The tagging SNPs associated in GWAS implicate a haplotype, but not the specific alleles therein that give rise to the phenotype of autism. Low-frequency alleles may segregate on the haplotype, or multiple genetic effects may exist in the same locus.
The GWAS result typically resides on tissue-specific enhancers distal from the protein-altering region. Sometimes, the effect is not a dysfunctional protein product, but rather a quantitative variation in expression or the cell type-specific expression pattern. Noncoding regions show less evolutionary conservation, complicating their detection. Epigenetics is also proceeding to help the scientific community understand the chromatin states in diverse cell types.
Rare Variants
Rare variants are likely very important, as the alleles that strongly predispose individuals to autism are likely kept at rare frequencies by purifying selection. There are many variants that alter or terminate protein sequence on the order of thousands per genome, complicating assignment of causality to certain variants. Rare variants may have only partial penetrance to the autism phenotype, resulting in subclinical impact on Intellectual Quotient or cognition, and contaminating control populations that may not be as carefully screened as case populations for GWAS[54•].
De novo variants also play a strong role, although they are exceedingly rare to find recurrently and must be placed in context of the gene-based mutation rate.
Common and rare alleles from autism GWAS have limited penetrance, act in concert with multiple other alleles in a genetic background, and are pleiotropic by contributing to multiple phenotypes.
Conclusions
We need progress from the narrow pathophysiological hypotheses from small, disconnected pieces of literature into a more holistic and systematic view of information to derive insights into neurobiological systems [55•].
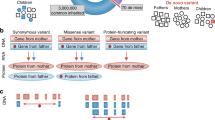
We seek common biological themes and pathways to converge on successful therapeutic targets (Fig. 1) [56]. Autdb (gene.sfari.org/autdb/) keeps a database of CNV reports associated with autism. Major recent CNV GWAS are listed in Table 3.

Genome-wide association studies (GWAS) of autism studies results. (a) STRING[57] protein–protein interaction network of genome-wide significant GWAS genes; (b) the complex puzzle of autism is partly assembled based on GWAS; (c) count copy number variation (CNV) associations made at each genomic locus. Red indicates deletions, blue indicates duplications, and black indicates both deletions and duplications
The unbiased genome-wide search for genetic variants predisposing individuals to autism still remains the mainstay approach to uncover relevant biology. However, we must embrace the reality that hundreds of modest, common, and rare/strong impact/partially penetrant variants abound in each genome.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome Biol. 2012;13:247.
Saemundsen E, Magnússon P, Georgsdóttir I, Egilsson E, Rafnsson V. Prevalence of autism spectrum disorders in an Icelandic birth cohort. BMJ open. 2013;3(6):e002748.
Idring S, Rai D, Dal H, Dalman C, Sturm H, Zander E, et al. Autism spectrum disorders in the Stockholm Youth Cohort: design, prevalence and validity. PLoS ONE. 2012;7:e41280.
Kim YS, Leventhal BL, Koh Y-J, Fombonne E, Laska E, Lim E-C, et al. Prevalence of autism spectrum disorders in a total population sample. Am J Psychiatr. 2011;168:904–12.
Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L, et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics. 2011;128:e488–95.
Blumberg, S.J., Bramlett, M.D., Kogan, M.D., Schieve, L.A., Jones, J.R., and Lu, M.C. (2013). Changes in prevalence of parent-reported autism spectrum disorder in school-aged U.S. children: 2007 to 2011–2012. Natl Health Stat Report, 1–11, 11 p following 11.
Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–102.
Rosenberg RE, Law JK, Yenokyan G, McGready J, Kaufmann WE, Law PA. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med. 2009;163:907–14.
Hallmayer J, Glasson EJ, Bower C, Petterson B, Croen L, Grether J, et al. On the twin risk in autism. Am J Hum Genet. 2002;71:941–6.
Oswald DP, Sonenklar NA. Medication use among children with autism spectrum disorders. J Child Adolesc Psychopharmacol. 2007;17:348–55.
Ghosh A, Michalon A, Lindemann L, Fontoura P, Santarelli L. Drug discovery for autism spectrum disorder: challenges and opportunities. Nat Rev Drug Discov. 2013;12:777–90.
Myers SM, Johnson CP. Management of children with autism spectrum disorders. Pediatrics. 2007;120:1162–82.
Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383:896–910.
Veenstra-VanderWeele J, Cook Jr EH. Molecular genetics of autism spectrum disorder. Mol Psychiatry. 2004;9:819–32.
Gupta AR, State MW. Recent advances in the genetics of autism. Biol Psychiatry. 2007;61:429–37.
Veenstra-Vanderweele J, Christian SL, Cook Jr EH. Autism as a paradigmatic complex genetic disorder. Annu Rev Genomics Hum Genet. 2004;5:379–405.
Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–7.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–73.
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–85.
Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5:e1000536.
Elia J, Glessner JT, Wang K, Takahashi N, Shtir CJ, Hadley D, et al. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet. 2012;44:78–84.
Gai X, Xie HM, Perin JC, Takahashi N, Murphy K, Wenocur AS, et al. Rare structural variation of synapse and neurotransmission genes in autism. Mol Psychiatry. 2012;17:402–11.
Girirajan S, Dennis MY, Baker C, Malig M, Coe BP, Campbell CD, et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Genet. 2013;92:221–37. A recent major study of CNVs in autism.
Glessner JT, Connolly JJ, Hakonarson H. Rare genomic deletions and duplications and their role in neurodevelopmental disorders. Curr Top Behav Neurosci. 2012;12:345–60.
Glessner JT, Li J, Hakonarson H. ParseCNV integrative copy number variation association software with quality tracking. Nucleic Acids Res. 2013;41:e64.
Hadley D, Wu ZL, Kao C, Kini A, Mohamed-Hadley A, Thomas K, et al. The impact of the metabotropic glutamate receptor and other gene family interaction networks on autism. Nat Commun. 2014;5:4074. Shows metabatropic glutamate receptors are important across the diagnostic psychatric boundaries.
Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–97.
Matsunami N, Hadley D, Hensel CH, Christensen GB, Kim C, Frackelton E, et al. Identification of rare recurrent copy number variants in high-risk autism families and their prevalence in a large ASD population. PLoS ONE. 2013;8:e52239.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72.
Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–5.
O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–50.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–41.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99.
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–33.
Kerin T, Ramanathan A, Rivas K, Grepo N, Coetzee GA, Campbell DB. A noncoding RNA antisense to moesin at 5p14.1 in autism. Sci Transl Med. 2012;4:128ra140.
Ma D, Salyakina D, Jaworski JM, Konidari I, Whitehead PL, Andersen AN, et al. A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann Hum Genet. 2009;73:263–73.
Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–8.
Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR, et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19:4072–82.
Cho SC, Yoo HJ, Park M, Cho IH, Kim BN, Kim JW, et al. Genome-wide association scan of korean autism spectrum disorders with language delay: a preliminary study. Psychiatry Investig. 2011;8:61–6.
Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, et al. Individual common variants exert weak effects on the risk for autism spectrum disorderspi. Hum Mol Genet. 2012;21:4781–92.
Connolly JJ, Glessner JT, Hakonarson H. A genome-wide association study of autism incorporating autism diagnostic interview-revised, autism diagnostic observation schedule, and social responsiveness scale. Child Dev. 2013;84:17–33. Autism subphenotypes are assessed using GWAS to dissect the variability in constellations of endophenotypes in the autism spectrum.
Smoller JW, Ripke S, Lee PH, Neale B, Nurnberger JI, Santangelo S, et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–9. GWAS meta-analysis of five major psychiatric disorders shows the genetic etiology of childhood psychiatric disease may overlap, as well as the promises and challenges of worldwide consortium.
Glessner JT, Reilly MP, Kim CE, Takahashi N, Albano A, Hou C, et al. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc Natl Acad Sci U S A. 2010;107:10584–9.
Xia, K., Guo, H., Hu, Z., Xun, G., Zuo, L., Peng, Y., Wang, K., He, Y., Xiong, Z., Sun, L., et al. (2013). Common genetic variants on 1p13.2 associate with risk of autism. Mol Psychiatry. TRIM33 is found in a meta-analysis including Chinese and European populations, further strengthening the role of E3 ubiquitin ligase in autism.
Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–55.
Klei, L., Sanders, S.J., Murtha, M.T., Hus, V., Lowe, J.K., Willsey, A.J., et al. (2012). Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism 3
Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–3.
Guernsey DL, Jiang H, Bedard K, Evans SC, Ferguson M, Matsuoka M, et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-Tooth disease. PLoS Genet. 2010;6.
Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet. 1999;8:567–74.
Tarpey PS, Raymond FL, O'Meara S, Edkins S, Teague J, Butler A, et al. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet. 2007;80:345–52.
Zou Y, Liu Q, Chen B, Zhang X, Guo C, Zhou H, et al. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet. 2007;80:561–6.
Chow ML, Pramparo T, Winn ME, Barnes CC, Li HR, Weiss L, et al. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet. 2012;8:e1002592.
Lionel AC, Tammimies K, Vaags AK, Rosenfeld JA, Ahn JW, Merico D, et al. Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum Mol Genet. 2014;23:2752–68.
McCarroll SA, Feng G, Hyman SE. Genome-scale neurogenetics: methodology and meaning. Nat Neurosci. 2014;17:756–63. A perspective on linkage, candidate gene, common/rare base and CNVs in autism and integrative neurogenetics.
Hu VW. From genes to environment: using integrative genomics to build a “systems-level” understanding of autism spectrum disorders. Child Dev. 2013;84:89–103. Integrative genomics gives a system-level view of autism biology and utlizes to a greater extent the GWAS SNP array data over single marker test statistics.
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94:677–94.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–15.
Pham J, Shaw C, Pursley A, Hixson P, Sampath S, Roney E, et al. Somatic mosaicism detected by exon-targeted, high-resolution aCGH in 10 362 consecutive cases. Eur J Hum Genet. 2014;22:969–78.
Correia CT, Conceicao IC, Oliveira B, Coelho J, Sousa I, Sequeira AF, et al. Recurrent duplications of the annexin A1 gene (ANXA1) in autism spectrum disorders. Mol Autism. 2014;5:28.
Karayannis T, Au E, Patel JC, Kruglikov I, Markx S, Delorme R, et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature. 2014;511:236–40.
Nguyen LS, Kim HG, Rosenfeld JA, Shen Y, Gusella JF, Lacassie Y, et al. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum Mol Genet. 2013;22:1816–25.
Walker S, Scherer SW. Identification of candidate intergenic risk loci in autism spectrum disorder. BMC Genomics. 2013;14:499.
Nagamani SC, Erez A, Ben-Zeev B, Frydman M, Winter S, Zeller R, et al. Detection of copy-number variation in AUTS2 gene by targeted exonic array CGH in patients with developmental delay and autistic spectrum disorders. Eur J Hum Genet. 2013;21:343–6.
Lionel AC, Vaags AK, Sato D, Gazzellone MJ, Mitchell EB, Chen HY, et al. Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum Mol Genet. 2013;22:2055–66.
Prasad A, Merico D, Thiruvahindrapuram B, Wei J, Lionel AC, Sato D, et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda). 2012;2:1665–85.
Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, Chung RH, et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet. 2012;21:3513–23.
Olson HE, Shen Y, Poduri A, Gorman MP, Dies KA, Robbins M, et al. Micro-duplications of 1q32.1 associated with neurodevelopmental delay. Eur J Med Genet. 2012;55:145–50.
Vaags AK, Lionel AC, Sato D, Goodenberger M, Stein QP, Curran S, et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am J Hum Genet. 2012;90:133–41.
Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–7.
Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S, et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am J Hum Genet. 2012;90:879–87.
Chen YZ, Matsushita M, Girirajan S, Lisowski M, Sun E, Sul Y, et al. Evidence for involvement of GNB1L in autism. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:61–71.
Acknowledgments
The work was funded in part by an Institutional Development Award to the Center for Applied Genomics from The Children’s Hospital of Philadelphia, Adele and Daniel Kubert donation, University of Pennsylvania Biomedical Graduate Studies training grant, the Cotswold foundation, and by 1R01MH097284-01 from the National Institute of Mental Health (NIMH).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
The authors have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Glessner, J.T., Connolly, J.J. & Hakonarson, H. Genome-Wide Association Studies of Autism. Curr Behav Neurosci Rep 1, 234–241 (2014). https://doi.org/10.1007/s40473-014-0023-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40473-014-0023-0